

Abstract
Aims
Activation of vascular GPER has been linked to vasodepressor effects in animals. However, the significance of GPER regulation on chronic blood pressure control in humans is unknown.
Methods
To examine this question we determined the functional significance of expression of a common missense single nucleotide variant of GPER, P16L in vascular smooth muscle cells, and its association with blood pressure in humans. Further, to validate the importance of carrying GPER P16L in the development of hypertension we assessed allele frequency in a cohort of hard-to-treat hypertensive patients referred to a tertiary care clinic.
Results
Expression of the GPER P16L variant (V) vs. wild type (WT) in rat aortic vascular smooth muscle cells, was associated with a significant decrease in G1 (1 μm, a GPER agonist)-mediated ERK phosphorylation (slope of the function of G1-stimulated ERK phosphorylation: GPER content WT: 16.2, 95% CI 9.9, 22.6; V: 5.0, 95% CI 1.0, 9.0; P < 0.005) and apoptosis (slope of the function of G1-stimulated apoptosis: GPER content: WT: 4.4, 95% CI: 3.4, 5.4; V: 2.5, 95% CI 1.6, 2.3 P < 0.005). Normotensive female subjects, but not male subjects, carrying this hypofunctional variant (allele frequency 22%) have increased blood pressure [mean arterial pressure: P16/P16: 80 ± 1 mmHg (n = 204) vs. P16L carriers: 82 ± 1 mmHg (n = 127), 95% CI for difference: 0.6, 4.0 mmHg, P < 0.05], [systolic blood pressure: P16/P16: 105 ± 1 mmHg vs. P16L carriers: 108 ± 1 mmHg, 95% CI for difference:1.0, 5.1 mmHg, P < 0.05], [diastolic blood pressure: P16/P16: 66 ± 0.5 mmHg vs. P16L carriers 68 ± 0.7, 95% CI for difference: 0.2, 3.6 mmHg, P < 0.05]. Further, the P16L allele frequency was almost two-fold higher in female vs. male hypertensive patients (31% vs. 16%, allele ratio 0.5, 95% CI 0.32, 0.76, P < 0.05).
Conclusions
The common genetic variant, GPER P16L, is hypofunctional and female carriers of this allele have increased blood pressure. There was an increased prevalence in a population of hard-to-treat hypertensive female patients. Cumulatively, these data suggest that in females, impaired GPER function might be associated with increased blood pressure and risk of hypertension.
Keywords: GPER, hypertension
What is already known about this subject
GPER is a newly recognized G protein coupled receptor linked to the actions of oestradiol.
GPER activation mediates vasodilation and regulates vascular cell growth.
However, the significance of GPER regulation in the regulation of human cardiovascular function is unknown.
What this study adds
We have identified that a common missense genetic variant of GPER, P16L GPER, is hypofunctional when expressed in vascular smooth muscle cells.
Further females carrying this genetic variant have increased blood pressure and have a higher allelic prevalence in a highly selected population of hard-to-treat hypertensive patients.
In total these studies support an important role of GPER in vascular regulation.
Introduction
The G protein coupled oestrogen receptor (GPER, aka GPR30) is a recently recognized G-protein coupled receptor (GPCR). GPER is widely expressed in a variety of tissues including the vasculature 1. GPER was first characterized as an orphan GPCR 2. GPER was subsequently demonstrated to mediate non-oestrogen receptor, rapid effects of oestradiol. The initial studies of the functional impact of GPER activation most frequently focused on its effects on growth regulatory mechanisms. Both proliferative/anti-apoptotic 3–5 as well as antiproliferative/pro-apoptotic effects 6,7 have been shown in various cell models. In rat aortic vascular smooth muscle cells 8 and endothelial cells 9 we have shown that GPER activation stimulates apoptosis, via an ERK-dependent mechanism.
The role of GPER has been increasingly appreciated in haemodynamic regulation. GPER stimulation has generally been reported to mediate endothelial-dependent vasodilation 10.
Although, the effects of GPER activation on vascular reactivity and regulation of vascular and endothelial cell growth have been established in cell models and in animals, the significance of GPER regulation in chronic blood pressure control in humans is unclear. In rat models, GPER activation lowers blood pressure both acutely 11 and chronically 12,13 although not universally 14. In mice, genetic deletion of GPER has been associated with an age-dependent increase in blood pressure in female (but not male) mice in one model 15. However, these blood pressure changes were not consistently observed in the three other mouse GPER knockout models 16. It is notable that in at least one of those models, although there was no reported difference in blood pressure, mice with genetic deletion of GPER had increased body weight and visceral obesity 11. The impact of chronic GPER regulation on cardiovascular function and/or body weight in humans is unknown.
