

Abstract
Aims
To provide model-based clinical development decision support including dose selection guidance for empagliflozin, an orally administered sodium glucose cotransporter 2 inhibitor, through developed exposure−response (E−R) models for efficacy and tolerability in patients with type 2 diabetes mellitus (T2DM).
Methods
Five randomized, placebo-controlled, multiple oral dose studies of empagliflozin in patients with T2DM (n = 974; 1–100 mg once daily, duration ≤12 weeks) were used to develop E−R models for efficacy (glycosylated haemoglobin [HbA1c], fasting plasma glucose [FPG] and urinary glucose excretion). Two studies (n = 748, 12 weeks) were used to evaluate tolerability E−R.
Results
The efficacy model predicted maximal decreases in FPG and HbA1c of 16% and 0.6%, respectively, assuming a baseline FPG concentration of 8 mm (144 mg dl−1) and 10–25 mg every day empagliflozin targeted 80–90% of these maximums. Increases in exposure had no effect on incidence rates of hypoglycaemia (n = 4), urinary tract infection (n = 17) or genital/vulvovaginal-related (n = 16) events, although low prevalence rates may have precluded more accurate evaluation.
Conclusions
E−R analyses indicated that 10 and 25 mg once daily empagliflozin doses achieved near maximal glucose lowering efficacy.
Keywords: BI 10773, empagliflozin, exposure−response analyses, SGLT2 inhibitor, type 2 diabetes
What is Already Known about this Subject
Model-based drug development adds quantitative understanding to many of the complicating factors included in development decisions.
A population pharmacokinetic (PK) model for empagliflozin has been developed but population PK−pharmacodynamic (PK−PD) models to describe the exposure−response relationship of empagliflozin in patients with type 2 diabetes mellitus (T2DM) have not been previously reported.
What this Study Adds
This study provided a description of the magnitude of empagliflozin exposure-related responses, including estimated doses to target 80–90% of maximal efficacy, and expected time courses (when relevant).
This study provided model-based decision support for empagliflozin clinical development including guidance for dose selection and it serves as a benchmark for similar T2DM treatment candidates, and further substantiates model-based decision support in drug development.
Introduction
Treatment of type 2 diabetes mellitus (T2DM) essentially consists of three components: lifestyle changes, oral anti-diabetic drugs and insulin therapy. Use of oral hypoglycaemic agents may be limited by side effects such as hypoglycaemia, weight gain and oedema 1. There is a need for efficacious anti-diabetic agents that can be used alone or in combination with available drugs to lower blood glucose and glycosylated haemoglobin (HbA1c) without clinically limiting side effects 2,3.
The sodium glucose cotransporter 2 (SGLT2), located in the brush border membrane of the proximal convoluted tubule of the nephron, is responsible for the reabsorption of glucose from the glomerular filtrate 2. It has the capacity to transport glucose across the membrane against a concentration gradient while sodium is simultaneously transported down its concentration gradient. Under normoglycaemic conditions, glucose is completely reabsorbed. However the re-uptake capacity of the kidney is saturated at glucose concentrations of about 11.0 mm (198 mg dl−1) resulting in glucosuria 4. This threshold concentration can be decreased by inhibition of SGLT2 2. Due to their insulin-independent mode of action, SGLT2 inhibitors are expected to be associated with a low risk of hypoglycaemia and have the potential to be combined with other anti-diabetic drugs to improve glycaemic control in patients with T2DM 2.
Empagliflozin is a potent and highly selective oral SGLT2 inhibitor that, in a rodent model, has been shown to inhibit reabsorption of glucose from the renal filtrate leading to a rapid appearance of glucose in the urine 5. Phase I clinical studies have confirmed the ability of empagliflozin to increase excretion of glucose in the urine and decrease plasma glucose concentrations in patients with T2DM 6,7.
In these studies, empagliflozin was rapidly absorbed following oral administration and increases in empagliflozin exposure were dose-proportional following multiple oral dosing. Peak plasma concentrations were observed approximately 1.5 h after dosing. Mean terminal elimination half-life at steady-state was approximately 14 h. Up to 22% accumulation was observed from first dose to steady-state based on the area under the concentration−time curve (AUC). Apparent clearance after oral administration (CL/F) at steady-state was similar to corresponding single dose values, suggesting time-independent pharmacokinetics (PK) 6,7. No major metabolites of empagliflozin were detected in human plasma and the most abundant metabolites were glucuronide conjugates. Systemic exposure of each metabolite was less than 10% of total drug-related material (unpublished data). Following oral administration in patients with T2DM (NCT00558571), approximately 18% of drug was excreted unchanged in urine. Steady-state renal clearance was approximately 36 ml min−1.
The aim of the analyses reported herein was to develop exposure−response (E−R) models for efficacy (HbA1c, fasting plasma glucose [FPG] and urinary glucose excretion [UGE]) and adverse events associated with empagliflozin. These results were intended to provide model-based support during the transition from phase 2 to phase 3 clinical development.
Methods
Study designs
Data from five clinical studies that included oral, once daily administration of empagliflozin were used for the efficacy E−R evaluations. All clinical trial protocols were approved by the relevant local Independent Ethics Committee and were carried out in compliance with the Ethical Principles for Medical Research Involving Human Subjects of the Declaration of Helsinki (October, 1996), the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use tripartite harmonized guideline E6(R1) ‘Good Clinical Practice’ and the standard operating procedures of Boehringer Ingelheim. Subjects provided written informed consent prior to participation.
Study A (EudraCT no. 2007-000654-32) 6 was a phase I randomized, double-blind, placebo-controlled trial conducted in 48 patients with T2DM that investigated the PK and pharmacodynamics (PD) of multiple doses of empagliflozin (2.5, 10, 25 and 100 mg once daily; n = 9 in each group) or placebo (n = 12) over 8 days. The study was conducted in Germany. Data collection included intensive PK evaluations on study days 1 and 9. FPG was measured daily on study days −2 to day 13. Urine collections (24 h) on study days −2, −1, 1, 8, and 9 were used to determine the amount (mg) of glucose excreted in urine from 0 to 24 h post-dose (UGE).
