

Abstract
Aims
The pharmacokinetic (PK) similarity between PF-05280014, a proposed trastuzumab biosimilar, trastuzumab sourced from European Union (trastuzumab-EU) or from United States (trastuzumab-US) was evaluated. Safety and immunogenicity were also assessed.
Methods
In this phase 1, double-blind trial (NCT01603264), 105 healthy male volunteers were randomized 1:1:1 to receive a single 6 mg kg−1 intravenous dose of PF-05280014, trastuzumab-EU, or trastuzumab-US, and evaluated for 70 days. Drug concentration–time data were analyzed by non-compartmental methods. PK similarity for the comparisons of PF-05280014 to each of trastuzumab-EU and trastuzumab-US, and trastuzumab-EU to trastuzumab-US were determined using the standard 80.00% to 125.00% bioequivalence criteria.
Results
Baseline demographics for the 101 subjects evaluable for PK were similar across all arms. The three products exhibited similar PK profiles with target-mediated disposition. The 90% CIs for the ratios of Cmax, 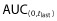 and AUC(0,∞) were within 80.00% to 125.00% for all three pairwise comparisons. Adverse events (AEs) were similar across all arms with treatment-related AEs reported by 71.4%, 68.6% and 65.7% subjects in the PF-05280014, trastuzumab-EU, and trastuzumab-US arms, respectively. The most common AEs were infusion-related reactions, headache, chills, pyrexia and nausea. The AE term ‘pyrexia’ was numerically greater in the PF-05280014 arm. All post-dose samples, except 1, tested negative for anti-drug antibodies (ADA).
and AUC(0,∞) were within 80.00% to 125.00% for all three pairwise comparisons. Adverse events (AEs) were similar across all arms with treatment-related AEs reported by 71.4%, 68.6% and 65.7% subjects in the PF-05280014, trastuzumab-EU, and trastuzumab-US arms, respectively. The most common AEs were infusion-related reactions, headache, chills, pyrexia and nausea. The AE term ‘pyrexia’ was numerically greater in the PF-05280014 arm. All post-dose samples, except 1, tested negative for anti-drug antibodies (ADA).
Conclusions
This study demonstrates PK similarity among PF-05280014, trastuzumab-EU and trastuzumab-US. The safety and immunogenicity profiles observed for the three products in this study are consistent with previous reports for trastuzumab.
Keywords: biosimilar, immunogenicity, PF-05280014, pharmacokinetics, safety, trastuzumab
What Is Already Known about This Subject
Trastuzumab, a humanized recombinant monoclonal antibody, targets HER2.
It is approved for treatment of HER2-overexpressing breast and gastric cancers.
What This Study Adds
This is the first clinical report of a potential trastuzumab biosimilar, PF-05280014.
These data demonstrate pharmacokinetic similarity between PF-05280014 and trastuzumab sourced from the US and EU.
These data also demonstrate the low immunogenicity of PF-05280014.
Introduction
A biosimilar is a biologic product that is highly similar to an approved biologic drug with demonstrated similarity to the reference product in key parameters such as structural identity, quality, pharmacokinetics (PK), immunogenicity, efficacy and safety 1–3. Demonstrating similarity in PK is generally the first step in the clinical assessment of biosimilarity.
Trastuzumab (Herceptin®, Genentech, San Francisco, CA, USA) is a humanized recombinant monoclonal antibody that selectively inhibits signalling via human epidermal growth factor receptor 2 (HER2) 4. Although its mechanism of action is still not fully understood 4,5, trastuzumab is known to block intracellular signalling, arrest the cell cycle in G1, induce antibody-dependent cellular cytotoxicity, induce apoptosis in human tumour cell lines overexpressing HER2 and reduce the size of tumour xenograft implants in athymic mice 6–8. It is approved for the treatment of HER2-overexpressing breast and gastric cancers 9.
PF-05280014 is being developed as a potential biosimilar of trastuzumab. Like trastuzumab, PF-05280014 is a recombinant humanized IgG1 monoclonal antibody directed against the extracellular domain of the HER2 receptor. It has an identical primary amino acid sequence to the trastuzumab products marketed in and sourced from the European Union (trastuzumab-EU) and United States (trastuzumab-US), and the same characteristics as these compounds with respect to in vitro binding assays and biological functional assays (Hurst et al., manuscript in preparation).
In this article, we report on the results of the first-in-human study designed to evaluate PK, safety and immunogenicity of PF-05280014, trastuzumab-EU and trastuzumab-US in healthy subjects.
Methods
Study population and design
A total of 105 healthy male volunteers 18–55 years old were enrolled in this phase 1 double-blind randomized trial (ClinicalTrials.gov #NCT01603264) conducted in accordance with the Declaration of Helsinki as well as applicable local laws and regulatory requirements. All subjects provided informed consent. The trial was limited to healthy male volunteers to control variability.