To address this question we assessed the functional significance of expression of a common missense single nucleotide variant and consequently assessed the impact on blood pressure regulation of carrying the allele for this change-in-function variant of GPER.
Three single nucleotide variants in the GPER gene have now been reported (http://www.ncbi.nlm.nih.gov/SNP). One of these variants is the relatively common (allele frequency ∼20%), namely GPER1 missense variant P16L, which results in substitution of leucine for a proline at amino acid residue 16 (http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=11544331). Correlation of expression of this variant with histopathological characteristics of human breast cancers has been reported 17. However, the impact on cardiovascular function of carrying this genetic variant is unknown.
Based on these considerations we assessed association of the GPER P16L variant with subphenotypes related to the potential effects of GPER regulation on human cardiovascular function. We demonstrate that this genetic variant of GPER is hypofunctional when expressed in vascular smooth muscle cells. Further we show that female humans carrying this variant have higher blood pressure.
Methods
Vascular smooth muscle cells (VSMC) culture
Rat aortic smooth muscle cells were isolated from six male rats as described previously, and cultured in DMEM with 10% fetal bovine serum supplemented with gentamicin and amphotericin B (Invitrogen™, Carlsbad, CA, USA) 8. Rats utilized in our studies as a source of aortic smooth muscle cells were maintained at Western University London, Canada and experiments were performed following the guidelines and protocols approved by the University Council on Animal Care (UCAC) for animal research. Notably these cells have abundant expression of GPER when freshly isolated which is quickly down-regulated when maintained in culture and with the shift of these cells from a ‘contractile’ to a ‘synthetic’ phenotype 18. Thus we previously have utilized these cultured rat aortic VMSC as a ‘null’ background in which we could modulate relative GPR30 expression with adenoviral-mediated gene transfer 8.
Assessment of the functionality of the P16L GPER genetic variant vs. wild type (WT) GPER
In order to determine the functional impact of the expression of the GPER P16L variant, rat vascular smooth muscle cells were transduced with either WT GPER or GPER P16L using adenoviral constructs at varying gene doses. Western blots were used to verify the extent of GPER protein expression. ERK phosphorylation and apoptosis were both assessed to determine wild type vs. variant receptor functionality.
GPER gene transfer in vascular smooth muscle cells
Rat vascular smooth cells were infected with adenoviral constructs, adenoWTGPER or adenoP16LGPER, for 24 h at 37°C following which infection media were replaced with fresh phenol red-free media, as phenol red has been reported to have oestrogenic activity 19. Cells were utilized for experimentation 48 h post-infection with serum deprivation for the last 24 h.
Assessment of ERK phosphorylation by immunoblotting
Vascular smooth muscle cells were infected with either WT GPER or variant GPER P16L adenovirus for 24 h, the medium was replaced with phenol red-free DMEM and incubated for an additional 24 h. Cells were then treated with the GPER agonist, G1 20 (1 μm; Calbiochem-Novabiochem Corporation, San Diego, CA, USA) for 15 min and subsequently lysed. Whole cell lysates were resolved on SDS-PAGE, transferred to PVDF membrane and blotted with anti-phospho-ERK antibody (at a dilution of 1:1000, Cell Signaling Technology, Danvers, MA, USA) to assess the extent of phospho-ERK expression or use of the anti-flag antibody, anti-M2 (at a dilution of 1:1000, Sigma-Aldrich Canada Ltd, Oakville, ON), to assess the extent of GPER expression as we have previously described 8.