Study B (NCT00558571; phase I) 7 and study C (NCT00885118; phase II) 8 were 4 week, randomized, double-blind, placebo-controlled, parallel-group trials that investigated the safety, tolerability, PK and PD of once daily treatment with empagliflozin vs. placebo in patients with T2DM. For study B, 78 patients were randomized to receive empagliflozin (10, 25 or 100 mg once daily; n = 16, 16 and 30, respectively) or placebo (n = 16). The study was conducted in Germany. Study C was conducted in Japan and included only Japanese patients with T2DM, who were randomized to receive empagliflozin (1, 5, 10 or 25 mg once daily; n = 19, 21, 20 and 19, respectively) or placebo (n = 21). All participants completed study B and 97 of 100 completed study C. Data collection for both studies included intensive PK evaluations on study days 1 and 28. FPG was measured on study days −2, −1, 1, 2, 3, 4, 7, 14, 21, 26, 27, 28 and 29, UGE was measured on study days −2, −1, 1, 27 and 28 and HbA1c was measured on study days −1 and 28.
Study D (NCT00789035) 9 was a phase IIb randomized, double-blind, 12 week, multinational trial comparing empagliflozin (5, 10 or 25 mg once daily; n = 79, 81 and 82, respectively), placebo (n = 82) and open-label metformin (500 mg twice daily for 4 weeks, then 1000 mg twice daily or the maximum tolerated dose, n = 80). Overall, 408 patients with T2DM were randomized, of whom two did not receive study medication. Two patients from the empagliflozin 5 mg once daily treatment group did not contribute PK samples and so were excluded from the E−R analyses. In addition, patients from the open label metformin arm were excluded. Thus a total of 324 patients contributed data from study D. Plasma samples were collected for PK evaluations just prior to dosing on study days 1, 28, 56 and 84, with additional samples taken on study day 84 at 1.5 h post-dose and (optionally) between 2 and 24 h post-dose. HbA1c and FPG were measured on study days −14, 1, 28, 56 and 84.
Study E (NCT00749190) 10 was a phase IIb, randomized, double-blind, 12 week, multinational, parallel group study comparing empagliflozin (1, 5, 10, 25 and 50 mg once daily, n = 71, 71, 71, 69 and 71, respectively), placebo (n = 71) and open label sitagliptin (n = 71) in patients with T2DM on metformin therapy. Patients from the open label sitagliptin arm were not included in the E−R analysis. Data collections were as described for study D.
Adverse events (AEs) reported from studies D 9 and E 10 were used for the E−R evaluations of tolerability endpoints. Adverse events of interest for these evaluations included hypoglycaemia, urinary tract infections, and genital candidiasis/vulvovaginitis.
Model-based analysis
Population PK and E−R (PK−PD) analyses for repeated-measures endpoints were conducted using the non-linear mixed-effects modelling (nonmem®) software, Version VI, Level 2.0 (ICON Development Solutions, Hanover, MD, USA). Models were developed on a computer grid with multiple compute nodes. Each node runs the Mac OS X operating system and utilizes the GNU Fortran compiler, GCC-3.4.0 (Mac OS X version, also known as G77; GNU Project, http://www.GNU.org/). NMQual 6.3.2 or greater was used to track all code patches/options and install the nonmem software. The first order conditional estimation method with η- ε interaction (FOCEI) was employed for all model runs 11.
Individual PK estimates from the population PK model were used to generate individual exposure (AUC) estimates used to evaluate E−R. AUCi,j (AUC of the dosing interval for each individual [i] following each dose time [j] in each study [k]) was used for efficacy E−R evaluation, and AUC(0,τ)i (AUC of the steady-state dosing interval for each individual [i]) was used for tolerability E−R evaluation.
The adequacy of the final model and its parameter estimates were investigated with a predictive check method. This is similar to a posterior predictive check, but assumes that parameter uncertainty is negligible, relative to inter-individual and residual variance 12,13. The basic premise is that a model and parameters derived from an observed data set should produce simulated data that are similar to the original observed data.
Following PK model development, the efficacy PK−PD model was used to simulate the expected percentage of subjects achieving HbA1c reductions from baseline of 0.5%, 0.7% and 1.0% at 12 and 24 weeks (Monte Carlo simulations), with a target reduction of ≥0.7 percentage points (HbA1c ≤7.3). A baseline FPG of 9.4 mm (169 mg dl−1) and a baseline HbA1c of 8.0% were used for these simulations. Variables included empagliflozin dose (0 [placebo], 5, 10 and 25 mg), weight (60, 85 and 110 kg) and the estimate of the AUC that resulted in 50% of the maximal stimulation of FPG removal (AUC50). The doses were intended to reflect the range expected to produce moderate to maximal efficacy. The body weight variables were intended to evaluate the effects of expected weight-related exposure (AUC) differences on efficacy response. The different AUC50 values were used to explore the sensitivity of the efficacy simulations to inter-study differences in this estimate. The simulations, based on the point estimates of the fixed effects and the random effects for inter-individual and residual variance, provided the expected percentages of subjects achieving longitudinal HbA1c changes from baseline. Semi-parametric modelling methods 14,15 (gam 1.01 in version 2.10.1 of R [http://r-project.org]) were used to explore the exposure−tolerability relationships. The tolerability endpoints were identified as dichotomous flags (0 = patient did not experience this adverse event, 1 = patient did experience this adverse event). Individual estimates of AUC(0,τ,ss) at each individual's last PK observation were used as the exposure predictor. A non-parametric smoothing technique was used to describe the shape of each E−R relationship for the three tolerability endpoints. Gender was also considered as a covariate for these relationships.
Results
Efficacy markers (UGE, FPG and HbA1c): empagliflozin dose−response
UGE measurements were available from patients in studies A, B and C. Baseline mean UGE observations were similar for studies A and B and were slightly greater for study C. An empagliflozin dose−response for UGE was apparent from day 1 and was sustained for 4 weeks, the longest observation period in these studies (Figure 1A). An increase in UGE appeared to occur with the lowest dose of empagliflozin, with a dose-dependent increase in UGE thereafter, reaching a plateau at approximately 10 to 25 mg.
Figure 1.

Observed dose−response for (A) urinary glucose excretion and (B) fasting plasma glucose (FPG). In (B), horizontal lines at 2.5, 1.25, 0, −1.25 and −2.5 mm are included for reference. In the box and whisker plots, median values are designated by a solid black circle within the box. Boxes indicate the inter-quartile range (IQR). Whiskers represent 1.5*IQR. Observations outside the whiskers are marked as circles.  , study day 1;
, study day 1;  , study day 27
, study day 27
Baseline FPG values were available for all studies (Table 1). The maximal observed decrease in FPG appeared to occur within 3–4 weeks after initiation of empagliflozin treatment and was dependent on dose (Figure 1B), reaching a plateau at approximately 10 to 25 mg. A near maximal decrease in FPG was observed in the 5 mg group in study D and in the 10 mg dose group in study E, whereas considerable decreases in FPG from baseline were observed even in lowest dose group (1 mg) from study C (Figure 1B). Study-specific estimates of the AUC50 were therefore considered.