To be included in the trial, subjects needed to have a body mass index of 17.5–30.5 kg m−2, a total body weight >50 kg and left ventricular ejection fraction (LVEF) within the normal range as measured by an echocardiogram within 8 weeks of randomization. Key exclusion criteria included history of significant clinical disease or evidence thereof at screening, previous history of cancer other than adequately treated basal cell or squamous cell carcinoma of the skin, hypertension, autoimmune disorders, previous exposure to monoclonal antibodies or current use of biologic drugs. The sample size of 105 subjects was estimated to ensure that at least 93 subjects (31 subjects per group) would complete the PK assessment. With 31 subjects per group, the study was expected to provide at least 81% power for showing PK similarity simultaneously for all the three pair-wise comparisons.
The enrolled subjects were randomized 1:1:1 to receive a single 6 mg kg−1 dose of PF-05280014, trastuzumab-EU or trastuzumab-US via intravenous infusion (Figure 1). Infusions were required to be completed at approximately 90 min, but not exceeding 3 h. All assessments were conducted for 70 days.
Figure 1.
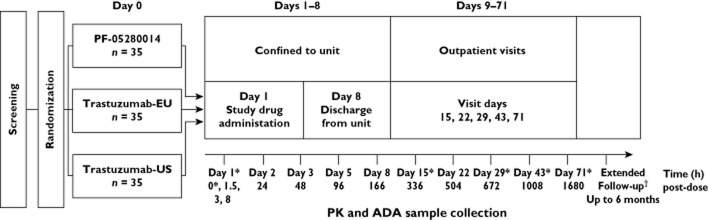
Study design. *Time/day on which samples were collected for ADA; samples for PK were collected at all times shown. †Subjects having an unresolved adverse event that was possibly related to ADA formation were asked to return for ADA and drug concentration blood sampling at up to 3 month intervals until the adverse event or its sequelae resolved or stabilized at a level acceptable to the investigator and the sponsor concurs with the investigator's assessment, up to 6 months from the visit on day 71 or the day of early withdrawal. ADA, anti-drug antibodies; PK, pharmacokinetics
The primary objective of this trial was to demonstrate the PK similarity of PF-05280014 to both trastuzumab-EU and trastuzumab-US, and of trastuzumab-EU to trastuzumab-US. Secondary objectives included assessing safety, tolerability and immunogenicity.
Pharmacokinetic evaluations
Samples for PK evaluation were collected at 0 (pre-dose), 1.5 (immediately before the end of infusion), 3, 8, 24, 48, 96, 168, 336, 504, 672, 1008 and 1680 h after the start of infusion and stored at −70°C until assayed. The samples were analyzed using a validated enzyme-linked immunosorbent assay. The lower limit of quantitation of the assay was 0.500 μg ml−1. The inter-run assay accuracy, expressed as percent relative error for quality control samples, ranged from −4.7% to 3.3%. The assay precision, expressed as the inter-run coefficients of variation for quality control samples, was less than 9.4%.
The drug concentration–time data were analyzed by standard non-compartmental methods 10, using internally validated software, eNCA (v2.2.3). Samples below the lower limit of quantitation were included in the analysis as zero unless otherwise specified. Actual sample collection times were used for the analysis. PK parameters estimated from the data analysis included the maximum serum drug concentration (Cmax), area under the serum concentration–time profile (AUC) from time zero to the last time point with quantifiable concentration (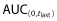 ) and extrapolated to infinite time (AUC(0,∞)), clearance (CL), volume of distribution at steady-state (Vss) and terminal half-life (t1/2).
) and extrapolated to infinite time (AUC(0,∞)), clearance (CL), volume of distribution at steady-state (Vss) and terminal half-life (t1/2).
Statistical analysis consisted of one way analyses of variance comparing the natural log transformed PK parameter for each pair-wise comparison: PF-05280014 vs. trastuzumab-EU, PF-05280014 vs. trastuzumab-US and trastuzumab-EU vs. trastuzumab-US. Estimates of adjusted mean difference and the confidence intervals (CIs) for the differences obtained from the model were exponentiated to provide estimates of the ratio of adjusted geometric means and the CIs for the ratios. PK similarity was considered demonstrated if 90% CIs for the test: reference ratios in AUC(0,t) and Cmax were within the 80.00% to 125.00% bioequivalence acceptance window.
Safety evaluations
The type, incidence, timing, seriousness and relatedness of adverse events (AEs) were documented. Severity was graded in accordance with the National Cancer Institute Common Terminology Criteria for Adverse Events (v4.03) (NCI CTCAE) 11. The Medical Dictionary for Regulatory Activities (version 15.1) was used to code AEs including serious AEs 12. Clinical parameters included infusion-related reactions (IRRs), laboratory analyses, vital signs, electrocardiograms (ECGs) and LVEF assessments.
Immunogenicity evaluations
Immunogenicity of the administered product was evaluated by measuring the incidence of antidrug antibodies (ADA) and neutralizing antibodies (NAb). Serum samples for detecting ADA and NAb were collected at 0 (day 1 pre-dose), 336, 672, 1008 and 1680 h following dose administration. ADA samples were analyzed using validated electrochemiluminescent (ECL) immunoassays by Intertek Pharmaceutical Services (San Diego, CA, USA) 13. Two parallel ECL assays were developed to detect ADA, one to detect antibodies against PF-05280014 and one to detect antibodies against trastuzumab. Both assays showed similar sensitivity, precision, accuracy and drug tolerance during the validation. Samples were first tested for ADA using the assay specific to the product administered. Confirmed positive samples were then assessed for cross-reactivity using the alternate assay.