Assessment of apoptosis by annexin V labelling
This was carried out using techniques that we have previously described 8,21. Vascular smooth cells were cultured 24 h before gene transfer and infected with adenoviral constructs expressing WT GPER, P16L GPER or GFP (as a control) for 24 h. The infection medium was then replaced with phenol red-free DMEM without serum. After 48 h of serum starvation, cells were treated with G1 (1 μm) for 24 h, detached with trypsin and washed in phosphate buffered saline (PBS). Pooled intact cells were suspended in annexin binding buffer containing fluorescein isothiocyanate–conjugated annexin V (0.25 μg ml−1) (BD Biosciences, Mississauga, ON, Canada) and propidium iodide (5 μg ml−1) (Sigma-Aldrich, Oakville, Canada) and incubated in the dark for 15 min. Annexin V binding was assessed using a BD FACScalibur flow cytometer (BD Biosciences). A total of 20 000 events were analyzed for double-stained positive cells for each sample with FlowJo software (Tree Star Inc.) by a blinded observer. Data were normalized relative to the control levels of annexin-positive staining determined for each experiment.
Human subject protocol
A) For normotensive healthy subjects
We studied 507 normal, healthy subjects, 18–39 years of age including both males and females. Informed consent was obtained for all analyses, with approval from the Western University Research Ethics Review Board. Recruitment was based on local advertizing and e-mail invitations for volunteers within the Robarts Research Institute and Western University. Exclusion criteria included: history of cardiovascular events, average alcohol intake over 2 units per day, pregnancy and use of anti-hypertensive drugs or anticoagulants.
Blood pressure was assessed while seated and the measurement protocol was in accordance with the Canadian Hypertension Education Programme (CHEP) recommendations for the measurement of ambulatory blood pressures 22. Blood pressures and heart rates were determined as the average of five sequential measurements of seated blood pressure and heart rate (BP Tru, VSM, Vancouver, British Columbia, Canada). Data on gender, weight, height, waist circumference and smoking status were also obtained. A blood sample was taken for genetic determinations.
Overall, these subjects had the following demographic characteristics: 65.3% were women; the mean age was 23.7, 95% CI 23.2, 24.2 years. The weight (kg), BMI (kg m−2) and waist circumference (cm) were 69.9, 95% CI 68.6, 71.2; 23.4, 95% CI 23.1, 23.8; 80.1, 95% CI 79.2, 81.2, respectively.
B) For hypertension clinic patients
One hundred and fifty patients with a presumptive diagnosis of primary hypertension were recruited from a tertiary care level hypertension clinic where they had been referred for management of difficult-to-treat hypertension. Informed consent was obtained for all analyses, with approval from the Western University Research Ethics Review Board. Hypertensive subjects studied had the following demographic characteristics: 39.3% were women and the mean age was 54.1, 95% CI 52.6, 55.7 years. The height (cm), weight (kg), BMI (kg m−2), waist circumference (cm) and heart rate (beats min−1) were 170, 95% CI 168, 172; 97, 95% CI 93, 99; 33, 95% CI: 31, 34; 104, 95% CI 102, 107 and 75, 95% CI 72, 77, respectively. Blood pressure determinations were performed using BP-Tru as described above.
Genotyping
Genomic DNA was extracted from whole blood and genotyped for GPER as previously described 23.
Data analysis
For the normotensive population screening, the statistical significance of differences in quantitative variables between wild type and GPER P16L variant groups was determined by Student's t-test for unpaired data (Prism 4.0, GraphPad Software, San Diego, CA, USA). P < 0.05 was taken as the minimum value of significance. Chi square test was used to compare the genotype frequencies in the healthy, normotensive vs. hypertension clinic populations. The association of GPER genotype with blood pressure was assessed by anova after adjusting for covariates using a general linear model as we have previously reported 24. This approach has been suggested to be an appropriate model especially in the setting of unbalanced study designs and reports significance after all assessed covariates are taken into account 24,25. The GPER genotype was introduced as a dichotomous variable CC (wild type-WT) or CT+TT (carrier) during the analysis. The dependent variable for all analysis was the blood pressure and the independent variables used were genotype, age, BMI and waist circumference. This analysis was done for the normotensive population and both males and females. SAS statistical software, version 9.1 (SAS institute, Inc., Cary, NC, USA) was used for all statistical analyses.
For determination of the differences in slopes for G1-mediated ERK phosphorylation and G1-mediated apoptosis for vascular smooth muscle cells expressing either the WT or variant, data was fit by linear regression and comparison of fits was determined by F-test based on the null hypothesis that the slopes were the same (Prism 4.0, GraphPad Software, San Diego, CA, USA).
All data are reported as either mean ± standard error of the mean (SEM) with the 95% confidence interval (CI) or where appropriate as the mean (95% CI).