Table 1.
Summary of baseline demographic, laboratory and pharmacokinetic exposure information from each study
| Study | A (n = 48) | B (n = 78) | C (n = 100) | D (n = 324) | E (n = 424) |
|---|---|---|---|---|---|
| Male/female, n | 39/9 | 67/11 | 84/16 | 172/152 | 212/212 |
| Race, n | |||||
| White | 47 | 76 | 0 | 212 | 416 |
| Black | 1 | 1 | 0 | 0 | 6 |
| Asian | 0 | 1 | 100 | 112 | 0 |
| Hawaiian/Pacific | 0 | 0 | 0 | 0 | 2 |
| Age (years) | 56.9 (8.9) (33–68) | 56.7 (8.7) (34–69) | 57.2 (9.2) (34–70) | 57.5 (10.0) (28–80) | 58.4 (8.6) (32–78) |
| Weight (kg) | 94.6 (14) (68–123) | 93.2 (15) (69–127) | 67.9 (13) (44–98) | 81.4 (17) (46–152) | 89.2 (16) (55–139) |
| Height (cm) | 175 (9.1) (146–198) | 175 (8.2) (158–198) | 166 (8.1) (142–184) | 167 (9.9) (145–196) | 168 (10.1) (138–196) |
| BMI (kg m−2) | 30.8 (3.5) (23.8–29.4) | 30.4 (4.5) (22.8–39.5) | 24.6 (3.8) (17.8–39.1) | 28.9 (4.5) (20.1–39.6) | 31.4 (4.5) (19.6–50.3) |
| BSA (m2) | 2.1 (0.2) (1.6–2.5) | 2.1 (0.2) (1.7–2.6) | 1.8 (0.2) (1.3–2.2) | 1.9 (0.2) (1.3–2.8) | 2.0 (0.2) (1.5–2.6) |
| Scr (mg dl−1) | (0.1) (0.9–1.2) | 0.9 (0.1) (0.5–1.2) | 0.8 (0.1) (0.7–1.0) | 0.9 (0.1) (0.7–1.8) | 0.9 (0.2) (0.6–1.4) |
| CLcr (ml min−1) | 103 (27.2) (64–182) | 117 (30.8) (70–264) | 94 (23.7) (51–159) | 99 (29.3) (39–202) | 107 (31.1) (47–264) |
| FPG (mmol l−1) | 8.3 (1.8) (5.5–12.8) | 8.4 (2.1) (2.8–14.3) | 8.9 (1.6) (5.8–13.3) | 9.7 (2.4) (5.2–21.0) | 9.7 (2.1) (3.5–18.0) |
| HbA1c (%) | 7.1 (0.5)* | 7.1 (0.8) (5.6–8.8) | 8.1 (0.8) (6.7–9.6) | 7.9 (0.8) (6.0–10.4) | 7.9 (0.7) (6.3–10.0) |
| AUCss (nmol l−1 h) | 1970 (354) (1160–2930) | 1830 (354) (1080–3100) | 2480 (414) (1660–3540) | 2280 (810) (960–8050) | 2130 (663) (925–8230) |
Not included in the E−R analyses. Data are mean (SD) and (range [min−max]) unless specified. AUCss, estimated area under empagliflozin plasma concentration−time curve at steady-state normalized to a 10 mg oral dose; BMI, body mass index; BSA, body surface area; CLcr, estimated creatinine clearance; FPG, fasting plasma glucose; HbA1c, glycosylated haemoglobin; Scr, serum creatinine.
Baseline HbA1c values were available for studies B, C, D and E (Table 1). The maximal HbA1c decrease from baseline, also dependent on dose, was approached by 8–12 weeks after initiation of empagliflozin treatment (Figure 2), reaching a plateau at approximately 10 to 25 mg. Comparing the dose−response across the 12 week studies (studies D and E), a near maximal decrease in HbA1c was observed in the 10 mg groups in both studies, while the mean decrease in HbA1c from baseline with the 5 mg dose was greater in study D than in study E (Figure 2).
Figure 2.

Observed dose−response and longitudinal effects (weeks 4, 8 and 12) for HbA1c. Horizontal lines at 0 (grey dashed) and −0.6 (green dotted) percentage points are included for reference. In the box and whisker plots, median values are designated by a solid black line within the box. Boxes indicate the inter-quartile range (IQR). Whiskers represent 1.5*IQR. Observations outside the whiskers are marked as circles.  , study B;
, study B;  , study C;
, study C;  , study D;
, study D;  , study E
, study E
Efficacy markers (UGE, FPG and HbA1c): relationships with empagliflozin exposure
Empagliflozin exposures were estimated from a population PK model that included two compartmental disposition with lagged first order absorption and first order elimination (Figure 3). Population estimates (inter-individual variance [IIV] estimates, coefficient of variation [CV%]) of CL/F, central and peripheral volumes of distribution and inter-compartmental clearance were 9.87 l h−1 (26.9%), 3.02 l, 60.4 l (30.8%) and 5.16 l h−1, respectively. The typical calculated steady-state AUC values for once daily doses of 1, 3, 10 and 25 mg were 225, 674, 2250 and 5620 nmol l−1 h, respectively (molecular weight = 450.9 g mol−1).
Figure 3.
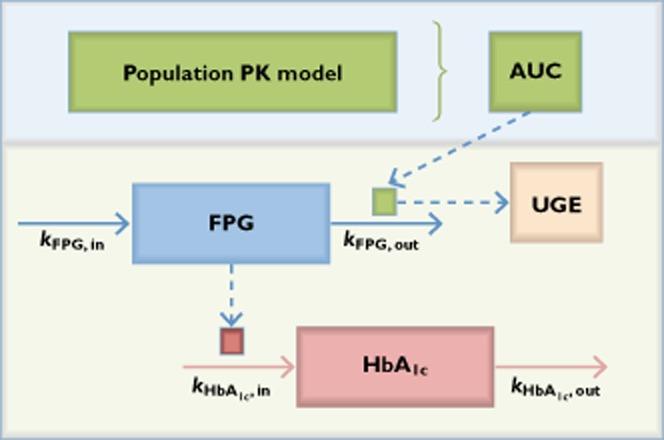
Schematic of population pharmacokinetic and exposure−response model.  , stimulation (STIM) lowered FPG and increased UGE;
, stimulation (STIM) lowered FPG and increased UGE;  , decreased FPG decreased HbA1c production
, decreased FPG decreased HbA1c production
Weight was included allometrically on each of these parameters. There were no other estimated PK differences considered to be clinically relevant. Following a 0.5 h lag, the typical oral absorption rate constant was estimated to be 0.224 h−1 (IIV, 15.2%). The terminal elimination half-life derived from these parameters was approximately 12 h (unpublished data on file, Boehringer Ingelheim Pharmaceuticals, Inc., Ridgefield, CT, USA).