Samples confirmed to be positive for ADA were further tested for NAb using a validated competitive ligand-binding assay 13. Two assays with the same ECL platform were developed and validated to detect NAb against PF-05280014 and against trastuzumab, respectively. The two assays showed similar performance with the positive and negative controls during validation.
Results
Subject disposition
All 105 subjects were male and received the assigned study product. Of the 105 subjects, 101 completed the study per protocol (Figure 2) and were included in the PK analyses. Four subjects were excluded, one from the PF-05280014 arm and three from the trastuzumab-US arm. The subject from the PF-05280014 arm and one subject from the trastuzumab-US arm were excluded for incomplete dosing due to IRR and infusion not being completed within the 3 h limit. One subject from the trastuzumab-US arm was excluded for infusion site infiltration and error in manual programming of the infusion pump. The last subject, from the trastuzumab-US arm, was excluded for infusion site extravasation. The baseline demographic characteristics were similar among the three arms of PK-eligible subjects (per protocol population; Table 1). Data from all 105 subjects were evaluated for safety and immunogenicity.
Figure 2.
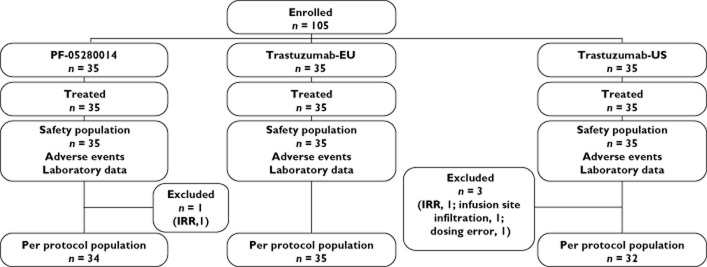
Subject disposition. IRR, incomplete dosing due to infusion-related reaction
Table 1.
Baseline demographics of enrolled subjects eligible for pharmacokinetic similarity assessment
| PF-05280014 | Trastuzumab-EU | Trastuzumab-US | |
|---|---|---|---|
| Number of subjects | 34 | 35 | 32 |
| Mean age ± SD (years) | 34.5 ± 10.7 | 36.1 ± 9.5 | 35.3 ± 9.2 |
| Race | |||
| White | 13 | 7 | 8 |
| Black | 14 | 22 | 15 |
| Other | 7 | 6 | 9 |
| Mean weight ± SD (kg) | 79.7 ± 11.2 | 86.3 ± 11.3 | 81.4 ± 11.0 |
| Mean height ± SD (cm) | 175.9 ± 6.7 | 177.8 ± 8.0 | 177.3 ± 7.4 |
| Mean BMI ± SD (kg m−2) | 25.8 ± 3.1 | 27.2 ± 2.6 | 25.9 ± 3.0 |
BMI, body mass index [weight (0.01 height)−2; with weight in kg and height in cm]; SD, standard deviation.
Pharmacokinetic evaluations
Of the 101 subjects included in the PK evaluation, 34 subjects received PF-05280014, 35 subjects received trastuzumab-EU and 32 subjects received trastuzumab-US (Figure 2). Evaluation of individual subject serum concentration–time data showed that the three products administered exhibited similar profiles (Figure 3A–C). The mean serum concentration–time profiles for the three products were superimposable (Figure 3D). There was a rapid decrease in the serum concentrations of each product at the end of the infusion, reflecting the drug distribution into the extravascular space. There was a slow elimination phase with an accelerated decline of serum concentration of the administered product towards the latter part, possibly due to the target-mediated disposition.
Figure 3.
