

Abstract
Aims
AMG 181 pharmacokinetics/pharmacodynamics (PK/PD), safety, tolerability and effects after single subcutaneous (s.c.) or intravenous (i.v.) administration were evaluated in a randomized, double-blind, placebo-controlled study.
Methods
Healthy male subjects (n= 68) received a single dose of AMG 181 or placebo at 0.7, 2.1, 7, 21, 70 mg s.c. (or i.v.), 210 mg s.c. (or i.v.), 420 mg i.v. or placebo. Four ulcerative colitis (UC) subjects (n= 4, male : female 2:2) received 210 mg AMG 181 or placebo s.c. (3:1). AMG 181 concentration, anti-AMG 181-antibody (ADA), α4β7 receptor occupancy (RO), target cell counts, serum C-reactive protein, fecal biomarkers and Mayo score were measured. Subjects were followed 3–9 months after dose.
Results
Following s.c. dosing, AMG 181 was absorbed with a median tmax ranging between 2–10 days and a bioavailability between 82% and 99%. Cmax and AUC increased dose-proportionally and approximately dose-proportionally, respectively, within the 70–210 mg s.c. and 70–420 mg i.v. ranges. The linear β-phase t1/2 was 31 (range 20–48) days. Target-mediated disposition occurred at serum AMG 181 concentrations of less than 1 μg ml−1. The PD effect on α4β7 RO showed an EC50 of 0.01 μg ml−1. Lymphocytes, eosinophils, CD4+ T cells and subset counts were unchanged. AMG 181-treated UC subjects were in remission with mucosal healing at weeks 6, 12 and/or 28. The placebo-treated UC subject experienced colitis flare at week 6. No ADA or AMG 181 treatment-related serious adverse events were observed.
Conclusions
AMG 181 has PK/PD, safety, and effect profiles suitable for further testing in subjects with inflammatory bowel diseases.
Keywords: α4β7 integrin, AMG 181, PK/PD, T cell trafficking, ulcerative colitis
What is already known about this subject
The clinical pharmacology and safety of AMG 181 in humans were unknown before we conducted the first-in-human single ascending dose study.
The pharmacokinetic ( PK) characterization of AMG 181 in healthy subjects was required for AMG 181 dose selection for phase 2 trials in subjects with inflammatory bowel disease (IBD).
What this study adds
AMG 181 is safe and well tolerated in healthy subjects.
AMG 181 PK and target α4β7 receptor occupancy are directly correlated in healthy humans, with no effects on α4β7 receptor bearing target cell counts.
AMG 181 has PK/PD and safety characteristics suitable for further testing in IBD subjects.
Introduction
Crohn's disease (CD) and ulcerative colitis (UC) are chronic, relapsing, inflammatory disorders of the gastrointestinal tract collectively termed inflammatory bowel diseases (IBDs). They affect approximately 1.4 million people in the United States (USA) and 2.2 million people in Europe with an incidence rate ranging between 10 and 30 per 100 000. Regions with traditionally lower incidence or prevalence of IBDs (e.g. Asia) have shown increasing trends in recent decades suggesting that IBDs are emerging as a global disease 1–3. The reported mean age at diagnosis is generally similar between Western and Asian populations with peak incidence occurring between 20 and 40 years 1,2,4. Although the pathophysiology is not fully understood, accumulating epidemiologic data suggest that IBDs may result from an inappropriate inflammatory response to alterations in intestinal microbiome in a genetically susceptible sub-group of the population 5–7.
Due to the autoimmune nature of IBDs, initial treatment typically includes conventional anti-inflammatory agents such as 5-aminosalicylic acids (5-ASA), corticosteroids and immunomodulators 8. After failure with the aforementioned conventional agents, the mainstay of the current approach is the use of anti-tumour necrosis factor antibodies (anti-TNFs) including the chimeric immunoglobulin G1 (IgG1) infliximab 9–11 and the human IgG1 adalimumab 12–14, both of which are indicated for treating CD and UC. In addition, the IgG4-binding pegylated humanized Fab (fragment, antigen binding) certolizumab 15,16 and the human IgG1 golimumab 17 are indicated for treating CD and UC, respectively. Despite the proven therapeutic efficacy of the anti-TNFs in inducing and maintaining clinical remission in IBDs, meta-analyses of randomized clinical trials (RCT) have shown that the rate of subjects whose disease is not improved by treatment (non-responders) remains high 18. For example, in RCTs for treating CD, the overall induction of remission rate was 18 to 48% at week 4, with 25 to 41% of those subjects remaining in remission after 1 year 19. In RCTs for treating UC, the overall induction and maintenance of remission rates were 19 to 39% and 26 to 37%, respectively 18. These data indicate that approximately 80 to 95% of the IBD patients were either non-responders to anti-TNFs or had lost anti-TNF treatment benefit within 1 year. From a safety perspective, anti-TNFs have been associated with an increased risk of serious infections and malignancy 20. Therefore, an unmet medical need remains for developing safer and more efficacious therapies for IBD patients.
In recent years, studies have been focusing on agents that block the influx of pro-inflammatory cells into the intestinal mucosa (gut-homing specific) through integrin-mediated leukocyte tethering, rolling and arrest 21. For example, blocking lymphocyte gut-homing integrin α4β7 22–24, integrin β7 25–27 or α4β7 target ligand MAdCAM-1 (mucosal addressin cell adhesion molecule-1) 28,29 has demonstrated efficacy in various preclinical IBD models. In fact, some of these targets are being investigated in clinical trials to evaluate safety and efficacy of therapeutic candidates such as vedolizumab (anti-α4β7), AMG 181 (anti-α4β7), etrolizumab (anti-β7) and PF-00547659 (anti-MAdCAM-1) 30.
Natalizumab, a pleiotropic pan-α4 antagonist blocking both α4β1 and α4β7 integrins, has shown clinical efficacy in treating CD and is approved for this indication in the USA 31,32. Additionally, natalizumab has been approved in the USA and Europe for treating relapsing and remitting forms of multiple sclerosis (MS) 33. Under certain conditions, and especially in the MS population, natalizumab has been found to increase the risk of developing progressive multifocal leukoencephalopathy (PML) due to re-activation of the latent human John Cunningham (JC) polyomavirus and subsequent infection of the central nervous system (CNS) 34. It is hypothesized that inhibition of α4β1-expressing cells by natalizumab may lead to broad impact on leukocyte trafficking to various tissues, including the brain and, by compromising local immune surveillance, may enable JC virus replication and ensuing PML 35–37.
Natalizumab and vedolizumab are humanized IgGs, and RCTs have shown that both antibodies have greater than 10% immunogenicity in IBD patients leading to reduced clinical efficacy 31,38–40. In order to reduce or eliminate the impact of immunogenicity and the safety implications associated with targeting α4β1 expressing leukocytes, Amgen has developed AMG 181, a human monoclonal IgG2 antibody that specifically binds to the α4β7 integrin on gut-tropic lymphocytes, which blocks interaction with the α4β7 target ligand MAdCAM-1 and thus prevents lymphocyte trafficking into mucosal tissue and alleviates inflammation. We have previously reported the in vitro pharmacology, as well as PK/PD and safety characteristics of AMG 181 in cynomolgus monkeys that support its testing in humans 41.
In this report, we present results from the first-in-human study conducted in healthy subjects and subjects with UC. Our specific aims for this phase 1 clinical trial were to evaluate single-dose PK/PD, safety, tolerability, and effects of AMG 181 after s.c. or i.v. administration 42. The results from this study have suggested that, given its pharmacological potency, apparent lack of immunogenicity and gut-specificity, AMG 181 has the potential for providing IBD patients with another treatment option. AMG 181 is currently being tested in subjects with UC or CD in phase 2 clinical trials 43,44.
Methods
Investigational product
AMG 181 (∼144 kD) was manufactured at Amgen Inc., Thousand Oaks, California (CA), USA, by expression in Chinese hamster ovary (CHO). The investigational AMG 181 substance was formulated at 70 mg ml−1 with pharmaceutically acceptable excipients, pH 5.2, and stored at −20°C to −70°C in a non-frost freezer, until ready for s.c. or i.v. administration. AMG 181 placebo (matching vehicle control) was formulated with the same excipients and pH, filled, packaged and stored to match the active AMG 181. The placebo was also used as the active s.c. dose diluent, while 5% dextrose (US Pharmacopeia) was used for i.v. infusion bags.
Study design and ethical considerations
This was a randomized, double-blind, placebo-controlled single ascending dose (SAD) study to evaluate the safety, tolerability, PK/PD, and effects of AMG 181 after s.c. or i.v. dose in healthy volunteers and subjects with UC (http://www.clinicaltrials.gov, identifier NCT01164904). The study design and dose escalation schedule are summarized in Figure 1.
Figure 1.

Study design and dose escalation schedule. Blue arrows represent dosing; black arrows represent pharmacokinetic, pharmacodynamic, immunogenicity, safety and/or effect assessment visits. Numbers in parenthesis indicate AMG 181: placebo randomization ratio per study protocol and the number of subjects (N) completing the study. s.c. = subcutaneous; i.v. = intravenous; UC = ulcerative colitis subjects else, healthy subjects
The study was carried out in accordance with the ethical principles set forth in the Declaration of Helsinki and its revisions (the most recent in 2008), the International Conference on Harmonization (ICH) E6 Guidance for Good Clinical Practice (CPMP/ICH/135/95) and all other applicable country-specific regulatory requirements and site-specific standard operating procedures (SOPs). The study portion in healthy volunteers was conducted at California Clinical Trials Medical Group, Inc. Glendale, CA, USA (approved by the Aspire Institutional Review Board), while the UC portion was conducted at the Royal Adelaide Hospital, Adelaide, South Australia (approved by the Royal Adelaide Hospital Research Ethics Committee) and at the Royal Brisbane and Women's Hospital, Herston, Queensland, Australia (approved by the Royal Brisbane and Women's Hospital Human Research Ethics Committee). Before subjects entered into the study, Amgen obtained a copy of the sites' written institutional review board's (IRB's) or ethics committees' (ECs') approval of the protocol and informed consent form, and all other subject information and/or recruitment material. All subjects or legally acceptable representatives personally signed and dated the IRB/EC-approved consent form before initiation of any screening procedures.
Subject enrolment criteria
Inclusion criteria for enrolment followed the standard practice for first-in-human studies. Briefly, eligible healthy men and women were 18 to 45 years of age (inclusive), with a body mass index (BMI) of 18 to 34 kg m−2, and in good health based on physical and neurological examinations and were sero-positive for Epstein-Barr virus. Subjects had normal laboratory values and normal limits for 12-lead electrocardiogram (ECG). All male subjects practiced a highly effective method of birth control during the study. No female subjects were enrolled in the healthy subject portion of the study.
Subjects who met any one of the following criteria were excluded from the study: history or evidence of clinically significant disorders, conditions or diseases that posed a risk to the subject's safety or interfered with study evaluation, procedures or completion; history of malignancy of any type, men who had pregnant partners, known history of drug or alcohol abuse within 1 year of screening or positive screen for alcohol and/or drugs with a high potential for abuse at screening or day −1, positive for HIV antibodies, hepatitis B surface antigen, hepatitis C antibodies, tuberculosis, or JC viraemia determined by PCR at screening, sero-negative for Epstein-Barr virus, use of any investigational biologic ≤ 6 months or use of prescription or non-prescription drugs (with the exception of paracetamol/acetaminophen [up to 2 g day−1]) within 14 days prior to investigational product administration.
The UC subjects met the above inclusion/exclusion criteria for healthy subjects but were allowed to be up to 65 years of age. Women were of either child bearing or non-child-bearing potential, with those of child-bearing potential having a negative pregnancy test on day −1 and agreeing to use highly effective methods of birth control for the duration of the study. Subjects with UC had a documented diagnosis of at least 2 months of active UC defined by an Ulcerative Colitis Disease Activity Index (UCDAI, or Mayo score) score 45 of 4 to 9 (inclusive), with a minimum sigmoidoscopy score of 1.