The dose-dependent increase in UGE and corresponding decrease in FPG with empagliflozin treatment did not affect the underlying relationship between UGE and FPG. A similar pattern of increased UGE concentrations as a result of increased FPG was seen with placebo and all empagliflozin doses (Figure 4). The relationships of UGE with FPG and exposure both appeared to be non-linear, leading to the development of a model (Equation 1, Figure 3) that described the UGE, in the ith individual in the jth study at the kth time, as a function of a baseline parameter (BASEi) that was normalized to a FPGijk value of 8 mm (144 mg dl−1). This baseline effect increased exponentially (γbase) with increased FPG. A hyperbolic effect of empagliflozin exposure was added to this baseline (Equation 1) driven by a stimulation (STIM) function (Equation 2). The hyperbolic expression asymptotes to a maximum (Umax,k) and is at its half maximum when 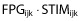 equals Ustim50,k. The drug effect STIM function was a product of the observed baseline FPG (FPGbaseline,ik), normalized and increased exponentially
equals Ustim50,k. The drug effect STIM function was a product of the observed baseline FPG (FPGbaseline,ik), normalized and increased exponentially 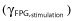 , and a drug exposure (AUCijk)-driven hyperbolic expression. For estimation stability, an alternative parameterization for the maximal effect (Emax) and AUC leading to the half maximal (AUC50ijk) was used for model parameter estimation 16,17.
, and a drug exposure (AUCijk)-driven hyperbolic expression. For estimation stability, an alternative parameterization for the maximal effect (Emax) and AUC leading to the half maximal (AUC50ijk) was used for model parameter estimation 16,17.
Figure 4.
Relationship of 24 h urinary glucose excretion with fasting plasma glucose. Observed values are shown as separate symbols for each treatment; population predicted values are shown as separate lines for each treatment.  , placebo/baseline;
, placebo/baseline;  , empagliflozin 1 mg once daily;
, empagliflozin 1 mg once daily;  , empagliflozin 2.5 mg once daily;
, empagliflozin 2.5 mg once daily;  , empagliflozin 5 mg once daily,
, empagliflozin 5 mg once daily,  , empagliflozin 10 mg once daily,
, empagliflozin 10 mg once daily,  , empagliflozin 25 mg once daily;
, empagliflozin 25 mg once daily;  , empagliflozin 100 mg once daily
, empagliflozin 100 mg once daily
In addition to affecting UGE (equation 1), this STIM function (equation 2) directly affected the removal rate of FPG (equation 3) according to individual dosing interval exposures (AUC), thereby causing a decrease in FPG over time. This removal rate was described using a first order rate constant 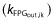 countered by zero order FPG input
countered by zero order FPG input 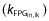 . Study-specific parameters described inter-study differences (Table 2).
. Study-specific parameters described inter-study differences (Table 2).
Table 2.
Parameter estimates from population pharmacokinetic/pharmacodynamic model used to describe urinary glucose excretion (UGE), fasting plasma glucose (FPG) and HbA1c exposure−response
| Non-parametric bootstrap | |||||
|---|---|---|---|---|---|
| Point estimate | RSE% | Median | 95% CI | Residual | |
| Parameter | |||||
| θ12: baseline UGE (g per 24 h) [Study A] | 3.71 | 1.79 | |||
| θ20: baseline UGE [Study B] = θ12 · θ20 | 0.320 | 18.6 | |||
| θ16: baseline UGE [Study C] = θ12 · θ16 | 0.632 | 51.3 | |||
| θ13: γbase = θ13 | 5.31 | 5.22 | |||
| θ17: γbase = θ13 · θ17 [Study C] | 1.16 | 118 | |||
| θ10: Umax (g 24 h−1) | 121 | 1.04 | |||
| θ14: Umax [Study C] = θ10 · θ14 | 1.11 | 177 | |||
| θ11: Ustim50 | 0.590 | 69.8 | |||
| θ15: Ustim50 [Study C] = θ11 · θ15 | 1.58 | 105 | |||
| θ1: baseline FPG (mmol l−1) [Study A] | 7.85 | 1.29 | |||
| θ2: baseline FPG (mmol l−1) [Study B] | 8.50 | 1.38 | |||
| θ3: baseline FPG (mmol l−1) [Study D] | 9.30 | 0.493 | |||
| θ4: baseline FPG (mmol l−1) [Study E] | 9.49 | 0.44 | |||
| θ18: baseline FPG (mmol l−1) [Study C] | 8.76 | 0.81 | |||
| θ5: kFPG, out (h−1) | 0.0407 | 18 | |||
| θ8: βFPG stimulation | 0.795 | 30.4 | |||
| θ9: γFPG stimulation | 1.47 | 30.8 | |||
| θ6: Emax,truncated [FPG stimulation] | 0.0701 | 18.2 | |||
| θ7: C*50 (nmol l−1 h) [FPG stimulation] | 498 | FIXED | |||
| θ19: C*50 [Study C, FPG stimulation] = θ7 · θ19 | 0.169 | 22.8 | |||
| θ21: C*50 [Study E, FPG stimulation] = θ7 · θ21 | 1.93 | ||||
| θ23: baseline HbA1c (%) [Study B] | 7.18 | 7.16 | (6.78, 7.39) | ||
| θ24: baseline HbA1c (%) [Study D] | 7.85 | 7.85 | (7.74, 7.92) | ||
| θ25: baseline HbA1c (%) [Study E] | 7.89 | 7.89 | (7.74, 7.95) | ||
| θ28: baseline HbA1c (%) [Study C] | 7.85 | 7.85 | (7.85, 7.85) | ||
| θ22: HbA1c physiologic limit parameter (%) | 3.34 | 3.52 | (2.92, 7.83) | ||
| θ26: kHbA1c,out (week−1) | 0.167 | 0.167 | (0.167, 0.167) | ||
| θ27: kHbA1c,in (% week−1 mm−1) | 0.078 | 0.0743 | (0, 0.0869) | ||
| θ29: shared eta (ηkHbA1c,out = θ29 · ηkHbA1c,in) | 2.7 | 2.59 | (–22.8, 48.9) | ||
| Calculated parameters | |||||
| FPG maximal decrease (proportional) | 0.158 | ||||
| FPG AUC50 (nmol l−1 h) | 626 | ||||
| FPG AUC50 (nmol l−1 h) [Study C] | 106 | ||||
| FPG AUC50 (nmol l−1 h) [Study E] | 1210 | ||||
| Residual variance | |||||
| UGE CV% | 0.380 | 11.9 | 67.9 (CV%) | ||
| FPG CV% | 0.01461 | 12.1 | (11.4, 12.9) | 12.1 (CV%) | |
| HbA1c CV% | 0.001287 | 3.59 | (3.3, 6.78) | 3.59 (CV%) | |
CI, confidence interval; CV, coefficient of variation; FPG AUC50, AUC that resulted in 50% of the maximal stimulation of FPG removal; FPG, fasting plasma glucose (mmol l−1); HbA1c, glycosylated haemoglobin (%); RSE, relative standard error = standard error of the estimate/point estimate; UGE, 24 h urine glucose excretion (g).