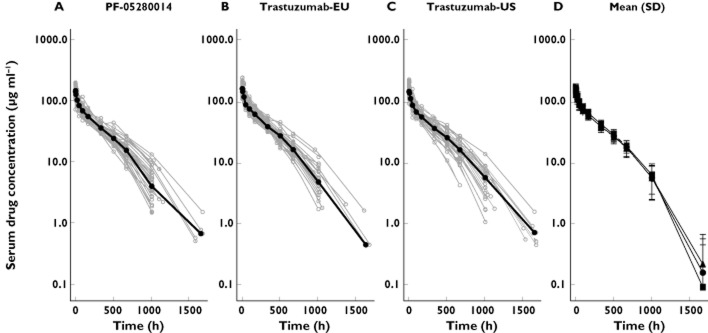
Individual (A–C) and mean ± SD (D) serum concentration–time profiles of PF-05280014, trastuzumab-EU and trastuzumab-US following a single dose of 6 mg kg−1 in healthy subjects. SD, standard deviation. A, B and C:  , median;
, median;  , individual; D: •, PF-02580014; ▪, trastuzumab-EU; ▴, trastuzumab-US
, individual; D: •, PF-02580014; ▪, trastuzumab-EU; ▴, trastuzumab-US
The PK parameters evaluated (Cmax, 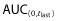 , AUC(0,∞), CL, Vss and t1/2) were similar in both the mean values and the inter-subject variability across all treatment arms (Table 2, Figure 4). The coefficients of variation were 16–19% for Cmax, 17–19% for
, AUC(0,∞), CL, Vss and t1/2) were similar in both the mean values and the inter-subject variability across all treatment arms (Table 2, Figure 4). The coefficients of variation were 16–19% for Cmax, 17–19% for 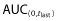 and 17–18% for AUC(0,∞) for the three administered products. The 90% CIs for the ratios of PF-05280014 to either trastuzumab-EU or trastuzumab-US for Cmax,
and 17–18% for AUC(0,∞) for the three administered products. The 90% CIs for the ratios of PF-05280014 to either trastuzumab-EU or trastuzumab-US for Cmax, 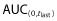 and AUC(0,∞) were within the bioequivalence window of 80.00% to 125.00% (Table 3). Furthermore, the 90% CIs of the ratios for Cmax,
and AUC(0,∞) were within the bioequivalence window of 80.00% to 125.00% (Table 3). Furthermore, the 90% CIs of the ratios for Cmax, 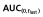 and AUC(0,∞) for the comparison of trastuzumab-EU to trastuzumab-US were also within the bioequivalence window of 80.00% to 125.00% (Table 3).
and AUC(0,∞) for the comparison of trastuzumab-EU to trastuzumab-US were also within the bioequivalence window of 80.00% to 125.00% (Table 3).
Table 2.
Mean ± SD pharmacokinetic parameter estimates
| PF-05280014 | Trastuzumab-EU | Trastuzumab-US | |
|---|---|---|---|
| Subjects (n) | 34 | 35 | 32 |
| Cmax (μg ml−1) | 159 ± 26 | 174 ± 31 | 164 ± 31 |
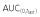 (μg ml−1 h)*
(μg ml−1 h)*
|
35700 ± 6287 | 38510 ± 6569 | 35870 ± 6878 |
| AUC(0,∞) (μg ml−1 h) | 37130 ± 6305 | 40330 ± 6994 | 37310 ± 6728 |
| CL (ml h−1 kg−1) | 0.166 ± 0.026 | 0.153 ± 0.025 | 0.166 ± 0.032 |
| Vss (ml kg−1) | 56.1 ± 8.2 | 51.7 ± 6.9 | 55.7 ± 8.8 |
| t1/2 (h) | 213 ± 42 | 220 ± 42 | 212 ± 47 |
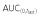 was ≥80% of corresponding AUC(0,∞) in all PK eligible subjects.
was ≥80% of corresponding AUC(0,∞) in all PK eligible subjects. 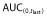 , area under the concentration–time curve from time 0 to last time point of measurable concentration; AUC(0,∞), area under the concentration–time curve from time 0 extrapolated to infinite time; CL, clearance; Cmax, maximum drug concentration; SD, standard deviation; t1/2, terminal half-life; Vss, volume of distribution at steady-state.
, area under the concentration–time curve from time 0 to last time point of measurable concentration; AUC(0,∞), area under the concentration–time curve from time 0 extrapolated to infinite time; CL, clearance; Cmax, maximum drug concentration; SD, standard deviation; t1/2, terminal half-life; Vss, volume of distribution at steady-state.
Figure 4.
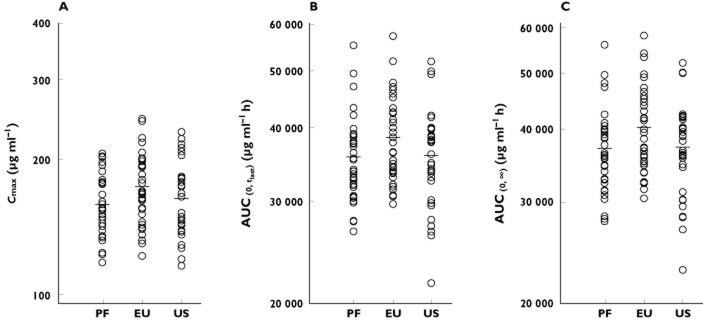
Individual and mean estimates of (A) Cmax, (B) 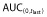 and (C) AUC(0,∞) of PF-05280014, trastuzumab-EU and trastuzumab-US. AUC(0,∞), area under the concentration–time curve from time 0 extrapolated to infinite time;
and (C) AUC(0,∞) of PF-05280014, trastuzumab-EU and trastuzumab-US. AUC(0,∞), area under the concentration–time curve from time 0 extrapolated to infinite time; 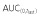 , area under the concentration-time curve from time 0 to last time point of measurable concentration; Cmax, maximum drug concentration; EU, trastuzumab EU; PF, PF-05280014; US, trastuzumab-US
, area under the concentration-time curve from time 0 to last time point of measurable concentration; Cmax, maximum drug concentration; EU, trastuzumab EU; PF, PF-05280014; US, trastuzumab-US
Table 3.