The drug/molecular target nomenclature used in this manuscript conforms to the British Journal of Pharmacology's The Concise Guide to PHARMACOLOGY 2013/2014 26.
Results
The GPER P16L variant is hypofunctional
The extent of GPER-mediated ERK phosphorylation is decreased in cells expressing the P16L variant of GPER
Our previous studies have demonstrated that in vascular smooth muscle cells GPER activation mediates ERK phosphorylation and ERK-dependent apoptosis 8. Notably, in these previous studies we demonstrated that the effect of the selective GPER agonist, G1, to mediate ERK phosphorylation and apoptosis was GPER-dependent (as shown using both pharmacological and shRNA approaches) 8. As depicted in Figure 1A, with increasing GPER protein expression levels (as assessed by immunoblotting and based on empirically increasing the viral dose) the extent of GPER-mediated ERK phosphorylation was increased (as assessed by response to stimulation by the GPER agonist G1 [1 μm for 15 min]). Across a greater than three-fold range of GPER protein expression levels the extent of G1-mediated stimulation of ERK phosphorylation was statistically significantly attenuated in cells transduced with the P16L variant of GPER vs. the WT (as assessed by the slope of the function of GPER-mediated ERK phosphorylation/GPER expression; wild type: 16.2, 95% CI 9.9, 22.6; P16L; variant: 5.0, 95% CI 1.0, 9.0, P < 0.05) (Figure 1A).
Figure 1.
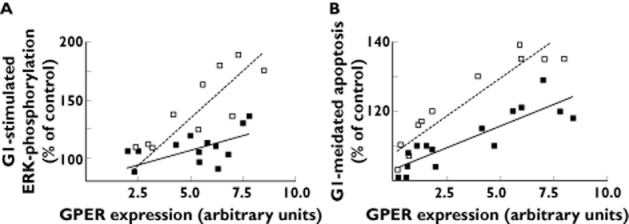
GPER agonist G1 (1 μm)-mediated ERK activation and stimulation of apoptosis in vascular smooth muscle cells transduced with either GPER wild type or P16L adenoviral construct. (A) Effect on G1-mediated ERK activation in vascular smooth muscle cells transduced with increasing viral doses of either GPER or P16L adenovirus. (B) Effect on G1-mediated apoptosis in vascular smooth muscle cells transduced by increasing viral doses of either wild type GPER or P16L GPER adenovirus. Data represent percentage of control vs. (flag-tagged) GPER expression as assessed by immunoblots using the anti-flag antibody (M2) normalized to a common GPER control (a pool of GPER-transduced vascular smooth cell lysates). Each point represents the extent of G1-mediated effect for a specific level of GPER protein expression of either WT or GPER P16L. * P < 0.05 based on a comparison of fits (Prism 4.0, GraphPad Software, San Diego California). - - -, WT GPER; —, P16L GPER
The extent of GPER-mediated apoptosis is decreased in cells expressing the P16L variant of GPER
As depicted in Figure 1B, the extent of both GPER receptor expression and GPER-mediated apoptosis was increased with increasing protein expression of WT GPER, as assessed by response to stimulation by the GPER agonist G1 (1 μm for 24 h). Further, the extent of GPER-mediated apoptosis was significantly attenuated in cells transduced with the P16L variant of GPER vs. WT GPER, viz. the slope of the function of {the extent of GPER-mediated apoptosis/extent of GPER expression} was statistically significantly lower in cells expressing the GPER P16L variant compared with the wild type (wild type: 4.4, 95% CI 3.4, 5.4; P16L variant: 2.5, 95% CI 1.6, 3.3, P < 0.05; Figure 1B).