Equation 1
Equation 2
 |
Equation 3
The model estimated a baseline UGE of approximately 1–3 g day−1 for a subject with a baseline FPG of 8 mm (144 mg dl−1) (Table 2). Baseline UGE was approximately doubled (e.g. from 2 to 4 to 8 to 16 g day−1) with baseline FPG increases from 8 mm (144 mg dl−1) to 9.1 mm (164 mg dl−1), 10.4 mm (187 mg dl−1) and 11.8 mm (213 mg dl−1), respectively in the placebo group (Figure 4). Patients with higher baseline FPG were observed to achieve greater increases in UGE (Figure 4) and decreases in FPG (not shown). The influence of baseline FPG on the maximal possible decrease in FPG was included in the STIM function (Equation 2). The estimated maximal stimulation represented a 16% decrease from a baseline FPG of 8 mm (144 mg dl−1) (Table 2). IIV for baseline UGE (CV%) was estimated to be 158.4%.
This maximum effect in the STIM function was carried through for UGE (Figure 3). The influence of FPG at each corresponding visit (FPGij) was also included (Equation 1) to account for potential FPG-related visit-to-visit differences in UGE. The STIM function itself was embedded within an Emax relationship to describe UGE E−R. This adjusted for apparent non-linearity between UGE and FPG stimulations. In this case, it appeared that the maximal FPG effect described through STIM was achieved at exposures in excess of those affecting the maximal UGE effect, i.e. UGE changes did not solely explain FPG changes and so the model required more than linking empagliflozin exposure to UGE to FPG.
The maximal UGE drug effect was estimated to be nearly 120 g day−1, but this maximum is attained in the mathematical expression for UGE (Equation 2) only with higher FPG values. For example, half the maximal increase in UGE (approximately 60 g day−1) was estimated to occur at the approximate typical steady-state exposure from a 3 mg empagliflozin once daily dose for a FPG of 8 mm (144 mg dl−1), whereas an increase in UGE of approximately 80 g day−1 was expected from this same exposure for a FPG of 10.5 mm (189 mg dl−1) (Table 2). These estimates assumed an AUC50 of 626 nmol l−1 h for the FPG STIM function. For an FPG of 8 mm (144 mg dl−1), empagliflozin doses of 10 mg (AUC = 2250 nmol l−1 h) and 25 mg (AUC = 5620 nmol l−1 h) were expected to result in UGE increases of approximately 72 and 75 g day−1, respectively. This increases to 80 and 88 g day−1, respectively, for an FPG of 10.5 mm (189 mg dl−1). These predictions were consistent with the observed data (Figures 1A and 4).
Translating these glucose effects, the formation rate of HbA1c was assumed to be a first order function of FPG. The drug-related stimulation of FPG removal therefore led to a time-dependent decrease in HbA1c (Equation 4).
Equation 4
The estimated AUC50 (626 nmol l−1 h) for the FPG STIM function using combined data from studies A, B and D corresponded to an empagliflozin exposure from a once daily dose of approximately 3 mg. AUC50 values estimated for studies E (1210 nmol·h l−1) and C (106 nmol l−1 h) (Table 2) corresponded to empagliflozin exposures from once daily doses of approximately 5 and 0.5 mg, respectively. The HbA1c half-life calculated from the point estimate of  was approximately 4.3 weeks. IIV estimates (CV%) for baseline HbA1c and
was approximately 4.3 weeks. IIV estimates (CV%) for baseline HbA1c and  were 9.53% and 8.23%, respectively, with a correlation estimate of −0.310.
were 9.53% and 8.23%, respectively, with a correlation estimate of −0.310.
As with UGE, the FPG and corresponding HbA1c responses were dependent on drug exposure and the baseline FPG (Equations 2–4). For example, the predicted maximal decreases (steady-state) in FPG and HbA1c at the reference baseline FPG (8 mm, 144 mg dl−1) were 1.3 mm (23 mg dl−1) (16%) and 0.6 percentage points, respectively, decreasing to 1.0 mm (18 mg dl−1) (FPG) and 0.5 percentage points (HbA1c) for a baseline FPG of 7.4 mm (133 mg dl−1). Correspondingly, greater maximal FPG decreases of 1.7 (31 mg dl−1) and 2.2 mm (40 mg dl−1) and maximal HbA1c decreases of 0.81 and 1.0 percentage points were expected with baseline FPG values of 9.1 (164 mg dl−1) and 10 mm (180 mg dl−1), respectively.
Targets of 80% and 90% of the maximal response after 12 weeks of treatment for FPG and HbA1c were obtained by empagliflozin doses of approximately 10 and 25 mg, respectively, based on the AUC50 estimate from studies A, B and D. These same doses would provide approximately 65% and 82% of the maximal response using the AUC50 estimate specific to study E. Therefore, a 25 mg once daily dose of empagliflozin was expected to represent a dose that will target 80–90% of the maximal response.
The possible effect of renal function on the empagliflozin efficacy E-R was investigated graphically (Supplementary Figures S1, S2). There was no apparent influence of creatinine clearance (CLcr), calculated using Cockcroft−Gault method 18, on either FPG or HbA1c response down to the minimum observed CLcr of approximately 50 ml min−1.
General goodness-of-fit diagnostics (not shown) indicated that the final population efficacy E−R model appropriately described the UGE, FPG and HbA1c observations from the five studies investigated. The results of the predictive checks were also consistent with the observed data. For example, the model appropriately described the median and variance (10th and 90th percentiles) for the FPG and HbA1c dose−response (Figure 5).
Figure 5.