Statistical comparison of pharmacokinetic exposure parameters between test and reference products
| Test | Reference | Parameter* | Geometric mean | Ratio (%)‡ | 90% CI (%) | |
|---|---|---|---|---|---|---|
| Test | Reference | |||||
| PF-05280014 | Trastuzumab-EU | Cmax | 157 | 171 | 91.49 | 85.32, 98.09 |
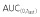 |
35210 | 38000 | 92.66 | 86.44, 99.34 | ||
| AUC(0,∞) | 36650 | 39770 | 92.15 | 86.03, 98.69 | ||
| PF-05280014 | Trastuzumab-US | Cmax | 157 | 161 | 97.41 | 90.71, 104.62 |
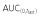 |
35210 | 35230 | 99.94 | 93.08, 107.31 | ||
| AUC(0,∞) | 36650 | 36710 | 99.83 | 93.06, 107.09 | ||
| Trastuzumab-EU | Trastuzumab-US | Cmax | 171 | 161 | 106.48 | 99.20, 114.30 |
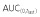 |
38000 | 35230 | 107.85 | 100.50, 115.75 | ||
| AUC(0,∞) | 39770 | 36710 | 108.34 | 101.05, 116.16 | ||
Cmax, 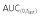 and AUC(0,∞) were in units of μg ml−1, μg ml−1 h and μg ml−1 h, respectively.
and AUC(0,∞) were in units of μg ml−1, μg ml−1 h and μg ml−1 h, respectively.
Ratio of adjusted geometric means.  , area under the concentration–time curve from time 0 to last time point of measurable concentration; AUC(0,∞), area under the concentration–time curve from time 0 extrapolated to infinite time; CI, confidence interval; Cmax, maximum drug concentration.
, area under the concentration–time curve from time 0 to last time point of measurable concentration; AUC(0,∞), area under the concentration–time curve from time 0 extrapolated to infinite time; CI, confidence interval; Cmax, maximum drug concentration.
Safety evaluations
Data from all 105 subjects enrolled in the study were evaluated for safety. Treatment-emergent AEs (TEAEs) were reported by 89 (81.9%) subjects of which 28 (80.0%), 29 (82.9%) and 29 (82.9%) subjects were from the PF-05280014, trastuzumab-EU and trastuzumab-US treatment arms, respectively. Treatment-related AEs (TRAEs) were reported by 72 (68.6%) subjects, of which 25 (71.4%), 24 (68.6%) and 23 (65.7%) subjects were from the PF-05280014, trastuzumab-EU and trastuzumab-US treatment arms, respectively (Table 4). There were no serious AEs, deaths or discontinuation due to an AE. The majority of AEs were mild. Grade 1 TEAEs were reported by 20, 21 and 24 subjects from the PF-05280014, trastuzumab-EU and trastuzumab-US treatment arms, respectively. Grade 1 TRAEs were reported by 20, 21 and 19 subjects from the PF-05280014, trastuzumab-EU and trastuzumab-US treatment arms, respectively. A total of 19 subjects reported grade 2 TEAEs (eight, six and five subjects from the PF-05280014, trastuzumab-EU and trastuzumab-US treatment arms, respectively) and 11 subjects reported grade 2 TRAEs (five, two and four subjects from the PF-05280014, trastuzumab-EU and trastuzumab-US treatment arms, respectively). There were only two subjects with grade 3 laboratory abnormality AEs, both in the trastuzumab-EU arm. One of these subjects experienced treatment-related elevated transaminase and the other experienced unrelated creatine kinase increase. Both events resolved without sequelae or intervention. There were no AEs of grade 4 or higher grade severity.
Table 4.
Summary of treatment-related adverse events
| PF-05280014 | Trastuzumab-EU | Trastuzumab-US | |
|---|---|---|---|
| Subjects (n) | 35 | 35 | 35 |
| Number of AEs reported | 93 | 85 | 95 |
| Subjects with AEs | 25 | 24 | 23 |
| Subjects with SAEs | 0 | 0 | 0 |
| Subjects with AEs by grade | |||
| Grade 1 | 20 | 21 | 19 |
| Grade 2 | 5 | 2 | 4 |
| Grade 3 | 0 | 1 | 0 |
| ≥Grade 4 | 0 | 0 | 0 |
| IRR | 13 | 10 | 7 |
| Subjects with permanent discontinuation due to AEs | 1 | 0 | 1 |
| Subjects with temporary discontinuation due to AEs | 1 | 0 | 3 |
AE, adverse event; IRR, infusion-related reaction; SAE, serious adverse event.
Common TEAEs were IRR (30 subjects, 28.6%), headache (30 subjects, 28.6%), chills (21 subjects, 20.0%), pyrexia (15 subjects, 14.3%) and nausea (13 subjects, 12.4%). The majority of all-causality TEAEs were evenly distributed among the three treatment arms (Table 5). With the small study size and low occurrence of individual AEs, numerical imbalances were observed across the three treatment arms. There was a higher incidence of the AE term ‘pyrexia’ in the PF-05280014 arm (n = 10) compared with trastuzumab-EU (n = 3) and trastuzumab-US (n = 2). However, the severity of pyrexia in the PF-05280014 arm was mild (grade 1) in all but one subject (grade 2), and was well managed with antipyretics. Thus, pyrexia was not considered a safety concern. Other safety parameters including laboratory results, vital signs, ECGs and LVEF values were unremarkable, with no safety issues identified.