The main exclusion criteria for UC subjects included disease limited to the rectum, women planning to become pregnant during the study, current use of oral steroids in excess of 20 mg prednisone or equivalent per day, tuberculosis or fungal infection within 3 months of screening or during screening, immunosuppressive therapy with either azathioprine, methotrexate, or mercaptopurine, within 3 months of day 1, immunosuppressive therapy with either ciclosporin A, tacrolimus, mycophenolate mofetil within 2 months of day 1, exposure to anti-TNF agents within 2 months, or five times the respective elimination half-life (whichever was longer) prior to day 1, prior exposure to natalizumab, rituximab or efalizumab, change of dose of oral mesalamine within the past weeks of screening or use of topical mesalamine or steroids within 2 weeks prior to study day 1.
Dosing and dose escalation procedures
After informed consent and within a period of 21 days prior to dosing, all screening tests establishing subject eligibility were performed. Subjects were admitted to the study clinic on the day prior to dosing (day −1) to perform day −1 procedures. On day 1, after completion of all pre-dose procedures, subjects received a single dose of AMG 181 or placebo and remained in the clinic until completion of study procedures on day 3 (48 h after dosing). Subjects were discharged from the study clinic on day 3 and returned for safety follow-up and PK/PD assessments.
AMG 181 was administered to healthy volunteers as a single dose of 0.7, 2.1, 7, 21, 70 or 210 mg s.c. or 70, 210 or 420 mg i.v. (1 h infusion) and, to UC subjects, 210 mg s.c., corresponding to cohorts 1 to 10, respectively (Figure 1). Within each single dose cohort, subjects were to be randomized such that six received AMG 181 and two received matching placebo (except for cohorts 1 and 2, where four received AMG 181 and two received placebo). A sentinel dosing design was applied for the 0.7 and 2.1 mg s.c. and 70 mg i.v. cohorts, where the first two subjects received AMG 181 treatment or placebo in a blinded fashion. In cohort 10, four staggered UC subjects received a single 210 mg AMG 181 or placebo s.c. dose (3:1 ratio).
S.c. doses were administered to the anterior abdominal wall of the subject. For individual s.c. dose volumes that exceeded 1.5 ml, the dose was split into separate syringes and administered into different sites on the subject's anterior abdominal wall. The s.c. injections were administered in a consecutive fashion with the injections separated by no more than 30 s. The i.v. dosing was performed through infusion in 5% dextrose in a volume of 100 ml over a 1 h period using a volumetric infusion pump through an i.v. access.
With the previous regimen(s) being well tolerated, no stopping rules being met, and under an unanimous dose level review meeting recommendation, escalation to the next higher dose cohort (cohorts 2 through 10) proceeded based on a safety data review through day 15 for all subjects in the current cohort and all available safety data from all subjects in the previous cohort(s). Subjects who dropped out of the study for reasons other than adverse events after dosing were replaced at the discretion of Amgen in consultation with the investigator or their designee. Replacement subjects received the identical treatment as the assigned treatment for the subject to be replaced. Completion of the end of study (EOS) assessments occurred on day 169 for cohorts 1–5 and 7, day 197 for cohorts 6, 8 and 10 and day 225 for cohort 9.
Safety and laboratory assessments
A detailed summary of the safety and laboratory study procedures and schedules is presented in Supplementary Table S1.
Adverse events and concomitant medication were monitored throughout the study. All adverse events were reported according to the United States National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) v4.0 46. The study sites recorded use of concomitant medication and all adverse events (treatment-emergent and treatment-related as assessed by the investigator) that occurred during or after dosing through the end of the study, as observed by the investigator or reported by the subject. All treatment emergent adverse events or laboratory abnormalities were followed until the resolution of the abnormality or until the condition was considered clinically stable in the opinion of the investigator. Descriptive statistics were calculated per dose level and tabulated by the system organ class and preferred term. Due to scarcity of the adverse events in this study, data using the preferred term and the treatment-emergent adverse events occurring in > 1 of healthy subjects are presented.
Blood samples were collected for CD4+ T cell subset enumeration and α4β7 RO/expression, and for JC viraemia determination. Other laboratory assessments included pregnancy test, follicle-stimulating hormone (FSH) measurement, human immune deficiency virus (HIV) and hepatitis B and C screening, and stool culture and stool ova and parasites, and drug and alcohol screening.
For UC subjects, the effects of AMG 181 treatment were assessed through the Mayo score, partial Mayo score, serum C-reactive protein, and fecal biomarkers including calprotectin and lactoferrin. The Mayo score has four components, including stool frequency, rectal bleeding, endoscopic findings and physician's global assessment. Each of the four components has an integer score range of 0 to 3, with the total score ranging from 0 to 12 45. When recto-sigmoidoscopy is not preformed, the partial Mayo score with a total score ranging from 0 to 9 can be assessed. Empirically and in most recent clinical trials, remission has been defined as Mayo score ≤ 2, with no individual subscore > 1 point, response as Mayo score decrease from baseline of ≥ 3 and ≥ 30%, plus rectal bleeding score decrease of ≥ 1 point or absolute reading of 0 or 1, mucosal healing as recto-sigmoidoscopy score of ≤ 1 14,47. Up to two stools were collected on each of 3 consecutive days during 5 different weeks to support assessment of fecal calprotectin and lactoferrin levels. Subjects returned stool specimens to the clinical sites for processing and subsequent testing within 72 h of collection. The assay limits of detection for fecal calprotectin and lactoferrin were 15.4 μg g−1 and 31 μg ml−1, respectively.
Serum AMG 181 concentration and immunogenicity assays
Serum AMG 181 concentrations were determined with a validated immunoassay utilizing the Meso Scale Discovery (Gaithersburg, MD, USA) electrochemiluminescence (ECL) platform with a lower limit of quantification (LLOQ) of 10.0 ng ml−1.
Serum was analyzed for anti-AMG 181 antibodies (ADAs) with a validated immunoassay utilizing the Meso Scale Discovery ECL platform with a lower limit of reliable detection (LLRD) of 20 ng ml−1 of affinity purified rabbit ADAs in neat pooled normal human serum. Detection of ADAs at the LLRD may have been prevented, creating a false negative result, if a test sample AMG 181 concentration was > 25 μg ml−1. Detailed information on the serum AMG 181 concentration and immunogenicity assays can be found in the supplementary material.
Pharmacokinetic analyses
Non-compartmental PK analysis was performed using WinNonlin® 5.02 (Pharsight® Corporation, Mountain View, CA, USA). Actual sampling times elapsed from dosing time for each individual subject were used for the calculation of AMG 181 PK parameters. These included maximum observed AMG 181 concentration in serum (Cmax and dose-normalized value: Cmax/Dose), time to Cmax (tmax) and area under the concentration–time curve from time zero to the last observed concentration (AUC(0,t)) and extrapolated to infinity (AUC(0,∞) and dose-normalized value, AUC(0,∞)/Dose). AUC values were calculated by the linear trapezoidal rule. AMG 181 bioavailability was calculated by dividing the cohort AUC(0,∞) means between 70 mg s.c. and i.v. or 210 mg s.c. and i.v..
AMG 181 exhibited target-mediated disposition, where capacity-limited binding to the target α4β7 integrin resulted in non-linear PK. This non-linear PK was obvious at low AMG 181 concentrations of < 1 μg ml−1, where AMG 181 disposition was predominantly driven by high affinity low capacity binding between AMG 181 and the limited amount of α4β7 receptors. At higher AMG 181 concentrations of > 1 μg ml−1, the saturable AMG 181 disposition through binding to α4β7 receptors was masked by the lower affinity, high capacity linear non-specific binding of AMG 181 to neonatal Fc receptors (FcRns).
Because AMG 181 exhibited target-mediated disposition at the terminal elimination phase (γ-phase), individual linear β-phase rate constant (β), a relevant measure of effective half-life for repeated dosing, was determined in the linear portion of the individual concentration−time profiles by visual inspection. At least three data points were in the linear portion with the coefficient of correlation for the linear regression of ≥ 0.90. The linear β-phase half-life (t1/2) was calculated as ln(2)/β. Other calculated linear PK parameters included: clearance (CL) and apparent clearance (CL/F) for i.v. and s.c., respectively, volume (Vz) and apparent volume of distribution (Vz/F) at the terminal elimination phase for i.v. and s.c., respectively, and volume of distribution at steady-state for i.v. (Vss). Descriptive statistics were calculated per cohort: n, mean (median for tmax) and standard deviation (SD) (range for tmax).
To explore the effects of demography on the AMG 181 PK, dose-normalized Cmax and AUC(0,∞) were plotted against body weight, BMI, age and race, with the latter being categorized into White, Black and others.
Pharmacodynamic assays and analyses
To characterize the direct PD effects of AMG 181, α4β7 receptor occupancy (RO) and changes in target cell counts in the selected lymphocyte subset populations were investigated.
A semi-quantitative flow cytometric assay was developed to assess coverage of α4β7 receptors by AMG 181 on CD4+ T cell subsets namely, naïve, central memory and effector memory T cells, in whole blood specimens. The principle of this assay was that exposure to AMG 181 was expected to reduce staining intensity of a fluorochrome-labelled anti-α4β7 RO antibody 1 (anti-α4β7-ROA1) which competes with AMG 181 for binding on target cell surface. To identify the floor of the assay, a separate aliquot of each blood specimen was pre-incubated with a saturating dose of AMG 181 (500 μg ml−1) ex vivo. For each specimen, the number of anti-α4β7-ROA1 molecules bound per CD4+ T cell subsets was calculated relative to that in the presence of saturating AMG 181, thereby providing an estimate of the fractional occupancy of α4β7 molecules per cell, where percent fractional receptor occupancy (%RO) = 100 − free fraction*100. A second, non-competing, anti-β7 RO antibody 2 (anti-β7-ROA2) was also used to quantify total α4β7 receptor levels on CD4+ T cell subsets after exclusion of CD103 (αE integrin, which binds to β7 integrin and forms αEβ7 heterodimer) positive cells.
CD4+ T cell subsets were enumerated by flow cytometric assay in conjunction with absolute lymphocyte count from complete blood counts (CBCs). In addition, α4β7 expressing CD4+ memory T cell subsets in blood were enumerated by multiplying the α4β7 high or low CD4+ memory T cell event count as a fraction of total lymphocyte event count by the total lymphocyte count determined by CBC. CD4+ α4β7 high or low memory T cell event count was measured by the anti-β7-ROA2. Other AMG 181 target cells expressing α4β7 receptors such as eosinophils and total lymphocytes were enumerated directly through CBCs.
The relationships between serum AMG 181 concentration and the subject/time-matched α4β7 RO on total CD4+, naïve, central memory and effector memory T cells were quantitatively assessed using a simple Emax PD model:
where E0 stands for the baseline, Emax is the maximal RO, C is serum AMG 181 concentration and EC50 is AMG 181 concentration at which the RO is half-maximal. An Emax PD model with Hill coefficient was tested but did not result in a significantly improved model fit. The AMG 181 concentrations at which RO was 90% (EC90) or 99% (EC99) maximum were calculated using the above equation. RO data from all subjects pre-dose and from all placebo subjects were pooled across the regimens with the assigned AMG 181 concentration of zero. Because the assay for the AMG 181 concentration had a lower limit of quantification (LLOQ) of 0.01 μg ml−1, RO data corresponding to AMG 181 concentrations that were below the assay LLOQ (BQL) and collected after AMG 181 treatment were not used for modelling (but were plotted with AMG 181 concentrations assigned a value at half of the LLOQ in Figure 5).
Figure 5.