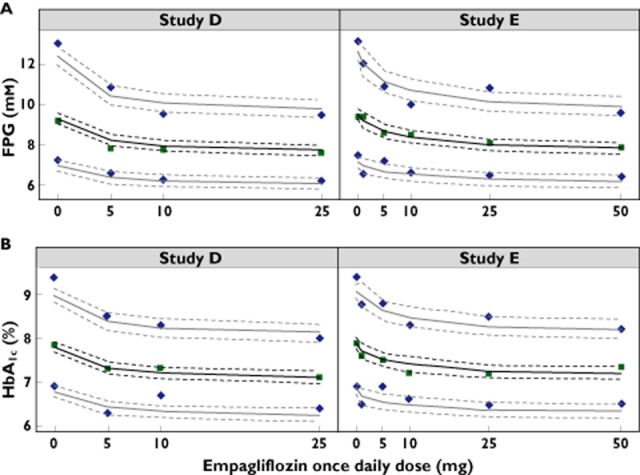
Predictive check showing the observed dose−response for (A) fasting plasma glucose (FPG) and (B) HbA1c from studies D and E. The median (solid green squares) along with the 10th and 90th percentiles (blue diamonds) represent distribution of last observed measurements per patient. The solid lines represent the predicted median (black) along with 10th and 90th percentiles (grey). Dashed lines represent the 80% confidence intervals for the respective predictions
Monte Carlo simulations were used to evaluate the expected percentage of subjects achieving HbA1c reductions from baseline of −0.5, −0.7 and −1.0 percentage points. These simulations indicated that 26% of subjects administered placebo would experience a decrease in HbA1c from baseline of 0.7 percentage points. Achievement of this same target for 10 mg and 25 mg doses of empagliflozin was expected for approximately 57% and 63% of subjects, respectively, assuming a body weight of 85 kg. The rates of achieving this target decrease in HbA1c were expected to increase by approximately 2–3 percentage points for a 60 kg subject and decrease by approximately 4–5 percentage points for a 110 kg subject relative to the rates for an 85 kg subject. These simulated values were consistent with data from studies D and E.
The tolerability endpoints included for E−R evaluation were hypoglycaemia (n = 4), urinary tract infection (n = 17) and genital/vulvovaginal-related (n = 16) events (Supplementary Table S1). The general additive models applied to evaluate the E−R for the tolerability data showed that there was no increase in the probability of experiencing an adverse event with increased exposure up to 50 mg (Figure 6).
Figure 6.
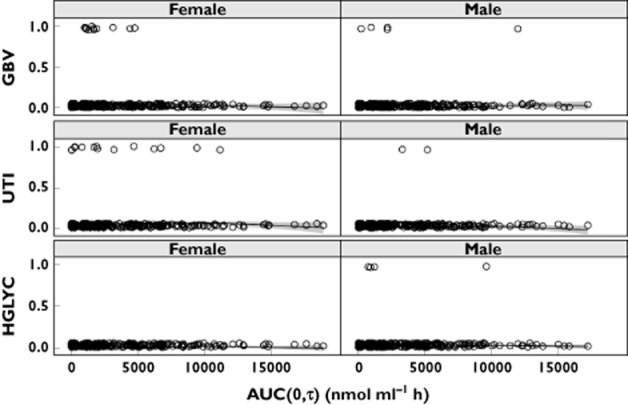
Exposure−response evaluation of tolerability adverse events separated for females and males. bottom: hypoglycaemia (HGLYC); middle: urinary tract infection, (UTI); top: genital/vulvovaginal-related (GBV). Observations (one per individual) are jittered at 0 (adverse event not reported) or 1 (adverse event was reported). AUC(0,τ) (area under the plasma concentration−time curve at the last pharmacokinetic [PK] observation) was estimated for each individual using their dosing history and PK parameter estimates
Discussion
SGLT2 inhibitors reduce the threshold concentration for renal glucose reabsorption leading to increased UGE and reduced plasma glucose concentrations in patients with T2DM 6,7,19,20. Empagliflozin exposure increases dose-proportionally and demonstrates linear pharmacokinetics with respect to time 6,7, and pharmacodynamic analyses have shown significant increases in UGE reaching a plateau at doses of 10 to 25 mg 6,7. Dapagliflozin exposure increases approximately dose-proportionally and dose dependent increases in UGE have been observed 19. Canagliflozin exposure increases dose-proportionally and pharmacodynamic effects have been shown to be dose- and exposure-dependent 20.
In this analysis, the population PK of empagliflozin in patients with T2DM was described by a two compartment model with lagged first order oral absorption that appropriately described the observed empagliflozin exposure in each of the studies (data not shown). The E−R results were consistent with known physiology associated with renal glucose excretion and appropriately described the longitudinal relationship between plasma glucose and HbA1c.
Approximately 180 g of glucose is filtered by the kidneys per day under normal physiological conditions, yet <0.5 g glucose day−1 is excreted in the urine in healthy individuals 21. In patients with T2DM, the elevated concentrations of glucose filtered through the glomerulus can exceed the threshold concentration for renal reabsorption in the proximal tubule, which is already above the level of non-diabetic subjects 22,23.
In this analysis, the baseline model for the effect of FPG on UGE was consistent with this characteristic physiology, predicting a typical range of <3 g day−1 for patients with T2DM with a baseline FPG ≤8 mm (144 mg dl−1), with exponential increases in UGE as FPG increases beyond 8 mm. For example, in the absence of empagliflozin treatment, UGE was expected to increase to approximately 32 g day−1 for an FPG of 12 mm (216 mg dl−1). Empagliflozin-mediated inhibition of SGLT2 was expected to substantially lower the glucose threshold, leading to even greater losses of glucose through the urine. As such, a dose-dependent increase in UGE with empagliflozin treatment would be expected to occur regardless of the plasma glucose concentrations. It is important to note that the relationship with FPG is retained with empagliflozin treatment, such that the magnitude of glucose removal (UGE) decreased as FPG decreased, thereby providing a degree of self-correction against hypoglycaemia. The estimated maximal increase in UGE was nearly 120 g day−1.
In addition, using the methodology previously described by Samtani 24, it was calculated that the final empagliflozin exposure-FPG-HbA1c model estimated a change in HbA1c from baseline at steady-state of 0.47% for every 1 mm (18 mg dl−1) change in FPG. For example, a decrease in FPG from 9.4 mm (169 mg dl−1) to 7.9 mm (142 mg dl−1) would be associated with an average decrease in HbA1c of 0.7%. The parameter estimates from this empagliflozin model for HbA1c production from FPG and for the first order HbA1c removal rate constant were strikingly consistent with those reported previously 24, which were based on numerous therapeutics, including sulphonylureas, meglitinides and thiazolidinediones, studied across 12 clinical trials. The physiological limit parameter (HbA1c,limit) was included to control for other homeostatic mechanisms that would prevent non-physiological drops in HbA1c 25.