Table 5.
Treatment-emergent, all-causality adverse events occurring in ≥5% of all subjects
| PF-05280014 (n = 35) | Trastuzumab-EU (n = 35) | Trastuzumab-US (n = 35) | |
|---|---|---|---|
| Subjects with any AE, n (%) | 28 (80.0) | 29 (82.9) | 29 (82.9) |
| Eye disorders, n (%) | |||
| Conjunctival hyperaemia | 4 (11.4) | 1 (2.9) | 2 (5.7) |
| Gastrointestinal disorders, n (%) | |||
| Diarrhoea | 3 (8.6) | 2 (5.7) | 1 (2.9) |
| Nausea | 5 (14.3) | 5 (14.3) | 3 (8.6) |
| General disorders and administration site conditions, n (%) | |||
| Pyrexia | 10 (28.6) | 3 (8.6) | 2 (5.7) |
| Chills | 9 (25.7) | 7 (20.0) | 5 (14.3) |
| Fatigue | 3 (8.6) | 3 (8.6) | 3 (8.6) |
| Infections and infestations, n (%) | |||
| Nasopharyngitis | 3 (8.6) | 3 (8.6) | 2 (5.7) |
| Pharyngitis | 1 (2.9) | 4 (11.4) | 2 (5.7) |
| Injury, poisoning and procedural complications, n (%) | |||
| Infusion-related reaction | 13 (37.1) | 10 (28.6) | 7 (20.0) |
| Musculoskeletal and connective tissue disorders, n (%) | |||
| Myalgia | 2 (5.7) | 2 (5.7) | 2 (5.7) |
| Nervous system disorders, n (%) | |||
| Headache | 10 (28.6) | 12 (34.3) | 8 (22.9) |
| Dizziness | 1 (2.9) | 4 (11.4) | 2 (5.7) |
| Respiratory, thoracic and mediastinal disorders, n (%) | |||
| Cough | 1 (2.9) | 4 (11.4) | 1 (2.9) |
AE, adverse event.
The most common TRAE was IRR (30 subjects, 28.6%). Of note, the investigator reported both the IRR AE event per NCI CTCAE criteria along with the associated signs and symptoms. Common IRR signs and symptoms were chills (21 subjects, 20.0%), pyrexia (15 subjects, 14.3%), headache (14 subjects, 13.3%) and nausea (eight subjects, 7.6%). All but five of the 30 IRRs reported were grade 1 severity (13, 10 and seven in the PF-05280014, trastuzumab-EU and trastuzumab-US arms, respectively; Table 4). The remaining five IRRs were grade 2 severity (three in the PF-05280014 arm and two in the trastuzumab-US arm). All IRRs occurred during or shortly after completion of product infusion and were typically associated with pyrexia and chills. They were treated with antipyretics and resolved within 24 h. Six subjects in total had treatment interruptions due to AEs, four of which were trastuzumab-related: two grade 1 chills in the PF-05280014 arm, and one grade 1 back pain and one grade 1 chills in the trastuzumab-US arm. The remaining two were not trastuzumab-related, one infusion site infiltration and the other infusion site extravasation.
Immunogenicity evaluations
Samples for immunogenicity assessment were collected from all 105 subjects as shown (Figure 1). Only two samples tested positive for ADA (Table 6), i.e. 522/524 (99.6%) of samples tested negative in the ADA assay specific for the administered product. One positive sample was collected at 1680 h after dosing from a subject who received trastuzumab-EU (subject 1). This sample had a low titre of ADA and tested negative against PF-05280014; the difference in testing results for this sample between the two assays likely reflected the between-run or between-assay variability for a low measurement signal associated with a low titre of ADA. There were no AEs attributable to the ADA finding for this subject. Furthermore, the PK profile of this subject was similar to those of other subjects receiving trastuzumab-EU. The other sample testing positive was collected before dosing from a subject who subsequently received PF-05280014 (subject 2). All subsequent samples collected from this subject tested negative. This low false-positive rate (1/105) of ADA at baseline was consistent with assay validation requirements to ensure a high probability of identifying all subjects who develop ADA. Both ADA-positive samples tested negative for NAb.
Table 6.