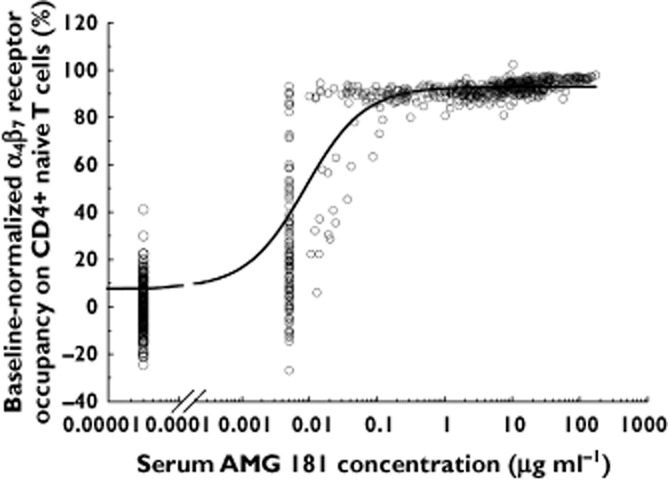
Individual healthy subject pre-dose baseline-normalized α4β7 receptor occupancy on CD4+ naïve T cell vs. serum AMG 181 concentrations. All data from pre-dose and placebo treated subjects are plotted at AMG 181 = 0 μg ml−1. The line represents the Emax pharmacodynamic model fit of the observed data. RO data with AMG 181 concentrations measured to be < 0.01 μg ml−1 (assay LLOQ) were plotted against 0.005 μg ml−1, corresponding to LLOQ/2. These data were not used for PD modelling because the exact AMG 181 concentrations for these samples could not be quantified
The relationship between serum AMG 181 concentration and the subject/time-matched α4β7 RO on total CD4+ T cells (including its three subsets: CD4+ naïve, central memory, and effector memory T cells) was further explored by plotting AUECall or area-under-the-effect (RO) − time curve vs. AMG 181 area under the concentration−time curve extrapolated to concentration of zero (AUCall) using data from all healthy subjects who were dosed AMG 181 or received placebo and completed the study. A simple Emax PD model as described above was used to fit the data for quantifying EAUCall,90 from EAUCall,50 (the AMG 181 AUCall at which the RO AUECall was half-maximum), to estimate an AUCall (or the associated dose) beyond which there would be no further meaningful α4β7 RO AUECall increases (e.g., ∼90% AUECall,max).
Statistical analyses
This was a phase 1 clinical trial, with sample size chosen based on practical considerations and consistent with conventions and, therefore, data have been either presented using descriptive statistics or individually (mostly UC data). We have conducted post hoc linear regression analyses of the demographic effects on dose-normalized AMG 181 peak (Cmax) and cumulative exposure (AUC) parameters. This will guide our future population analysis when more abundant data become available from the on-going phase 2 studies.
Results
Baseline demographics
Sixty-eight (68) healthy male subjects and four male and female UC subjects were enrolled in this single ascending dose study. Summary statistics of subject baseline demography by dosing regimen or cohort, as well as the overall summary across cohorts are provided in Table 1. The baseline characteristics were similar among dosing regimens regarding age, body weight, height and BMI, except for race, for which the percentage of White subjects ranged from 0 to 83%.
Table 1.
Mean (SD) baseline demographics
| Regimen | n | Age (years) | Weight (kg) | Height (cm) | BMI (kg m−2) | Race (%White) |
|---|---|---|---|---|---|---|
| 0.7 mg s.c. | 4 | 32.3 (9.1) | 78.7 (8.82) | 177.4 (4.1) | 25.0 (2.64) | 50 |
| 2.1 mg s.c. | 4 | 30.5 (3.4) | 80.5 (3.61) | 180.7 (7.8) | 24.8 (2.87) | 0 |
| 7 mg s.c. | 6 | 26.5 (6.3) | 79.4 (10.7) | 178.0 (8.0) | 25.1 (3.58) | 33 |
| 21 mg s.c. | 6 | 24.2 (3.1) | 76.4 (11.1) | 174.5 (5.2) | 25.2 (3.87) | 50 |
| 70 mg s.c. | 6 | 28.3 (6.2) | 72.7 (10.0) | 172.1 (3.8) | 24.5 (3.11) | 33 |
| 210 mg s.c. | 6 | 29.5 (5.9) | 80.3 (12.0) | 174.6 (5.4) | 26.4 (4.06) | 50 |
| 70 mg i.v. | 6 | 29.0 (4.9) | 82.6 (6.27) | 179.6 (3.7) | 25.7 (2.39) | 50 |
| 210 mg i.v. | 6 | 28.5 (7.8) | 84.4 (22.3) | 181.4 (15.2) | 25.4 (4.43) | 83 |
| 420 mg i.v. | 6 | 29.8 (5.7) | 79.9 (11.3) | 179.8 (7.5) | 24.7 (2.76) | 0 |
| 210 mg s.c. (UC) | 3 | 37.3 (5.1) | 66.5 (11.0) | 168.0 (3.1) | 23.5 (2.98) | 67 |
| Combined | 53 | 29.0 (6.2) | 78.7 (11.8) | 176.9 (7.8) | 25.1 (3.17) | 42 |
| Placebo | 19 | 29.9 (6.5) | 82.7 (12.1) | 176.3 (6.0) | 26.7 (4.24) | 63 |
| All | 72 | 29.3 (6.2) | 79.7 (12.0) | 176.7 (7.3) | 25.5 (3.52) | 47 |
Healthy subjects: all male; UC subjects: two female and one male were AMG 181-treated, one male received placebo. Combined = all AMG 181-treated, including healthy and UC subjects; Placebo = all placebo treated, including healthy and one UC subjects. BMI = body mass index; SD = standard deviation; s.c. = subcutaneous; i.v. = intravenous; UC = ulcerative colitis.
Of the 68 healthy subjects, 60 completed the study, two withdrew consent due to personal reasons and six were lost to follow-up. Of the four UC subjects, one subject who received placebo withdrew from the study after the day 43 visit due to colitis flare.
Safety, tolerability and immunogenicity
For healthy subjects, due to small cohort sizes, low subject incidences of adverse events, and absence of apparent trends in subject incidence of adverse events associated with dose or route of administration, data from all cohorts have been pooled by AMG 181 or placebo treatment (Table 2). The overall subject incidences for treatment-emergent adverse events were comparable between the AMG 181 group and the placebo group (60% vs. 50%). All adverse events were reported as grade 1 in severity in healthy subjects except for one adverse event of grade 2 hypertension (210 mg AMG 181 s.c. subject) and two adverse events of grade 3 syncope (70 mg AMG 181 i.v. and placebo s.c. subject). Mild injection site reactions were reported for two subjects who received AMG 181 and no infusion reactions were reported.
Table 2.
Treatment-emergent adverse events in healthy subjects (reported for more than subject)
| Preferred term | AMG 181 n = 50 n (%) | Placebo n = 18 n (%) |
|---|---|---|
| Any adverse event | 30 (60) | 9 (50) |
| Upper respiratory tract infection | 15 (30) | 1 (6) |
| Headache | 8 (16) | 1 (6) |
| Oropharyngeal pain | 3 (6) | 1 (6) |
| Diarrhoea | 1 (2) | 2 (11) |
| Nausea | 2 (4) | 1 (6) |
| Dizziness | 2 (4) | 0 (0) |
| Abdominal pain | 0 (0) | 2 (11) |
| Fatigue | 1 (2) | 1 (6) |
| Syncope | 1 (2) | 1 (6) |
| Viral infection | 1 (2) | 1 (6) |
All four subjects with UC had treatment-emergent adverse events. Two cases each of upper respiratory tract infection and headache occurred in UC subjects who received AMG 181 and abdominal pain and injection site erythema were each reported for two subjects (one AMG 181, one placebo). Among the four subjects with UC, there were a total of 37 adverse events (34 AMG 181, three placebo). These adverse events were reported as grade 1 in severity, except for a grade 2 malaise and a grade 3 ulcerative colitis flare, both of which occurred in one subject who received 210 mg s.c. and grade 2 arthralgia in another subject who received 210 mg s.c. In addition, a serious, grade 4 colitis flare was reported for the subject who received placebo leading to the subject's early discontinuation at the day 43. Aside from the serious adverse event of colitis, no other serious adverse events were reported.
For healthy subjects and subjects with UC, no trends indicative of clinically important treatment-related laboratory abnormalities, ECG changes or changes in vital signs were observed.
All subjects enrolled in this study tested negative for JC viraemia and ADAs at all pre-selected time points as outlined in Supplementary Table S1, with the exception of one subject who was mistakenly enrolled with a positive JC viraemia test at screening. This subject received 210 mg AMG 181 s.c. and tested negative for JC viraemia at all post-screening time points.
Based on review of the safety and immunogenicity data, the safety and tolerability profile of AMG 181 has been acceptable in healthy subjects receiving single s.c. dose of up to 210 mg or single i.v. dose of up to 420 mg and in UC subjects receiving single s.c. dose of up to 210 mg.
Pharmacokinetics
The mean (SD) serum AMG 181 concentration–time profiles from all 53 AMG 181-treated subjects (50 healthy and three UC) are presented in Figure 2. After a single 0.7 mg s.c. administration, a complete AMG 181 concentration–time profile was available only for one subject. The two other subjects each had only one measureable concentration.
Figure 2.
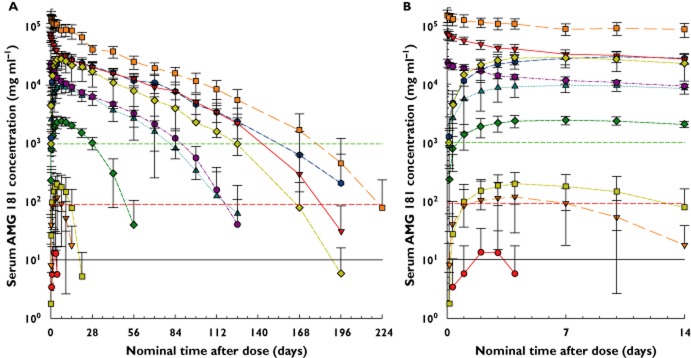
Mean (SD) serum AMG 181 concentration–time profiles after single subcutaneous or intravenous dose in healthy or ulcerative colitis subjects (UC). The full PK profile (A) and the absorption phase only (B) are shown. s.c. = subcutaneous; i.v. = intravenous; LLOQ = lower limit of quantification; RO = α4β7 receptor occupancy on CD4+ naïve T cells. LLOQ and EC50 = 0.01 μg ml−1 (—), EC90 = 0.09 μg ml−1 (– –), and EC99 = 0.99 μg ml−1 (- - -).  , Cohort 1: 0.7 mg s.c. (n = 3–4);
, Cohort 1: 0.7 mg s.c. (n = 3–4);  , Cohort 2: 2.1 mg s.c. (n = 4);
, Cohort 2: 2.1 mg s.c. (n = 4);  , Cohort 3: 7 mg s.c. (n = 5–6);
, Cohort 3: 7 mg s.c. (n = 5–6);  , Cohort 4: 21 mg s.c. (n = 5–6);
, Cohort 4: 21 mg s.c. (n = 5–6);  , Cohort 5: 70 mg s.c. (n = 5–6);
, Cohort 5: 70 mg s.c. (n = 5–6);  , Cohort 6: 210 mg s.c. (n = 5–6);
, Cohort 6: 210 mg s.c. (n = 5–6);  , Cohort 7: 70 mg i.v. (n = 5–6);
, Cohort 7: 70 mg i.v. (n = 5–6);  , Cohort 8: 210 mg i.v. (n = 4–6);
, Cohort 8: 210 mg i.v. (n = 4–6);  , Cohort 9: 420 mg i.v. (n = 4–6);
, Cohort 9: 420 mg i.v. (n = 4–6);  , Cohort 10: 210 mg s.c.UC (n = 3).
, Cohort 10: 210 mg s.c.UC (n = 3).
AMG 181 exhibited non-linear PK with rapid concentration decline when concentrations fell below approximately 1 μg ml−1. This is consistent with target-mediated disposition characteristics that have previously been reported for monoclonal antibodies. At low AMG 181 concentration, clearance is mainly driven by the low-capacity and highly specific saturable binding (and subsequent internalization) to the target cell surface receptor α4β7. At higher concentration, high-capacity and non-specific clearance through FcRn is more predominant and thus is masking the target-mediated disposition. For doses of ≥ 21 mg s.c., AMG 181 PK was characterized by a rapid absorption phase, a slow linear disposition phase, and a target-mediated disposition phase. The AMG 181 PK profiles after 70 and 210 mg i.v. were generally comparable with those through the s.c. route at the same doses, except for the initial rapid distribution phase typically observed for i.v. administration.