The consistency of the effects on HbA1c suggests that although empagliflozin affects plasma glucose changes through a different mechanism than previously studied anti-diabetic medications, it maintains the underlying dynamic relationship between plasma glucose and HbA1c production. Another consideration from these results is the time required to evaluate changes in FPG compared with HbA1c. Effects on FPG were typically stabilized within 2 to 4 weeks from the start of therapy, whereas effects on HbA1c, with an estimated half-life of >4 weeks, require several months to equilibrate fully. The consistency with which FPG changes after 4 weeks are predictive of longer term HbA1c changes suggests that FPG may serve as a useful translational marker during clinical development.
There was no apparent influence of CLcr on either FPG or HbA1c response down to the minimum observed CLcr of approximately 50 ml min−1 (mild renal impairment). However, effects in subjects with lower renal function are of interest to the SGLT2 inhibitor class in general. For example, pharmacokinetic and pharmacodynamic analyses have shown that systemic exposure of canagliflozin, dapagliflozin and empagliflozin increases with decreasing renal function, but that UGE decreases with increasing renal impairment 26–28. Limited UGE is observed in patients with severe renal impairment 26–28.
Inter-study differences in the estimate of AUC50 (Table 2) did not appear to be attributable to differences in baseline FPG or HbA1c by dose, within or between studies D and E. Additionally, there did not appear to be a difference between studies D and E in drug exposure (AUC) for corresponding doses (not shown), so an exposure difference was not expected to explain the estimated response difference. However, there were differences in the treatments administered between these studies. Study D was with empagliflozin alone and covered an active empagliflozin dose range of 5 to 25 mg once daily, whereas study E included metformin background therapy and covered an active empagliflozin dose range of 1 to 50 mg once daily. Empagliflozin plasma exposures by dose were consistently greater in study C compared with the other studies (Table 1), likely due to the lower body weights of the Japanese study population in study C. This may explain, at least in part, the increased response at lower doses in this study. Confounding factors, however, preclude an exact understanding of the inter-study differences. Despite this, empagliflozin doses of 10 mg and 25 mg were supported by the E−R analysis as these doses provided exposures associated with 80% to 90% of the maximal glucose-lowering effect, thereby covering the uncertainty in AUC50.
In contrast to the distinguishable E−R for efficacy, incidence rates of the evaluated tolerability endpoints did not increase with corresponding exposures from empagliflozin once daily doses up to 50 mg in the patients with T2DM. It should be noted, however, that the low adverse event prevalence rates may have precluded a more accurate evaluation.
In summary, E−R analyses indicated that empagliflozin once daily doses of 10 and 25 mg achieved near maximal (>80%) glucose-lowering efficacy.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare the data reported in this article were obtained from studies funded by Boehringer Ingelheim. SM, LS, AS, TM and HJW are employees of Boehringer Ingelheim and MR, WG, and MG are employees of the Metrum Research Group who served as paid consultants to Boehringer Ingelheim. There are no other relationships or activities that could appear to have influenced the submitted work.
Assays for plasma and urine concentrations of empagliflozin were performed by Bioanalytical Systems, Inc., West Lafayette, IN, USA. The authors acknowledge Lois Rowland for co-ordinating bioanalytical work. Medical writing assistance, supported financially by Boehringer Ingelheim, was provided by Isobel Lever and Elizabeth Ng of the Fleishman-Hillard Group Ltd during the preparation of this manuscript. The authors were fully responsible for all content and editorial decisions, were involved at all stages of manuscript development and have approved the final version.
Author contributions
The authors meet criteria for authorship as recommended by the International Committee of Medical Journal Editors (ICMJE). MR, SM, AS, WG and M.G. made substantial contributions to the conception and design, MR, SM, WG and MG were involved in the acquisition of data and MR, SMLS, AS, TM, WG, MG and HJW were involved in the analysis and interpretation of data. All authors were involved with drafting the article or revising it critically for important intellectual content and all authors have approved the final version to be published.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Observed change in FPG from baseline vs. estimated creatinine clearance
Figure S2 Observed change in HbA1c from baseline vs. estimated creatinine clearance
Table S1 Summary of selected adverse events from studies D and E (placebo- and empagliflozin-treated subjects): number of subjects reporting each event
References
- Nathan DM, Buse JB, Davidson MB, Ferrannini E, Holman RR, Sherwin R, Zinman B American Diabetes Association; European Association for the Study of Diabetes. Medical management of hyperglycaemia in type 2 diabetes mellitus: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetologia. 2009;52:17–30. doi: 10.1007/s00125-008-1157-y. [DOI] [PubMed] [Google Scholar]
- Bailey CJ. Renal glucose reabsorption inhibitors to treat diabetes. Trends Pharmacol Sci. 2011;32:63–71. doi: 10.1016/j.tips.2010.11.011. [DOI] [PubMed] [Google Scholar]
- Chao EC. A paradigm shift in diabetes therapy – dapagliflozin and other SGLT2 inhibitors. Discov Med. 2011;11:255–263. [PubMed] [Google Scholar]
- Moe OW, Wright SH, Palacín M. Renal handling of organic solutes. In: Brenner BM, editor. Brenner and Rector's The Kidney. Philadelphia: Saunders Elsevier; 2008. pp. 214–247. [Google Scholar]