Samples testing positive for antidrug antibodies (ADA)
| Subject | Treatment | Sampling time | Assay for ADA against PF-05280014 | Assay for ADA against references | ||
|---|---|---|---|---|---|---|
| ADA | Titre | ADA | Titre | |||
| Subject 1 | PF-05280014 | Pre-dose | Positive | 2.32 | Positive | 2.25 |
| Subject 2 | Trastuzumab-EU | 1680 h | Negative | <1.00 | Positive | 2.16 |
Discussion
Biosimilars are considered as ‘highly similar’ to their reference/innovator product but not identical (i.e. they should not be viewed as ‘bio-generics’). As defined by Section 351(i) of the Public Health Service Act, the term ‘biosimilar’ refers to a ‘biologic product that is highly similar to the reference product, notwithstanding minor differences in clinically inactive components’. There should be no clinically meaningful differences between biosimilars and reference biologics in terms of safety, purity and potency 1–3,14,15. Although they may vary in minor details, they generally include thorough in vitro, non-clinical in vivo and clinical assessments to demonstrate biosimilarity. The primary goal of this study was to demonstrate clinical PK similarity of PF-05280014 to the trastuzumab products approved in the EU and licensed in the US. Also, the PK similarity between the two marketed trastuzumab products was evaluated to provide the bridging data justifying the use of a single reference product in further comparative clinical trials in patient populations.
Trastuzumab PK, characterized by dose-dependent exposure consistent with target-mediated disposition, has been evaluated in female patients with metastatic breast cancer 9,16. Patients with higher baseline levels of circulating extracellular domain of the HER2 receptor (shed antigen) or a larger number of metastatic sites tend to have higher clearance 9,16. However, there was no prior experience of trastuzumab PK in healthy females. Thus, this trial was limited to healthy male volunteers.
The three products exhibited similar PK profiles consistent with those of monoclonal antibodies with target-mediated disposition. The three administered products showed a similar pattern of more rapid decrease in the later part of the terminal disposition phase, suggesting increased clearance due to complex formation with the target, HER2 17. This suggests that the three study products not only have similar disposition properties but also share similar drug–target interaction kinetic characteristics. The 90% CIs for the test: reference ratios of Cmax, and AUC(0,∞) were all within the prespecified limits of 80.00% to 125.00% for all three pairwise comparisons. These data demonstrate that the PK of PF-05280014 were similar to those of trastuzumab-US and trastuzumab-EU and that the PK of the two marketed trastuzumab products were similar to each other. It should be noted that the mean values for these PK parameters were 6–9% higher in the trastuzumab-EU group than in the other two treatment arms, which may be related to the higher mean body weight of 6–8% of the trastuzumab-EU group compared with the other two groups, which corresponded to a similar degree of reduction in Vss per kg body weight (see Table 2). The PK parameters including Cmax and AUC(0,∞) reported here were comparable with those previously reported for the marketed trastuzumab product administered as a single intravenous dose of 6 mg kg−1 in both healthy male volunteers and female patients with HER2-positive breast cancer 18.
The low incidence of ADA (one subject after receiving trastuzumab-EU out of 105 subjects) observed in this study was consistent with data reported for trastuzumab 9,18. Furthermore, there was no NAb detected in the samples testing positive for ADA. These data underscore the low immunogenicity of PF-05280014 and trastuzumab. Further comparative assessment of the immunogenicity between PF-05280014 and trastuzumab will be needed in the target cancer patient population following repeated administration with the dose and regimen approved for trastuzumab in the indication.
All three products were well tolerated, with no serious AEs reported in any of the subjects. The majority of AEs were evenly distributed among the three treatment arms. The most common AEs were IRRs, usually together with pyrexia and chills. Although there was a numerically higher incidence of the AE term ‘pyrexia’ in the PF-05280014 arm, the severity was generally mild, suggesting that pyrexia was not a safety issue. These data were comparable with the data reported for trastuzumab 9,18. In the previously reported open label, single dose phase 1 trial, the most commonly observed AEs across all cohorts were IRRs, headache, musculoskeletal pain, diarrhoea and upper respiratory tract infection, most of which were mild in severity 18. In that trial, the sole severe IRR occurred 5 h after infusion commencement and resolved within 2 days following treatment with antipyretics, analgesics and sedatives 18. In the current trial, as with the previous report, there were no deaths or serious AEs 9,18.
It is noteworthy to mention that, given the number of subjects in each arm and the primary objectives of this study, it is inconclusive whether the observed numerical difference in pyrexia was the result of a random event associated with evaluating multiple AEs in a relatively small study. A follow-up study in healthy subjects (NCT02015156) has been conducted to estimate the relative risk of pyrexia of PF-05280014 in comparison with trastuzumab and the results are forthcoming.
Overall, the data reported here demonstrate that the three products are similar in PK and have safety and immunogenicity properties that are consistent with previous reports for trastuzumab. These data support the continued development of PF-05280014 as a potential biosimilar to trastuzumab (Herceptin).
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare this study was funded by Pfizer Inc. All authors are employees of either Pfizer or of an organization contracted by Pfizer for this study and there are no other relationships or activities that could appear to have influenced the submitted work.
The authors would like to thank Sherry Cai from the Pfizer Clinical Assay Group for managing the analyses of drug concentration and immunogenicity samples and Drs Jim McNally and Boris Gorovits from Pfizer Pharmacokinetics, Dynamics, and Metabolism for developing the neutralizing ADA assay and conducting the neutralizing ADA sample analysis. Medical writing support was provided by Mukund Nori, PhD, MBA, CMPP, of Engage Scientific Solutions and was funded by Pfizer Inc, New York, NY, USA.