A summary of the AMG 181 PK parameters using non-compartmental analysis is provided in Table 3. Of the 50 healthy subjects who received AMG 181, two (one each for 0.7 and 7 mg s.c.) were excluded from non-compartmental analysis due to insufficient data. After s.c. administration, the tmax increased with dose and ranged between 2 to 10 days (2 to 14 days for individual subjects). A relative bioavailability of 82% and 99% was observed for 70 and 210 mg, respectively. As described above, AMG 181 PK profiles exhibited non-linearity due to target-mediated disposition at concentrations < 1 μg ml−1, resulting in greater than dose proportional increases in Cmax, AUC(0,∞) and β phase t1/2 when doses were between 0.7 and 21 mg s.c. AMG 181 Cmax was dose proportional across the 21 to 210 mg s.c. and 70 to 420 mg i.v. ranges. Across the 70 to 210 mg s.c. and 70 to 420 mg i.v. ranges, AMG 181 AUC(0,∞) was approximately dose proportional. The estimated mean AMG 181 linear β phase t1/2 was identical for 70 to 210 mg s.c. (31 days; n = 14, including UC) and for 70 to 420 mg i.v. (31 days, n = 17) administrations. The grand mean (SD) for t1/2 across all 70 to 420 mg s.c. and i.v. doses was 31 (7) days (n = 31, including UC; individual range 20 to 48) days. For 70 to 420 mg i.v. doses, the estimated AMG 181 CL and Vss grand mean (SD) values (n = 17) were 0.120 (0.025) l day−1 and 4.30 (0.82) l, respectively.
Table 3.
Mean (SD) pharmacokinetic parameters of AMG 181 after a single intravenous (i.v.) or subcutaneous (s.c.) administration in healthy subjects, or subjects with ulcerative colitis (UC)
| Regimen | tmax (days, h) | Cmax (μg ml−1) | Cmax/Dose (μg ml−1 mg−1) | AUC(0,t) (μg ml−1 day) | AUC(0,∞) (μg ml−1 day) | AUC(0,∞)/Dose (μg ml−1 day mg−1) | t1/2 (days) | Vz, Vz/F (l) | CL, CL/F (l day−1) | Vss (l) |
|---|---|---|---|---|---|---|---|---|---|---|
| 0.7 mg s.c. | 2.0 | 0.0225 | 0.0322 | 0.0433 | NC | NC | NC | NC | NC | NC |
| (n = 3) | (2.0, 3.0) | (0.0180) | (0.0256) | (0.0644) | NC | NC | NC | NC | NC | NC |
| 2.1 mg s.c. | 3.5 | 0.126 | 0.0598 | 1.01 | 2.84* | 1.35* | 11.0* | 11.7* | 0.739* | NC |
| (n = 4) | (2.0, 7.1) | (0.100) | (0.0476) | (0.95) | NC | NC | NC | NC | NC | NC |
| 7 mg s.c. | 4.0 | 0.245 | 0.0351 | 2.64 | 4.12* | 0.589* | 10.0* | 24.5* | 1.70* | NC |
| (n = 5) | (2.0, 4.0) | (0.046) | (0.0066) | (1.25) | NC | NC | NC | NC | NC | NC |
| 21 mg s.c. | 7.1 | 2.51 | 0.119 | 63.5† | 67.2† | 3.20† | 20.6† | 9.53† | 0.326† | NC |
| (n = 6) | (3.1, 10.0) | (0.51) | (0.024) | (13.6) | (15.0) | (0.71) | (9.0) | (3.92) | (0.078) | NC |
| 70 mg s.c. | 5.6 | 10.1 | 0.144 | 410† | 414† | 5.92† | 25.2† | 6.36† | 0.185† | NC |
| (n = 6) | (3.0, 10.0) | (2.45) | (0.035) | (149) | (155) | (2.22) | (7.0) | (1.16) | (0.056) | NC |
| 210 mg s.c. | 10.0 | 29.8 | 0.142 | 1690† | 1720 | 8.21 | 37.3 | 6.76 | 0.132 | NC |
| (n = 6) | (7.0, 14.0) | (6.2) | (0.030) | (520) | (487) | (2.32) | (7.43) | (1.08) | (0.046) | NC |
| 70 mg i.v. | 1.4 | 23.0 | 0.329 | 501 | 503 | 7.19 | 27.3 | 5.50 | 0.140 | 4.12 |
| (n = 6) | (1.4, 1.6) | (3.5) | (0.050) | (34) | (34) | (0.48) | (3.5) | (0.63) | (0.010) | (0.43) |
| 210 mg i.v. | 1.0 | 72.1 | 0.343 | 1720† | 1730† | 8.25† | 34.5† | 6.17† | 0.124† | 5.08† |
| (n = 6) | (1.0, 1.0) | (13.3) | (0.063) | (290) | (277) | (1.32) | (4.1) | (1.25) | (0.023) | (0.86) |
| 420 mg i.v. | 1.0 | 151 | 0.360 | 4380† | 4480 | 10.7 | 32.2 | 4.40 | 0.0965 | 3.83 |
| (n = 6) | (1.0, 94.8) | (35.3) | (0.084) | (860) | (802) | (1.91) | (6.6) | (0.78) | (0.0189) | (0.66) |
| 210 mg s.c. | 4.0 | 30.1 | 0.143 | 1270 | 1270 | 6.07 | 24.0 | 7.31 | 0.228 | NC |
| (UC; n = 3) | (3.9, 7.2) | (8.47) | (0.040) | (682) | (687) | (3.27) | (5.1) | (4.60) | (0.176) | NC |
n = 1
n = 5. Smaller ns indicated insufficient data for estimation due to BQL values or subjects withdrew consent early. All values are reported to three significant figures except for tmax, where values are reported to one decimal place. tmax: time to Cmax, presented as median (min–max); units: days for s.c., h for i.v.. Cmax: maximum observed AMG 181 concentration in serum; dose-normalized value: Cmax/Dose. AUC(0,t): area under the concentration–time curve from time zero to the last observed concentration. AUC(0,∞): area under the concentration–time curve from time zero extrapolated to infinity; dose-normalized value: AUC(0,∞)/Dose. t1/2: linear β-phase half-life. Vz and Vz/F: linear β-phase volume and apparent volume of distribution for i.v. and s.c., respectively; Vss: volume of distribution at steady-state for i.v.. F: bioavailability after s.c. administration. CL and CL/F: clearance and apparent clearance for i.v. and s.c., respectively. NC, not calculated.
Although this study was not prospectively designed to evaluate the potential effect of demographic parameters, plots of individual Cmax/Dose and AUC(0,∞)/Dose values vs. body weight, BMI, age and race were examined. Figure 3 shows that the larger the body weight, the lower the AMG 181 Cmax/Dose and AUC(0,∞)/Dose values, with a smaller effect observed for Cmax/Dose for s.c. doses. The body weight effect was apparent within each cohort and among all cohorts combined. Except for Cmax/Dose after dosing (Figure 3C), no other slopes of the linear regression lines were statistically significantly different from zero (P > 0.05). Results for BMI effect on AMG 181 Cmax/Dose and AUC(0,∞)/Dose were consistent with those for body weight, with the effect of BMI being much less pronounced. Age had virtually no effect on AMG 181 Cmax/Dose and AUC(0,∞)/Dose. Race, categorized by White, Black and others had no effect on AMG 181 Cmax/Dose and AUC(0,∞)/Dose (analysis of variance or anova for race effect: P > 0.05).
Figure 3.

Body weight effects on dose-normalized AMG 181 Cmax (Cmax/Dose) and AUC(0,∞) (AUC(0,∞)/Dose) for subcutaneous (A and B) or intravenous (C and D) administration. Symbols are observations and lines represent linear regressions. s.c. = subcutaneous; i.v. = intravenous. (A and B) •, Cohort 1: 0.7 mg s.c. (n = 1–3);  , Cohort 2: 2.1 mg s.c. (n = 2–4);
, Cohort 2: 2.1 mg s.c. (n = 2–4);  , Cohort 3: 7 mg s.c. (n = 5);
, Cohort 3: 7 mg s.c. (n = 5);  , Cohort 4: 21 mg s.c. (n = 5);
, Cohort 4: 21 mg s.c. (n = 5);  , Cohort 5: 70 mg s.c. (n = 5);
, Cohort 5: 70 mg s.c. (n = 5);  , Cohort 6: 210 mg s.c. (n = 5);
, Cohort 6: 210 mg s.c. (n = 5);  , Cohort UC (10): 210 mg s.c. (n = 3); —, linear regression line. (C and D) •, Cohort 7: 70 mg i.v. (n = 6);
, Cohort UC (10): 210 mg s.c. (n = 3); —, linear regression line. (C and D) •, Cohort 7: 70 mg i.v. (n = 6);  Cohort 8: 210 mg i.v. (n = 5–6);
Cohort 8: 210 mg i.v. (n = 5–6);  , Cohort 9: 420 mg i.v. (n = 5–6); —, linear regression line.
, Cohort 9: 420 mg i.v. (n = 5–6); —, linear regression line.
Pharmacodynamics
AMG 181 effects on free (measured by anti-α4β7-ROA1 with competitive binding against AMG 181) and total (measured by anti-β7-ROA2, binding non-competitively against AMG 181) α4β7 receptors on CD4+ naïve T cells and central memory T cells are presented in Figure 4A–D. The pre-dose normalized percent free α4β7 receptor profiles on circulating CD4+ naïve T cells were directly correlated with time-matched AMG 181 concentrations. The duration of this PD effect was dose-related and reversible upon elimination of AMG 181. For example, > 90% saturation of α4β7 receptors was achieved for ≥ 43 days, ≥ 71 days, and ≥ 127 days following a single dose of 21 mg, 70 mg (i.v. or s.c.), and 210 mg (i.v. or s.c.) AMG 181, respectively, reaching or approaching pre-dose baseline at the end of study (Figure 4A).
Figure 4.

Mean (SEM) percent of pre-dose baseline-normalized α4β7 receptor profiles on CD4+ naïve (A free, B total) and central memory (C free, D total) T cells after AMG 181 or placebo administration in healthy subjects. s.c. = subcutaneous; i.v. = intravenous. HV = healthy volunteers.  , Cohort 1: 0.7 mg s.c. (n = 3–4);
, Cohort 1: 0.7 mg s.c. (n = 3–4);  , Cohort 2: 2.1 mg s.c. (n = 4);
, Cohort 2: 2.1 mg s.c. (n = 4);  , Cohort 3: 7.0 mg s.c. (n = 5–6);
, Cohort 3: 7.0 mg s.c. (n = 5–6);  , Cohort 4: 21 mg s.c. (n = 5–6);
, Cohort 4: 21 mg s.c. (n = 5–6);  , Cohort 5: 70 mg s.c. (n = 4–6);
, Cohort 5: 70 mg s.c. (n = 4–6);  , Cohort 6: 210 mg s.c. (n = 4–6);
, Cohort 6: 210 mg s.c. (n = 4–6);  , Cohort 7: 70 mg i.v. (n = 5–6);
, Cohort 7: 70 mg i.v. (n = 5–6);  , Cohort 8: 210 mg i.v. (n = 4–6);
, Cohort 8: 210 mg i.v. (n = 4–6);  , Cohort 9: 420 mg i.v. (n = 4–6);
, Cohort 9: 420 mg i.v. (n = 4–6);  , placebo (n = 1–18)
, placebo (n = 1–18)
Similar dose-associated and concentration-correlated profiles were observed for circulating CD4+ central memory T cells (Figure 4C) and CD4+ effector memory and total memory T cells (data on file at Amgen Inc.). Total α4β7 levels on both CD4+ naïve and central memory T cells were decreased by approximately 40 to 60% at nadir but were reversible upon elimination of AMG 181 (Figure 4B and D). Data for other CD4+ T cell subsets showed similar profiles (on file at Amgen Inc.).