- Thomas L, Grempler R, Eckhardt M, Himmelsbach F, Sauer A, Klein T, Eickelmann P, Mark M. Long-term treatment with empagliflozin, a novel, potent and selective SGLT-2 inhibitor, improves glycaemic control and features of metabolic syndrome in diabetic rats. Diabetes Obes Metab. 2012;14:94–96. doi: 10.1111/j.1463-1326.2011.01518.x. [DOI] [PubMed] [Google Scholar]
- Heise T, Seman L, Macha S, Jones P, Marquart A, Pinnetti S, Woerle HJ, Dugi K. Safety, tolerability, pharmacokinetics and pharmacodynamics of multiple rising doses of empagliflozin in patients with T2DM. Diabetes Ther. 2013;4:331–345. doi: 10.1007/s13300-013-0030-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heise T, Seewaldt-Becker E, Macha S, Hantel S, Pinnetti S, Seman L, Woerle HJ. Safety, tolerability, pharmacokinetics and pharmacodynamics following 4 weeks' treatment with empagliflozin once daily in patients with type 2 diabetes. Diabetes Obes Metab. 2013;15:613–621. doi: 10.1111/dom.12073. [DOI] [PubMed] [Google Scholar]
- Kanada S, Koiwai K, Taniguchi A, Sarashina A, Seman L, Woerle HJ. Pharmacokinetics, pharmacodynamics, safety and tolerability of four weeks' treatment with empagliflozin in Japanese patients with type 2 diabetes mellitus. J Diabetes Investig. 2013;4:613–617. doi: 10.1111/jdi.12110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrannini E, Seman L, Seewaldt-Becker E, Hantel S, Pinnetti S, Woerle H. A phase IIb, randomised, placebo-controlled study of the SGLT2 inhibitor empagliflozin in patients with type 2 diabetes. Diabetes Obes Metab. 2013;15:721–728. doi: 10.1111/dom.12081. [DOI] [PubMed] [Google Scholar]
- Rosenstock J, Seman LJ, Jelaska A, Hantel S, Pinnetti S, Hach T, Woerle HJ. Efficacy and safety of empagliflozin, a sodium glucose cotransporter 2 (SGLT2) inhibitor, as add-on to metformin in type 2 diabetes with mild hyperglycaemia. Diabetes Obes Metab. 2013;15:1154–1160. doi: 10.1111/dom.12185. [DOI] [PubMed] [Google Scholar]
- Beal SL, Sheiner LB, Boeckmann AJE. NONMEM Users Guide: Part I–VII (1989–2006) Ellicott City, MD, USA: Icon Development Solutions; 2006. [Google Scholar]
- Gelman A, Carlin JB, Stern HS, Rubin DB. Bayesian Data Analysis. New York, USA: Chapman & Hall/CRC; 2004. [Google Scholar]
- Yano Y, Beal SL, Sheiner LB. Evaluating pharmacokinetic/pharmacodynamic models using the posterior predictive check. J Pharmacokinet Pharmacodyn. 2001;28:171–192. doi: 10.1023/a:1011555016423. [DOI] [PubMed] [Google Scholar]
- Hastie T, Tibshirani R. Generalized Additive Models. Boca Raton, FL, USA: Chapman & Hall/CRC; 1999. [Google Scholar]
- Ruppert D, Wand MP, Carroll RJ. Semiparametric Regression. Cambridge, UK: Cambridge University Press; 2003. [Google Scholar]
- Bachman WJ, Gillespie WR. Truncated sigmoid Emax models: a reparameterization of the sigmoids Emax model for use with truncated PK/PD data. Clin Pharmacol Ther. 1998;63:199. [Google Scholar]
- Schoemaker RC, van Gerven JM, Cohen AF. Estimating potency for the Emax-model without attaining maximal effects. J Pharmacokinet Biopharm. 1998;26:581–593. doi: 10.1023/a:1023277201179. [DOI] [PubMed] [Google Scholar]
- Cockcroft ADW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- Komoroski B, Vachharajani N, Feng Y, Li L, Kornhauser D, Pfister M. Dapagliflozin, a novel, selective SGLT2 inhibitor, improved glycemic control over 2 weeks in patients with type 2 diabetes mellitus. Clin Pharmacol Ther. 2009;85:513–519. doi: 10.1038/clpt.2008.250. [DOI] [PubMed] [Google Scholar]
- Devineni D, Curtin CR, Polidori D, Gutierrez MJ, Murphy J, Rusch S, Rothenberg PL. Pharmacokinetics and pharmacodynamics of canagliflozin, a sodium glucose co-transporter 2 inhibitor, in subjects with type 2 diabetes mellitus. J Clin Pharmacol. 2013;53:601–610. doi: 10.1002/jcph.88. [DOI] [PubMed] [Google Scholar]
- Wright EM, Hirayama BA, Loo DF. Active sugar transport in health and disease. J Intern Med. 2007;261:32–43. doi: 10.1111/j.1365-2796.2006.01746.x. [DOI] [PubMed] [Google Scholar]
- Rave K, Nosek L, Posner J, Heise T, Roggen K, van Hoogdalem EJ. Renal glucose excretion as a function of blood glucose concentration in subjects with type 2 diabetes – results of a hyperglycaemic glucose clamp study. Nephrol Dial Transplant. 2006;21:2166–2171. doi: 10.1093/ndt/gfl175. [DOI] [PubMed] [Google Scholar]
- Gerich JE. Role of the kidney in normal glucose homeostasis and in the hyperglycaemia of diabetes mellitus: therapeutic implications. Diabet Med. 2010;27:136–142. doi: 10.1111/j.1464-5491.2009.02894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samtani MN. Simple pharmacometric tools for oral anti-diabetic drug development: competitive landscape for oral non-insulin therapies in type 2 diabetes. Biopharm Drug Dispos. 2010;31:162–177. doi: 10.1002/bdd.700. [DOI] [PubMed] [Google Scholar]
- Yao Z, Krzyzanski W, Jusko WJ. Assessment of basic indirect pharmacodynamic response models with physiological limits. J Pharmacokinet Pharmacodyn. 2006;33:167–193. doi: 10.1007/s10928-006-9003-7. [DOI] [PubMed] [Google Scholar]
- Devineni D, Marbury TC, Curtin CR, Vaccaro N, Wexler D, Vandebosch A, Wajs E, Polidori D. Effects of renal function on canagliflozin (CANA) pharmacokinetics (PK) and pharmacodynamics (PD) in non-diabetic subjects. J Am Soc Nephrol. 2012;23:961A. [Google Scholar]
- Kasichayanula S, Liu X, Benito MP, Yao M, Pfister M, Lacreta FP, Humphreys WG, Boulton DW. The influence of kidney function on dapagliflozin exposure, metabolism and efficacy in healthy subjects and in patients with type 2 diabetes mellitus. Br J Clin Pharmacol. 2013;76:432–444. doi: 10.1111/bcp.12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macha S, Mattheus M, Halabi A, Pinnetti S, Woerle HJ, Broedl UC. Pharmacokinetics, pharmacodynamics and safety of empagliflozin, a sodium glucose cotransporter 2 (SGLT2) inhibitor, in subjects with renal impairment. Diabetes Obes Metab. 2014;16:215–222. doi: 10.1111/dom.12182. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Observed change in FPG from baseline vs. estimated creatinine clearance
Figure S2 Observed change in HbA1c from baseline vs. estimated creatinine clearance
Table S1 Summary of selected adverse events from studies D and E (placebo- and empagliflozin-treated subjects): number of subjects reporting each event