Contributors
All authors had roles in the design and conduct of the study, collection, management, analysis and interpretation of the data, and preparation, review and approval of the article.
References
- Committee for Medicinal Products for Human Use (CHMP) 2005. Guideline on similar biological medicinal products. European Medicines Agency,. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003517.pdf (last accessed 11 October 2012)
- Food and Drug Administration Center for Drug Evaluation and Research. 2012. Guidance for industry: scientific considerations in demonstrating biosimilarity to a reference product. U.S. Department of Health and Human Services,. Available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf (last accessed 1 March 2012)
- Committee for Medicinal Products for Human Use (CHMP) 2012. Guideline on similar biological medicinal products containing monoclonal antibodies – non-clinical and clinical issues. European Medicines Agency,. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf (last accessed 11 October 2012)
- Hudis CA. Trastuzumab – mechanism of action and use in clinical practice. N Engl J Med. 2007;357:39–51. doi: 10.1056/NEJMra043186. [DOI] [PubMed] [Google Scholar]
- Baselga J, Albanell J, Molina MA, Arribas J. Mechanism of action of trastuzumab and scientific update. Semin Oncol. 2001;28:4–11. doi: 10.1016/s0093-7754(01)90276-3. [DOI] [PubMed] [Google Scholar]
- Baselga J, Norton L, Albanell J, Kim YM, Mendelsohn J. Recombinant humanized anti-HER2 antibody (Herceptin) enhances the antitumor activity of paclitaxel and doxorubicin against HER2/neu overexpressing human breast cancer xenografts. Cancer Res. 1998;58:2825–2831. [PubMed] [Google Scholar]
- Pegram M, Hsu S, Lewis G, Pietras R, Beryt M, Sliwkowski M, Coombs D, Baly D, Kabbinavar F, Slamon D. Inhibitory effects of combinations of HER-2/neu antibody and chemotherapeutic agents used for treatment of human breast cancers. Oncogene. 1999;18:2241–2251. doi: 10.1038/sj.onc.1202526. [DOI] [PubMed] [Google Scholar]
- Pegram MD, Konecny G, Slamon DJ. The molecular and cellular biology of HER2/neu gene amplification/overexpression and the clinical development of herceptin (trastuzumab) therapy for breast cancer. Cancer Treat Res. 2000;103:57–75. doi: 10.1007/978-1-4757-3147-7_4. [DOI] [PubMed] [Google Scholar]
- HERCEPTIN® [trastuzumab\. US Prescription Information. San Francisco, CA, USA: Genentech Inc; 2010. [Google Scholar]
- Gibaldi M, Pharmacokinetics PD, editors. New York. NY: Marcel Dekker, Inc; 1982. [Google Scholar]
- National Cancer Institute. 2010. Common terminology criteria for adverse events (CTCAE); version 4.03. National Institutes of Health, U.S. Department of Health and Human Services,. Available at http://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_8.5x11.pdf (last accessed 20 May 2013)
- Brown EG, Wood L, Wood S. The medical dictionary for regulatory activities (MedDRA) Drug Saf. 1999;20:109–117. doi: 10.2165/00002018-199920020-00002. [DOI] [PubMed] [Google Scholar]
- Yin D, Cai C-H, Taylor CT, Zacharchuk CM, Rudin D, Reich SD, Barker KB, Li R, Ricart AD, Meng X. 2013. Immunogenicity assessment of PF-05280014, a potential biosimilar to trastuzumab, in healthy subjects (REFLECTIONS B327-01). Presented at European Cancer Congress, Amsterdam, The Netherlands, 27 September–1 October. [DOI] [PMC free article] [PubMed]
- Kozlowski S, Woodcock J, Midthun K, Sherman RB. Developing the nationx0027;s biosimilars program. N Engl J Med. 2011;365:385–388. doi: 10.1056/NEJMp1107285. [DOI] [PubMed] [Google Scholar]
- Expert Committee on Biological Standardization. 2010. Guidelines on evaluation of similar biotherapeutic products (SBPs). World Health Organization,. Available at http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf (last accessed 1 March 2013)
- Bruno R, Washington CB, Lu JF, Lieberman G, Banken L, Klein P. Population pharmacokinetics of trastuzumab in patients with HER2+ metastatic breast cancer. Cancer Chemother Pharmacol. 2005;56:361–369. doi: 10.1007/s00280-005-1026-z. [DOI] [PubMed] [Google Scholar]
- Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci. 2004;93:2645–2668. doi: 10.1002/jps.20178. [DOI] [PubMed] [Google Scholar]
- Wynne C, Harvey V, Schwabe C, Waaka D, McIntyre C, Bittner B. Comparison of subcutaneous and intravenous administration of trastuzumab: a phase I/Ib trial in healthy male volunteers and patients with HER2-positive breast cancer. J Clin Pharmacol. 2013;53:192–201. doi: 10.1177/0091270012436560. [DOI] [PubMed] [Google Scholar]