Individual healthy subject pre-dose baseline-normalized α4β7 RO on CD4+ naïve T cells vs. serum AMG 181 concentration profile and the Emax PD model fitted curve are presented in Figure 5. As with AMG 181 PK, PD was apparently not affected by race (data on file at Amgen Inc.). The majority of the data points between AMG 181 concentrations of 0.01 to 100 μg ml−1 corresponded to > 90% α4β7 RO, indicating a very potent PD effect of AMG 181. The PD model estimated EC50 value for α4β7 RO on CD4+ naïve T cells was 0.00884 μg ml−1 (%CV 13.7%) with the estimated Emax and E0 values of 93.1% and 7.73%, respectively. For the α4β7 RO on CD4+ central memory and effector memory T cells, the estimated EC50 values were 0.0140 (%CV 20.8%) and 0.0102 μg ml−1 (%CV: 29.2%), respectively. Because all three estimated RO EC50 values were close to the assay LLOQ of 0.01 μg ml−1, the EC50 value was set at 0.01 μg ml−1 and plotted, together with the estimated EC90 and EC99 lines, in Figure 2.
Individual healthy subject AUECall for pre-dose baseline-normalized α4β7 receptor occupancy on CD4+ naïve T cells vs. AMG 181 AUCall profile and the Emax PD model fitted curve are presented in Figure 6. The PD model estimated EAUCall,50 value for CD4+ naïve T cells was 411 μg ml−1 day (%CV 15%; Emax 19900 day·%), coinciding with a 70 mg dose. The estimated EAUCall,90 of 3460 μg ml−1 day corresponds to a 324 mg dose. For CD4+ central memory and effector memory T cells, the estimated EAUCall,50 values were 538 and 530 μg ml−1 day, respectively, neither of which was substantially different from that of CD4+ naïve T cells.
Figure 6.
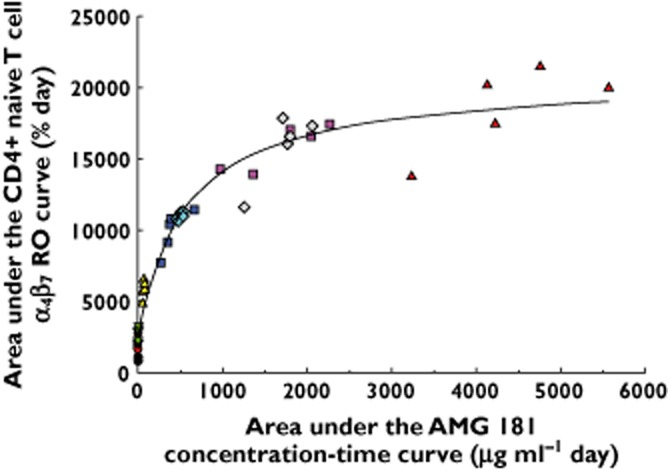
Individual healthy subject area under the effect−time curve for pre-dose baseline-normalized α4β7 receptor occupancy (RO) on CD4+ naïve T cells (AUECall) vs. serum AMG 181 area under the concentration−time curve (AUCall) profile (symbols) and the Emax PD model fitting of the observed data (line). s.c. = subcutaneous; i.v. = intravenous. •, Cohort 1: 0.7 mg s.c. (n = 3);  , Cohort 2: 2.1 mg s.c. (n = 4);
, Cohort 2: 2.1 mg s.c. (n = 4);  , Cohort 3: 7 mg s.c. (n = 5);
, Cohort 3: 7 mg s.c. (n = 5);  , Cohort 4: 21 mg s.c. (n = 5);
, Cohort 4: 21 mg s.c. (n = 5);  , Cohort 5: 70 mg s.c. (n = 5);
, Cohort 5: 70 mg s.c. (n = 5);  , Cohort 6: 210 mg s.c. (n = 5);
, Cohort 6: 210 mg s.c. (n = 5);  , Cohort 7: 70 mg i.v. (n = 6);
, Cohort 7: 70 mg i.v. (n = 6);  , Cohort 8: 210 mg i.v. (n = 5);
, Cohort 8: 210 mg i.v. (n = 5);  , Cohort 9: 420 mg i.v. (n = 5)
, Cohort 9: 420 mg i.v. (n = 5)
Figure 7 shows that the absolute mean (SEM) count profiles of lymphocytes, eosinophils, CD4+ T cells and their subsets were virtually identical between AMG 181 and placebo treatments in all subjects.
Figure 7.
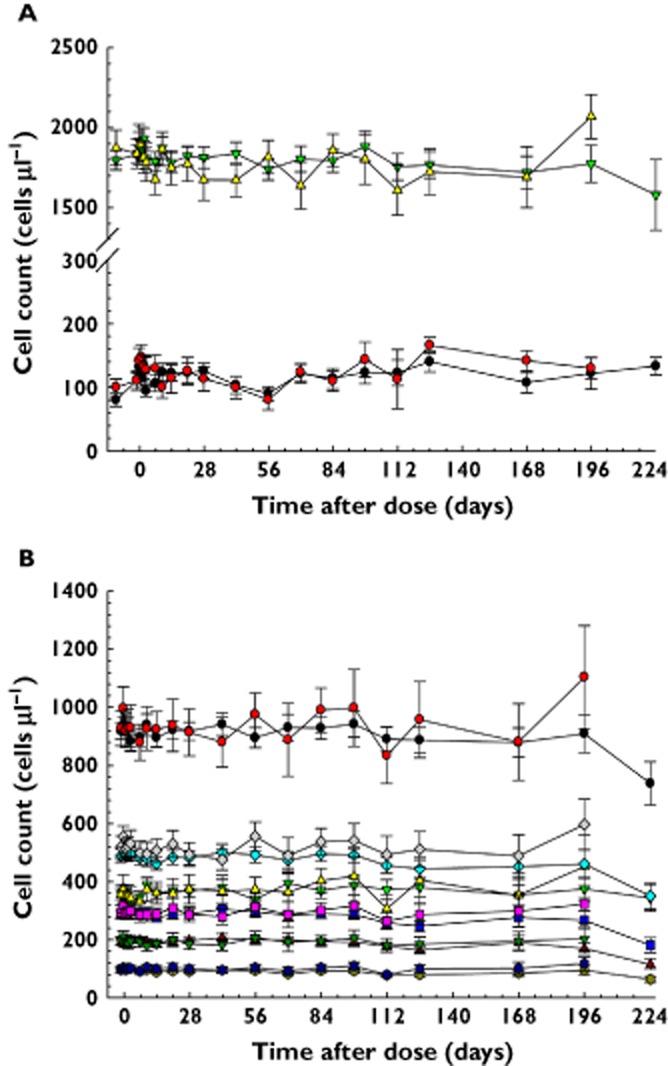
Mean count (SEM) profiles of lymphocytes and eosinophils (A) and CD4+ total, naïve, central memory (Tcm), and memory (mem) T cells, and Tcm cells expressing low or high levels of α4β7 receptors (B) in all subjects treated with AMG 181 or placebo. (A)  , Eosinophil, active;
, Eosinophil, active;  , Eosinophil, placebo;
, Eosinophil, placebo;  , Lymphocyte, active;
, Lymphocyte, active;  , Lymphocyte, placebo; Active (n = 5–50); Placebo (n = 6–19). (B)
, Lymphocyte, placebo; Active (n = 5–50); Placebo (n = 6–19). (B)  , CD4 + total, active;
, CD4 + total, active;  , CD4 + total, placebo;
, CD4 + total, placebo;  , CD4 + naive, active;
, CD4 + naive, active;  , CD4 + naive, placebo;
, CD4 + naive, placebo;  , CD4 + Tcm, active;
, CD4 + Tcm, active;  , CD4 + Tcm, placebo;
, CD4 + Tcm, placebo;  , CD4 + mem, active;
, CD4 + mem, active;  , CD4 + mem, placebo;
, CD4 + mem, placebo;  , α4β7 low CD4 + Tcm, active;
, α4β7 low CD4 + Tcm, active;  , α4β7 low CD4 + Tcm, placebo;
, α4β7 low CD4 + Tcm, placebo;  , α4β7 high CD4 + Tcm, active;
, α4β7 high CD4 + Tcm, active;  , α4β7 high CD4 + Tcm, placebo; Active (n = 5–52); Placebo (n = 5–18)
, α4β7 high CD4 + Tcm, placebo; Active (n = 5–52); Placebo (n = 5–18)
Pharmacokinetics, pharmacodynamics, and effects in ulcerative colitis subjects
AMG 181 PK profiles and the estimated PK parameters in two UC subjects were similar to healthy subjects receiving the same 210 mg s.c. dose. In the remaining actively treated UC subject, the AMG 181 PK profile was initially (up to day 8) similar to that of healthy subjects', but displayed a subsequent rapid decline and reached BQL on day 85 (Figure 8A). No ADAs were detected in any of this subject's serum samples. The PD profiles (free and total α4β7 levels) mirrored the PK profiles: The two UC subjects with PK profiles comparable with healthy subjects also had PD profiles comparable with healthy subjects. The subject with the rapid decline in AMG 181 PK also had a rapid reversal of the initial RO response (Figure 8A). Consistent with the findings in healthy subjects, AMG 181 concentration and the pre-dose baseline adjusted rate of free α4β7 receptor on CD4+ naïve T cells were directly correlated (Figure 8A). In addition, similar to healthy subjects, the CD4+ central memory T cell counts did not change in UC subjects after 210 mg AMG 181 or placebo s.c. dosing (Figure 8B).
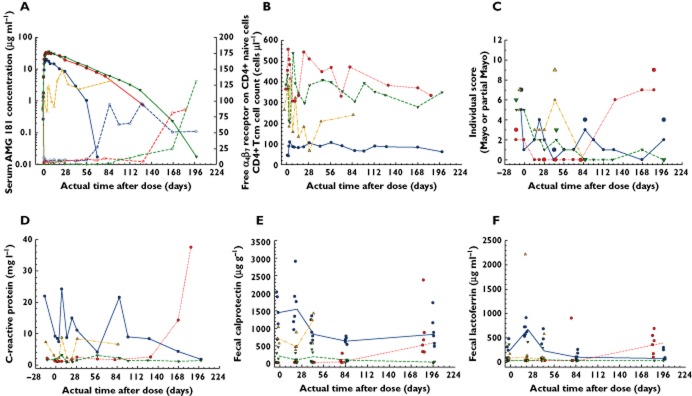
Figure 8.
Individual profiles in ulcerative colitis subjects: AMG 181 concentration (left y-axis; PK) and pre-dose baseline-normalized rate of free α4β7 receptor on CD4+ naïve T cells (right y -axis; PD) (A), CD4 + central memory T cell (Tcm) count (B), total (large symbols only) and partial (small symbols with lines connected) Mayo score (C), serum C-reactive protein (D), fecal calprotectin (E) and lactoferrin (F) presented as observed (symbols) and median (lines). Note: Subject 4 was on placebo; others were on 210 mg AMG 181s.c. (A) PK:  , Subject 1;
, Subject 1;  , Subject 2;
, Subject 2;  , Subject 3;
, Subject 3;  , Subject 4 PD:
, Subject 4 PD:  , Subject 1;
, Subject 1;  , Subject 2;
, Subject 2;  , Subject 3;
, Subject 3;  , Subject 4. (B and D)
, Subject 4. (B and D)  , Subject 1;
, Subject 1;  , Subject 2;
, Subject 2;  , Subject 3;
, Subject 3;  , Subject 4. (C) Mayo:
, Subject 4. (C) Mayo:  , Subject 1;
, Subject 1;  , Subject 2;
, Subject 2;  , Subject 3;
, Subject 3;  , Subject 4 Partial Mayo:
, Subject 4 Partial Mayo:  , Subject 1;
, Subject 1;  , Subject 2;
, Subject 2;  , Subject 3;
, Subject 3;  , Subject 4
, Subject 4  ,
,  ,
,  ,
,  . (E and F)
. (E and F)  , Subject 1;
, Subject 1;  , Subject 2;
, Subject 2;  , Subject 3;
, Subject 3;  , Subject 4;
, Subject 4;  ,
,  ,
,  ,
, 
At screening, the four UC subjects had a median Mayo score of 6.5 (range 3 to 7), with median score of 6 for the three subjects who received AMG 181 and a score of 7 for the subject who received placebo (Table 4). One UC subject with a Mayo score of 3 was enrolled after discussion between the investigator and Amgen and approval by the Amgen medical monitor. The screening recto-sigmoidoscopy scores for the three AMG 181-treated subjects were 2, 2 and 1, and 2 for the placebo-treated subject.
Table 4.
Effects of a single subcutaneous 210 mg AMG 181 or placebo administration in individual subjects with ulcerative colitis
| Subject | Regimen | Mayo score (Remission*, Response†, Mucosal Healing‡)§ | Concomitant therapy for UC treatment | |||
|---|---|---|---|---|---|---|
| Screening | Day 43 | Day 85 | Day 197 | |||
| 1 | AMG 181 | 7 | 1 (+, +, +) | 4 (−, +, +) | 4 (−, +, −) | None |
| 2 | AMG 181 | 3 | 0 (+, +, +) | 0 (+, +, +) | 9 (−, −, −)¶ | Mesalazine |
| 3 | AMG 181 | 6 | 3 (−, +, +) | 0 (+, +, +) | 0 (+, +, +) | Balsalazide sodium |
| 4 | Placebo | 7 | 9 (−, −, −)** | NA | NA | Aminosalicylic acid, prednisolone |
Note: Full Mayo score assessed on days 43, 85 and 197, corresponding to weeks 6, 12 and 28 visits.
Remission: Mayo Score ≤ 2, with no individual subscore > 1 point.
Response: Mayo Score ↓ from baseline ≥ 3 and ≥ 30% + rectal bleeding score ↓ ≥ 1 point or absolute reading of 0 or 1.
Mucosal healing: rectosigmoidoscopy score of 0 or 1.
‘+’ or ‘-’ = ‘Yes’ or ‘No’, respectively, for remission, response or mucosal healing.
Grade 3 ulcerative colitis flare.
Grade 4 ulcerative colitis flare. Subject experienced serious adverse event of colitis flare and withdrew from the study after day 43 visit. This subject received prednisolone, hydrocortisone and mesalazine as rescue therapies and recovered at days 85 and 197 visits. NA, Not Applicable.
After dosing, all three AMG 181-treated subjects completed the study, while the placebo-treated subject withdrew from the study after day 43 visit due to colitis flare and subsequently received rescue therapy (recovered from colitis on days 85 and 197 visits; Table 4). The three AMG 181-treated subjects achieved remission, response and mucosal healing at multiple time points of the study. In all, the eight instances of responses were matched with five instances of remissions and seven instances of mucosal healing (Table 4).
Figure 8C shows that the Mayo and partial Mayo score profiles were consistent throughout the study for each of the subjects. By day 43, subjects 1 and 2, which received AMG 181 had reached the lowest Mayo and partial Mayo scores and were in remission. Subject 3 (also received AMG 181) reached remission beginning on day 85 and sustained remission until the end of the 28 week (day 197) study period. Based on the partial Mayo score alone, and by defining clinical response as a reduction of ≥ 2 points and ≥ 25% change from baseline 48, sustained response was achieved for 10 (from week 2 to 12), 24 (from week 4 to 28), and 26 (from week 2 to 28) weeks for subjects 1, 2 and 3, respectively.
The two subjects who had the last measurable AMG 181 concentration on days 127 and 71 experienced Mayo and partial Mayo scores rebounding from nadir afterwards, while the third subject having an AMG 181 concentration measurable until day 197 had sustained response and remission to the end of the study on day 197 (Figure 8C). This indicates that maintaining AMG 181 exposure may be required for sustaining clinical effects.
Figure 8D to F presents serum C-reactive protein, fecal calprotectin and lactoferrin profiles for all four UC subjects. Compared with pre-dose baselines, the three biomarkers remained flat, trended lower or rebounded at different time points, generally matching the PK/PD profiles individually. Note that for subject 2, AMG 181 concentration reached LLOQ after week 20, with corresponding reduced α4β7 receptor coverage on CD4+ naïve T cells (Figure 8A), return of CD4+ central memory T cell count toward baseline (Figure 8B), rebound in Mayo and partial Mayo scores (Figure 8C) and elevation of CRP and fecal biomarkers (Figure 8D–F). Overall, due to small sample size and data variability, the effects of AMG 181 treatment on these biomarkers could not be definitively or quantitatively assessed.
Discussion
In this study we evaluated single dose PK/PD, safety, tolerability and effects of the gut-specific fully human monoclonal anti-α4β7 antibody AMG 181 after s.c. or i.v. administration in healthy subjects and subjects with mild to moderate UC. AMG 181 blocks the interaction of the α4β7 integrin with its target ligand MAdCAM-1 and thus prevents lymphocyte adhesion, extravasation and homing to the gut mucosal tissue.
After s.c. dosing, AMG 181 was extensively absorbed with relatively high bioavailability of 82 to 99% and a long half-life of ∼31 days. AMG 181 displayed non-linear PK with a rapid decline when concentrations fell below 1 μg ml−1. This is consistent with target-mediated disposition and is especially important for AMG 181 doses of ≤ 21 mg. AMG 181 doses of ≥ 70 mg resulted in saturation of this target-specific mechanism and the non-specific linear processes masked the target-mediated disposition. Consequently, approximately linear PK profiles were observed at higher AMG 181 doses, where the terminal non-linear phase had limited contribution to the AUC. The estimated AMG 181 linear CL and Vss of 0.120 (0.025) l day−1 and 4.30 (0.82) l indicate that AMG 181 was cleared slowly during the linear elimination phase, with distribution mostly contained within the central circulation. Because the 21, 70 and 210 mg s.c. cohorts maintained AMG 181 concentrations above the RO EC99 level of 0.9 μg ml−1 for approximately 4, 12 and 22 weeks, it may be possible to propose therapeutic regimens by dosing AMG 181 once every 1, 3 or 6 months, respectively, taking moderate accumulations after repeated dosing into consideration.
Based on non-compartmental analysis, the mean predicted extrapolation from AUC(0,t) for calculating AUC(0,∞) was ∼5% for cohorts ≥ 70 mg s.c. or i.v., which was virtually identical to the mean observed percent extrapolation, indicating that the terminal non-linear target-mediated disposition phase was negligible and did not influence the calculation of linear β-phase PK parameters such as t1/2, CL and Vss. Because the influence of target-mediated disposition was minor after a single dose of ≥ 70 mg, and because of the expected moderate accumulation after repeated dosing which would result in trough AMG 181 concentrations of ≥ 1 μg ml−1 at doses larger than 21 mg s.c., we expect that repeated dosing of 21, 70 and 210 mg s.c. once every 1, 3, and 6 months, respectively, would show linear PK characteristics at steady-state with an estimated effective t1/2 similar to the linear elimination t1/2 of 31 days. In a future report, we will examine this prediction by correlating the linear beta phase t1/2 estimated in this single dose study with the estimated effective t1/2 from the multiple ascending dose study in healthy and UC subjects 43.
Potential demographic effects on AMG 181 PK were explored by plotting and regressing individual Cmax/Dose and AUC(0,∞)/Dose values vs. body weight, BMI, age and race. While the latter three showed no statistically significant effects on AMG 181 exposures, the body weight had statistically significant effects on the Cmax/Dose after i.v. administration. The clinical relevance of this finding will need to be assessed in a larger sample of IBD subjects. Note that in the current study, heavier body weight, especially the one healthy subject weighing ∼120 kg in the i.v. cohort had contributed to the statistical significance of the linear regression for Cmax/Dose. Using target α4β7 RO as the guide post for selecting a therapeutic regimen, one might argue that with repeated dosing, the expected PK accumulation at steady-state should ensure adequate target α4β7 receptor coverage at the trough. Furthermore, the hysteresis phenomenon reported as prolonged therapeutic effects lasting longer than drug concentration and target coverage previously reported with natalizumab and vedolizumab suggests the potential for accommodating, at a population level, lower trough AMG 181 concentration due to heavier body weight 39,40,49.
No ADAs were detected in this study, so the potential effects of immunogenicity on AMG 181 PK/PD, safety and treatment effects could not be examined. Circulating serum AMG 181 concentrations in some subjects at certain time points were higher than the immunogenicity assay drug tolerance level of 25 μg ml−1. For example, on days 15 and 29 for subjects dosed 210 mg s.c. or i.v., and on days 15, 29, and 57 for subjects dosed 420 mg i.v., determination of ADAs may have been prevented, providing false negative results at those time points. However, the five to six sampling time points after day 29 or 57, where AMG 181 concentrations were < 25 μg ml−1, had provided a valid ADA negative result, suggesting that these subjects at least did not develop persistent ADAs.
Two of the three UC subjects exhibited AMG 181 PK similar to that of the healthy subjects given the same dose of 210 mg s.c. The one UC subject (subject 1) who had lower AMG 181 PK did not display unusually high target α4β7 receptor expression (data on file at Amgen Inc.) or develop immunogenicity. One might speculate that high disease activity, and/or gut-leaking protein losing enteropathy as suggested by low serum albumin level at screening (< 3.5 g dl−1) might have contributed to the faster AMG 181 clearance as has been previously reported for infliximab in IBD subjects 50,51. Also, AMG 181 PD and treatment effects in this subject were sub-optimal vs. the other two AMG 181-treated UC subjects. Due to the small sample size, we could not reach a conclusion for possible impact of disease status on AMG 181 PK/PD in UC, even though the correlation between PK and PD appeared to be consistent between healthy and UC subjects.
AMG 181 showed potent yet reversible effects on saturating α4β7 receptors on target T cells. One subset, the CD4+ naïve T cells, homogenously expressed α4β7 receptors, while the other subset, CD4+ central memory T cells, was further divided into two subsets, one of which expressed low while the other expressed very high levels of α4β7 receptors. The median flow cytometry measurements of α4β7 receptors on CD4+ central memory T cells included data from cells expressing low and high α4β7 receptors and were thus less homogenous than those from CD4+ naïve T cells. Therefore, we presented both free and total α4β7 receptors on these two cell types (Figure 4) and modelled their relationships with AMG 181 concentrations. The PD modelling results show that the EC50 values for the CD4+ naïve, central and effector memory T cells were similar, indicating similar potency of about 0.01 μg ml−1, the PK assay LLOQ. Thus using α4β7 RO on CD4+ naïve T cells as a surrogate representing RO values for central memory T cells was adequate.
Excluding data from all placebo-treated healthy subjects and all pre-dose PK samples from AMG 181-treated healthy subjects, a total of 111 out of 658 (16.9%) PK samples had AMG 181 concentrations < 0.01 μg ml−1, the assay LLOQ. These values were excluded from analysis but were plotted at half of the LLOQ (0.005 μg ml−1) in Figure 5, to provide information about the spread of RO. The calculated EC90 value of 0.09 μg ml−1 was a reasonable estimate, as the observations (symbols) in Figure 5 showed that RO was mostly > 90% when AMG 181 concentrations were > 0.1 μg ml−1. This consistency can also be observed by comparing the α4β7 RO of over 99% based on the duration of the PK profiles over the EC99 line (Figure 2) and those observed as presented in Figure 4A and C for doses ≥ 21 mg.
Two complimentary approaches were used to assess quantitatively the AMG 181 PK/PD relationship. One was the direct, time-matched, instant exposure (concentration) approach (Figure 5) and the other was the accumulative exposure (AUC) approach (Figure 6). The Emax model describing the relationship between serum AMG 181 concentration and the subject/time-matched α4β7 RO on total CD4+ T cells (including the subsets) was a simple and direct assessment of AMG 181 ex vivo potency through EC50. The disadvantages of this approach are that some PK/PD sampling time points did not match (Table 1), and that those AMG 181 concentrations that were BQL (below quantitative limit) were excluded from PD modelling as discussed above, even though RO were measurable. By assessing AMG 181 AUCall vs. RO AUECall, mismatched PK/PD observations and BQL measures were no longer an issue and AUECall was a useful guide for measuring the cumulative extent of RO. Figure 6 shows that data points associated with 70 mg i.v. or s.c. doses were concentrated at the inflection point of the curve fitting. To increase AUECall from ∼10000 to 20000 day % (∼two-fold increase), AMG 181 dose would have to increase from 70 to 420 mg i.v., a six-fold increase. Both 210 mg s.c. and i.v. were estimated to achieve 82% of the Emax value of 19900 day %, suggesting limited cumulative RO improvement beyond the 210 mg dose, which is also apparent from visual inspection of Figure 6. The model estimated AMG 181 AUCall leading to 90% maximal effect (EAUCall,90) was approximately 3460·μg ml−1 day, corresponding to a 324 mg dose. Therefore, a ∼1.5-fold dose increase from 210 to 324 mg, would only yield ∼8% increase in cumulative RO. These results imply that the maximum practically useful single dose would be 210 mg i.v. or s.c., with no anticipated gain in PD and efficacy from further dose escalation.
Previous reports showed that natalizumab has led to increases in lymphocyte, eosinophil, basophil, monocyte and detectible nucleated red blood cells 31. In the current study, we did not observe cell count changes in any of these cell types, consistent with the pharmacology of AMG 181, which specifically targets a small population of gut-homing T cells. There were no differences between cohorts, nor pooled AMG 181 vs. placebo data from all healthy and UC subjects. Both lymphocyte and eosinophil counts were well contained within the normal ranges of 1000 to 3500 and 30 to 600 cells μl−1, respectively. Even among CD4+ T cells and the subsets targeted by AMG 181, there were no apparent differences between cohorts or between pooled AMG 181 vs. placebo (Figure 7). Compared with natalizumab, the implication for lack of AMG 181 effects on various immune cells warrants further examination in large confirmatory clinical trials. It remains to be more carefully investigated whether cell counts might change in the IBD population given that circulation dynamics of gut-homing lymphocytes in that population might be different from that in healthy subjects.
Consistent with previous reports 52,53, our data have shown that the Mayo and partial Mayo score profiles were correlated throughout the study for each of the four UC subjects. The three AMG 181-treated UC subjects reached remission on study indicating favourable treatment effects of AMG 181. The onset of remission occurred as early as week 4 for subject 2 and as late as week 12 for subject 3. The lowest Mayo score was achieved within the week 4 to 12 window post-dose for the three AMG 181-treated subjects, suggesting a delayed onset of treatment effects relative to the median AMG 181 PK tmax of 4 days. Subject 1 did not take any concomitant UC therapies. Subjects 2 to 4 were taking aminosalicylic acid while on the study and subject 3 was also taking a steroid. Due to the small sample size, the potential contributions of these concomitant medications could not be assessed.
Greater inflammatory activities are expected in autoimmune diseases such as IBD when serum C-reactive protein, fecal calprotectin and lactoferrin are higher than 10 mg ml−1, 250 μg g−1, and 7 μg ml−1, respectively 54–56. Even though subjects with mild to moderate rather than moderate to severe UC were enrolled, both biological signs of active gut inflammation and clinical measures of disease activities were observed. Overall, serum C-reactive protein and fecal biomarker data were consistent with UC subjects' disease severity and AMG 181 treatment outcomes, even though small sample size and data variability precluded definitive or quantitative assessments.
In conclusion, this study has demonstrated that AMG 181 has an acceptable safety and tolerability profile at single doses of up to 210 mg s.c. or 420 mg i.v., and has the potential for convenient dosing intervals of every 1, 3, and 6 months at 21, 70 and 210 mg, respectively. Results from this study together with those from a repeated dosing study (to be reported separately) have supported advancement of AMG 181 into phase 2 clinical trials in UC and CD 43,44.
Competing Interests
This work and the study presented herein were financially funded by Amgen Inc., Thousand Oaks, CA, USA and MedImmune, LLC, Gaithersburg, MD, USA. AMG 181 is being co-developed by MedImmune, LLC (a wholly-owned subsidiary of AstraZeneca plc.) and Amgen Inc. and may be referred to as AMG 181 or MEDI7183.
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare that all authors had support from Amgen Inc. for the submitted work, J.M.A. received grants and personal fees from AbbVie, Janssen, Shire, Abbott, Ferring, and AstraZeneca in the previous 3 years and W.J.P., B.A.S., D.C.B. and W.A.R. have patents pending or issued that are broadly relevant to the work. All authors declare that they are either employees and shareholders or contractors of Amgen Inc. or consultants who have received research support from Amgen Inc. in the previous 3 years.
The authors would like to thank the Amgen Therapeutic Discovery, Inflammation Research, Medical Sciences, Biostatistics, Safety, Clinical Development, and Pharmacokinetics and Drug Metabolism departments, the California Clinical Trials Medical Group, Inc, the Royal Adelaide Hospital, the Royal Brisbane and Women's Hospital (especially Natalie Gerns, CN, IBD Clinical Trial Coordinator and Nanette Douglas, BN, RN, Clinical Trial Nurse, Q-Pharm Pty Limited), and other Amgen contractors for their kind assistance in the antibody generation, production, and purification, the conduct of the study, as well as data management and processing. The authors would also like to thank the healthy volunteers and the ulcerative colitis patients for study participation, and Keith Langley, PhD and Jon Nilsen, PhD, Amgen Inc. for medical writing support.
References
- Loftus EV., Jr Clinical epidemiology of inflammatory bowel disease: incidence, prevalence, and environmental influences. Gastroenterology. 2004;126:1504–1517. doi: 10.1053/j.gastro.2004.01.063. [DOI] [PubMed] [Google Scholar]
- Thia KT, Loftus EV, Jr, Sandborn WJ, Yang SK. An update on the epidemiology of inflammatory bowel disease in Asia. Am J Gastroenterol. 2008;103:3167–3182. doi: 10.1111/j.1572-0241.2008.02158.x. [DOI] [PubMed] [Google Scholar]
- Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, Benchimol EI, Panaccione R, Ghosh S, Barkema HW, Kaplan GG. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142:46–54. doi: 10.1053/j.gastro.2011.10.001. e42; quiz e30. [DOI] [PubMed] [Google Scholar]
- Asakura K, Nishiwaki Y, Inoue N, Hibi T, Watanabe M, Takebayashi T. Prevalence of ulcerative colitis and Crohn's disease in Japan. J Gastroenterol. 2009;44:659–665. doi: 10.1007/s00535-009-0057-3. [DOI] [PubMed] [Google Scholar]
- Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361:2066–2078. doi: 10.1056/NEJMra0804647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther J, Dave M, Higgins PD, Kao JY. Association between Helicobacter pylori infection and inflammatory bowel disease: a meta-analysis and systematic review of the literature. Inflamm Bowel Dis. 2010;16:1077–1084. doi: 10.1002/ibd.21116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, Leleiko N, Snapper SB, Bousvaros A, Korzenik J, Sands BE, Xavier RJ, Huttenhower C. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012;13:R79. doi: 10.1186/gb-2012-13-9-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein CN, Fried M, Krabshuis JH, Cohen H, Eliakim R, Fedail S, Gearry R, Goh KL, Hamid S, Khan AG, LeMair AW, Malfertheiner, Ouyang Q, Rey JF, Sood A, Steinwurz F, Thomsen OO, Thomson A, Watermeyer G. World Gastroenterology Organization Practice Guidelines for the diagnosis and management of IBD in 2010. Inflamm Bowel Dis. 2010;16:112–124. doi: 10.1002/ibd.21048. [DOI] [PubMed] [Google Scholar]
- Hanauer SB, Feagan BG, Lichtenstein GR, Mayer LF, Schreiber S, Colombel JF, Rachmilewitz D, Wolf DC, Olson A, Bao W, Rutgeerts P. Maintenance infliximab for Crohn's disease: the ACCENT I randomised trial. Lancet. 2002;359:1541–1549. doi: 10.1016/S0140-6736(02)08512-4. [DOI] [PubMed] [Google Scholar]
- Rutgeerts P, Sandborn WJ, Feagan BG, Reinisch W, Olson A, Johanns J, Travers S, Rachmilewitz D, Hanauer SB, Lichtenstein GR, de Villiers WJ, Present D, Sands BE, Colombel JF. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005;353:2462–2476. doi: 10.1056/NEJMoa050516. [DOI] [PubMed] [Google Scholar]
- Rutgeerts PJ. Review article: efficacy of infliximab in Crohn's disease – induction and maintenance of remission. Aliment Pharmacol Ther. 1999;13(Suppl. 4):9–15. doi: 10.1046/j.1365-2036.1999.00025.x. discussion 38. [DOI] [PubMed] [Google Scholar]
- Colombel JF, Sandborn WJ, Rutgeerts P, Enns R, Hanauer SB, Panaccione R, Schreiber S, Byczkowski D, Li J, Kent JD, Pollack PF. Adalimumab for maintenance of clinical response and remission in patients with Crohn's disease: the CHARM trial. Gastroenterology. 2007;132:52–65. doi: 10.1053/j.gastro.2006.11.041. [DOI] [PubMed] [Google Scholar]
- Cottone M, Mocciaro F, Scimeca D. Adalimumab induction for Crohn's disease. Gastroenterology. 2006;130:1929. doi: 10.1053/j.gastro.2006.03.051. [DOI] [PubMed] [Google Scholar]
- Reinisch W, Sandborn WJ, Hommes DW, D'Haens G, Hanauer S, Schreiber S, Panaccione R, Fedorak RN, Tighe MB, Huang B, Kampman W, Lazar A, Thakkar R. Adalimumab for induction of clinical remission in moderately to severely active ulcerative colitis: results of a randomised controlled trial. Gut. 2011;60:780–787. doi: 10.1136/gut.2010.221127. [DOI] [PubMed] [Google Scholar]
- Sandborn WJ, Feagan BG, Stoinov S, Honiball PJ, Rutgeerts P, Mason D, Bloomfield R, Schreiber S. Certolizumab pegol for the treatment of Crohn's disease. N Engl J Med. 2007;357:228–238. doi: 10.1056/NEJMoa067594. [DOI] [PubMed] [Google Scholar]
- Schreiber S, Khaliq-Kareemi M, Lawrance IC, Thomsen OO, Hanauer SB, McColm J, Bloomfield R, Sandborn WJ. Maintenance therapy with certolizumab pegol for Crohn's disease. N Engl J Med. 2007;357:239–250. doi: 10.1056/NEJMoa062897. [DOI] [PubMed] [Google Scholar]
- Sandborn WJ, Feagan BG, Marano C, Zhang H, Strauss R, Johanns J, Adedokun OJ, Guzzo C, Colombel JF, Reinisch W, Gibson PR, Collins J, Jarnerot G, Hibi T, Rutgeerts P. Subcutaneous golimumab induces clinical response and remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2014;146:85–95. doi: 10.1053/j.gastro.2013.05.048. [DOI] [PubMed] [Google Scholar]
- Peyrin-Biroulet L, Lemann M. Review article: remission rates achievable by current therapies for inflammatory bowel disease. Aliment Pharmacol Ther. 2011;33:870–879. doi: 10.1111/j.1365-2036.2011.04599.x. [DOI] [PubMed] [Google Scholar]
- Peyrin-Biroulet L, Deltenre P, de Suray N, Branche J, Sandborn WJ, Colombel JF. Efficacy and safety of tumor necrosis factor antagonists in Crohn's disease: meta-analysis of placebo-controlled trials. Clin Gastroenterol Hepatol. 2008;6:644–653. doi: 10.1016/j.cgh.2008.03.014. [DOI] [PubMed] [Google Scholar]
- Rosenblum H, Amital H. Anti-TNF therapy: safety aspects of taking the risk. Autoimmun Rev. 2011;10:563–568. doi: 10.1016/j.autrev.2011.04.010. [DOI] [PubMed] [Google Scholar]
- Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- Fedyk ER, Wyant T, Yang LL, Csizmadia V, Burke K, Yang H, Kadambi VJ. Exclusive antagonism of the alpha(4) beta(7) integrin by vedolizumab confirms the gut-selectivity of this pathway in primates. Inflamm Bowel Dis. 2012;18:2107–2119. doi: 10.1002/ibd.22940. [DOI] [PubMed] [Google Scholar]
- Hesterberg PE, Winsor-Hines D, Briskin MJ, Soler-Ferran D, Merrill C, Mackay CR, Newman W, Ringler DJ. Rapid resolution of chronic colitis in the cotton-top tamarin with an antibody to a gut-homing integrin alpha 4 beta 7. Gastroenterology. 1996;111:1373–1380. doi: 10.1053/gast.1996.v111.pm8898653. [DOI] [PubMed] [Google Scholar]
- Pan WJ, Lear SP, Patel SK, Prince PJ, Doherty DR, Tam CY, Sheckler CM, Hsu H, Rees WA, Anderson AA, Wisler JA, Reynhardt KO, Lynch JL, Brandvig JL, Wienkers LC, Borie DC. Pharmacokinetics and pharmacodynamics in cynomolgus monkeys of AMG 181, a fully human anti-α4β7 antibody for treating inflammatory bowel disease. J Crohns Colitis. 2012;6:pS14. [Google Scholar]
- Apostolaki M, Manoloukos M, Roulis M, Wurbel MA, Muller W, Papadakis KA, Kontoyiannis DL, Malissen B, Kollias G. Role of beta7 integrin and the chemokine/chemokine receptor pair CCL25/CCR9 in modeled TNF-dependent Crohn's disease. Gastroenterology. 2008;134:2025–2035. doi: 10.1053/j.gastro.2008.02.085. [DOI] [PubMed] [Google Scholar]
- Stefanich EG, Danilenko DM, Wang H, O'Byrne S, Erickson R, Gelzleichter T, Hiraragi H, Chiu H, Ivelja S, Jeet S, Gadkari S, Hwang O, Fuh F, Looney C, Howell K, Albert V, Balazs M, Refino C, Fong S, Iyer S, Williams M. A humanized monoclonal antibody targeting the beta7 integrin selectively blocks intestinal homing of T lymphocytes. Br J Pharmacol. 2011;162:1855–1870. doi: 10.1111/j.1476-5381.2011.01205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner N, Lohler J, Kunkel EJ, Ley K, Leung E, Krissansen G, Rajewsky K, Muller W. Critical role for beta7 integrins in formation of the gut-associated lymphoid tissue. Nature. 1996;382:366–370. doi: 10.1038/382366a0. [DOI] [PubMed] [Google Scholar]
- Picarella D, Hurlbut P, Rottman J, Shi X, Butcher E, Ringler DJ. Monoclonal antibodies specific for beta 7 integrin and mucosal addressin cell adhesion molecule-1 (MAdCAM-1) reduce inflammation in the colon of scid mice reconstituted with CD45RBhigh CD4+ T cells. J Immunol. 1997;158:2099–2106. [PubMed] [Google Scholar]
- Pullen N, Molloy E, Carter D, Syntin P, Clemo F, Finco-Kent D, Reagan W, Zhao S, Kawabata T, Sreckovic S. Pharmacological characterization of PF-00547659, an anti-human MAdCAM monoclonal antibody. Br J Pharmacol. 2009;157:281–293. doi: 10.1111/j.1476-5381.2009.00137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S, Baumgart DC. Targeting leukocyte migration and adhesion in Crohn's disease and ulcerative colitis. Inflammopharmacology. 2012;20:1–18. doi: 10.1007/s10787-011-0104-6. [DOI] [PubMed] [Google Scholar]
- Sandborn WJ, Colombel JF, Enns R, Feagan BG, Hanauer SB, Lawrance IC, Panaccione R, Sanders M, Schreiber S, Targan S, van Deventer S, Goldblum R, Despain D, Hogge GS, Rutgeerts P. Natalizumab induction and maintenance therapy for Crohn's disease. N Engl J Med. 2005;353:1912–1925. doi: 10.1056/NEJMoa043335. [DOI] [PubMed] [Google Scholar]
- Targan SR, Feagan BG, Fedorak RN, Lashner BA, Panaccione R, Present DH, Spehlmann ME, Rutgeerts PJ, Tulassay Z, Volfova M, Wolf DC, Hernandez C, Bornstein J, Sandborn WJ. Natalizumab for the treatment of active Crohn's disease: results of the ENCORE Trial. Gastroenterology. 2007;132:1672–1683. doi: 10.1053/j.gastro.2007.03.024. [DOI] [PubMed] [Google Scholar]
- Polman CH, O'Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, Phillips JT, Lublin FD, Giovannoni G, Wajgt A, Toal M, Lynn F, Panzara MA, Sandrock AW. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354:899–910. doi: 10.1056/NEJMoa044397. [DOI] [PubMed] [Google Scholar]
- Berger JR, Koralnik IJ. Progressive multifocal leukoencephalopathy and natalizumab–unforeseen consequences. N Engl J Med. 2005;353:414–416. doi: 10.1056/NEJMe058122. [DOI] [PubMed] [Google Scholar]
- Berger T, Deisenhammer F. Progressive multifocal leukoencephalopathy, natalizumab, and multiple sclerosis. N Engl J Med. 2005;353:1744–1746. doi: 10.1056/NEJMc052311. ; author reply 44–6. [DOI] [PubMed] [Google Scholar]
- Giovannoni G, Kinkel P, Vartanian T. Treating multiple sclerosis in the natalizumab era: risks, benefits, clinical decision making, and a comparison between North American and European Union practices. Rev Neurol Dis. 2007;4:184–193. [PubMed] [Google Scholar]
- Van Assche G, Van Ranst M, Sciot R, Dubois B, Vermeire S, Noman M, Verbeeck J, Geboes K, Robberecht W, Rutgeerts P. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn's disease. N Engl J Med. 2005;353:362–368. doi: 10.1056/NEJMoa051586. [DOI] [PubMed] [Google Scholar]
- Feagan BG, Greenberg GR, Wild G, Fedorak RN, Pare P, McDonald JW, Cohen A, Bitton A, Baker J, Dube R, Landau SB, Vandervoort MK, Parikh A. Treatment of active Crohn's disease with MLN0002, a humanized antibody to the alpha4beta7 integrin. Clin Gastroenterol Hepatol. 2008;6:1370–1377. doi: 10.1016/j.cgh.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Feagan BG, Greenberg GR, Wild G, Fedorak RN, Pare P, McDonald JW, Dube R, Cohen A, Steinhart AH, Landau S, Aguzzi RA, Fox IH, Vandervoort MK. Treatment of ulcerative colitis with a humanized antibody to the alpha4beta7 integrin. N Engl J Med. 2005;352:2499–2507. doi: 10.1056/NEJMoa042982. [DOI] [PubMed] [Google Scholar]
- Parikh A, Leach T, Wyant T, Scholz C, Sankoh S, Mould DR, Ponich T, Fox I, Feagan BG. Vedolizumab for the treatment of active ulcerative colitis: a randomized controlled phase 2 dose-ranging study. Inflamm Bowel Dis. 2012;18:1470–1479. doi: 10.1002/ibd.21896. [DOI] [PubMed] [Google Scholar]
- Pan W, Hsu H, Rees W, Lear S, Lee F, Foltz I, Rathanaswami P, Manchulenko K, Chan B, Zhang M, Xia X, Patel S, Prince P, Doherty D, Sheckler C, Reynhardt K, Krill C, Harder B, Wisler J, Brandvig J, Lynch J, Anderson A, Wienkers L, Borie D. Pharmacology of AMG 181, a human anti-alpha4 beta7 antibody that specifically alters trafficking of gut-homing T cells. Br J Pharmacol. 2013;169:51–68. doi: 10.1111/bph.12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIH. 2010. Safety, tolerability, pharmacokinetics and pharmacodynamics of AMG 181 in healthy subjects and subjects with mild to moderate ulcerative colitis ( NCT01164904). In, 7/1/2010 Edition, US National Institute of Health, Available at http://clinicaltrials.gov/ct2/results?term=01164904&Search=Search (last accessed 04 January 2014)
- NIH. 2012. AMG 181 phase 2 study in subjects with moderate to severe ulcerative colitis ( NCT01694485). In, 9/24/2012 Edition, US National Institute of Health, http://clinicaltrials.gov/ct2/results?term=01694485&Search=Search (last accessed 04 January 2014)
- NIH. 2012. AMG 181 in subjects with moderate to severe Crohn's disease ( NCT01696396). In, 9/27/2012 Edition, US National Institute of Health, http://clinicaltrials.gov/ct2/results?term=01696396&Search=Search (last accessed 04 January 2014)
- Schroeder KW, Tremaine WJ, Ilstrup DM. Coated oral 5-aminosalicylic acid therapy for mildly to moderately active ulcerative colitis. A randomized study. N Engl J Med. 1987;317:1625–1629. doi: 10.1056/NEJM198712243172603. [DOI] [PubMed] [Google Scholar]
- National Cancer Institute. 2009. NCI, NIH, DHHS Bethesda, MD. Common Terminology Criteria for Adverse Events v4.0.
- Sandborn WJ, Colombel JF, D'Haens G, Van Assche G, Wolf D, Kron M, Lazar A, Robinson AM, Yang M, Chao JD, Thakkar R. One-year maintenance outcomes among patients with moderately-to-severely active ulcerative colitis who responded to induction therapy with adalimumab: subgroup analyses from ULTRA 2. Aliment Pharmacol Ther. 2013;37:204–213. doi: 10.1111/apt.12145. [DOI] [PubMed] [Google Scholar]
- Sandborn W, Sands B, Rutgeerts P, Sankoh S, Rosario M, Milch C, Fox I. Sustained therapeutic benefit of vedolizumab throughout 1 year in ulcerative colitis in GEMINI I, a randomized, placebo-controlled, double-blind, multicenter trial. Inflamm Bowel Dis. 2012;18:S1. [Google Scholar]
- Ghosh S, Goldin E, Gordon FH, Malchow HA, Rask-Madsen J, Rutgeerts P, Vyhnalek P, Zadorova Z, Palmer T, Donoghue S. Natalizumab for active Crohn's disease. N Engl J Med. 2003;348:24–32. doi: 10.1056/NEJMoa020732. [DOI] [PubMed] [Google Scholar]
- Fasanmade AA, Adedokun OJ, Olson A, Strauss R, Davis HM. Serum albumin concentration: a predictive factor of infliximab pharmacokinetics and clinical response in patients with ulcerative colitis. Int J Clin Pharmacol Ther. 2010;48:297–308. doi: 10.5414/cpp48297. [DOI] [PubMed] [Google Scholar]
- Ordas I, Mould DR, Feagan BG, Sandborn WJ. Anti-TNF monoclonal antibodies in inflammatory bowel disease: pharmacokinetics-based dosing paradigms. Clin Pharmacol Ther. 2012;91:635–646. doi: 10.1038/clpt.2011.328. [DOI] [PubMed] [Google Scholar]
- Lewis JD, Chuai S, Nessel L, Lichtenstein GR, Aberra FN, Ellenberg JH. Use of the noninvasive components of the Mayo score to assess clinical response in ulcerative colitis. Inflamm Bowel Dis. 2008;14:1660–1666. doi: 10.1002/ibd.20520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner D, Seow CH, Greenberg GR, Griffiths AM, Silverberg MS, Steinhart AH. A systematic prospective comparison of noninvasive disease activity indices in ulcerative colitis. Clin Gastroenterol Hepatol. 2009;7:1081–1088. doi: 10.1016/j.cgh.2009.06.024. [DOI] [PubMed] [Google Scholar]
- Konikoff MR, Denson LA. Role of fecal calprotectin as a biomarker of intestinal inflammation in inflammatory bowel disease. Inflamm Bowel Dis. 2006;12:524–534. doi: 10.1097/00054725-200606000-00013. [DOI] [PubMed] [Google Scholar]
- Parsi MA, Shen B, Achkar JP, Remzi FF, Goldblum JR, Boone J, Lin D, Connor JT, Fazio VW, Lashner BA. Fecal lactoferrin for diagnosis of symptomatic patients with ileal pouch-anal anastomosis. Gastroenterology. 2004;126:1280–1286. doi: 10.1053/j.gastro.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Vermeire S, Van Assche G, Rutgeerts P. Laboratory markers in IBD: useful, magic, or unnecessary toys? Gut. 2006;55:426–431. doi: 10.1136/gut.2005.069476. [DOI] [PMC free article] [PubMed] [Google Scholar]