Healthy individuals carrying the GPER P16L variant have higher blood pressures
The surveyed population of normal healthy adults (n = 507) reflected the ethnic background of the region, i.e. predominantly White (87%), with a small subset of Asian (9%), a small subset of south Asian (3%) and Black (1%) subjects. The genotype frequency for GPER (WT [C] vs. GPER P16L variant [T]) were: wild type homozygotes (CC) 61.5%, heterozygotes (CT) 32% and P16L homozygotes (TT) 6.5%. The allele frequency of the P16L variant in this population was 22.5%. There were no significant differences in allele frequency between males and females (males 22.4%, females 22.5%). GPER P16L carriers (CT heterozygotes plus TT homozygotes) had significantly higher mean arterial pressure (MAP) (83 ± 0.6 mmHg in P16L carriers vs. 81 ± 0.5 mmHg in P16/P16, P < 0.05, 95% CI for BP difference 0.3, 3.2) (Figure 2A) and systolic blood pressure (SBP) (111 ± 1 mmHg in P16L carriers vs. 109 ± 1 mmHg in P16/P16, P < 0.05, 95% CI for BP difference 0.5, 4.2) than homozygotes for the WT allele (Figure 2B). The difference in diastolic blood pressure (DBP) (Figure 2C) in wild type (68 ± 1 mmHg) vs. GPER P16L carriers (69 ± 1 mmHg) was not statistically significant, P = 0.053, 95% CI −0.02, 2.8. Paralleling the increase in blood pressure, GPER P16L carriers had a higher BMI and increased waist circumference (Table 1A).
Figure 2.
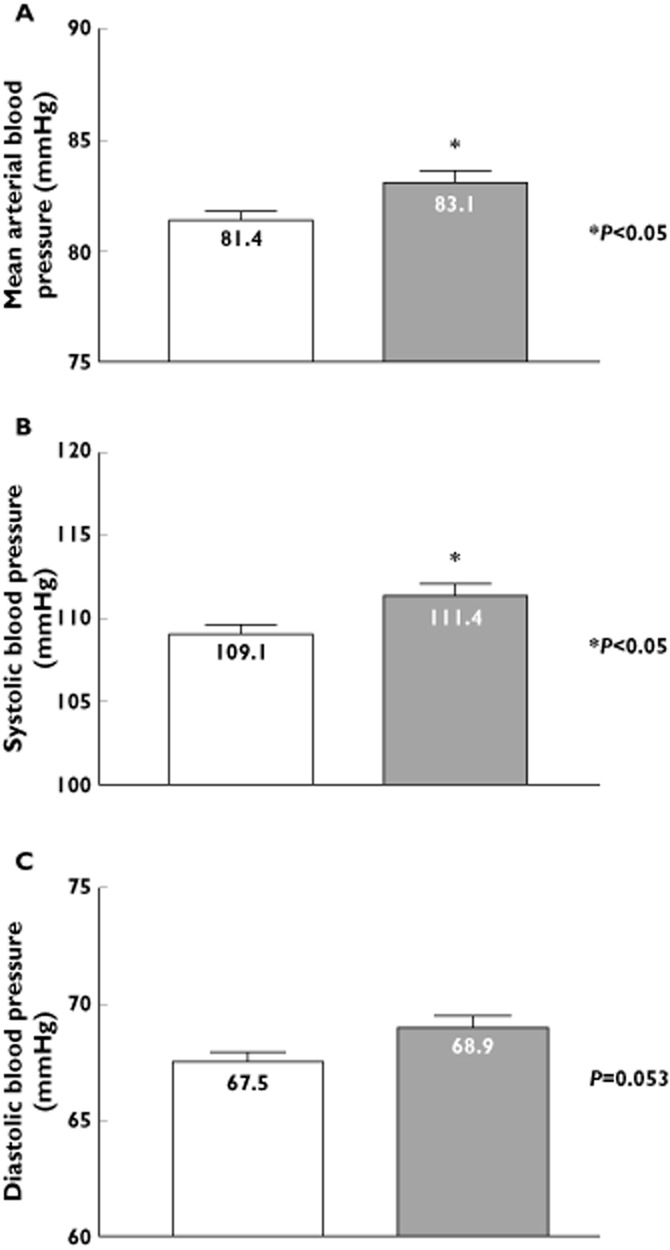
Impact of GPER P16L expression on blood pressure in normotensive subjects. Subjects were classified according to presence or absence of the P16L genotype, i.e. the P16L (V) variant group consists of both T/T homozygote C/T heterozygote subgroups. (A) Mean arterial blood pressure, (B) systolic blood pressure or (C) diastolic blood pressures were measured in wild type (CC) or GPER P16L variant (V) carriers. * represent P < 0.05 vs. subjects notcarrying the GPER P16L genetic variant (WT).  , WT (n = 312);
, WT (n = 312);  , P16L (n = 195)
, P16L (n = 195)
Table 1.
Subject demographics: normal healthy adults
| A | |||||
|---|---|---|---|---|---|
| All normotensive subjects | WT (n = 312) | WT (95% CI) | V (n = 195) | V (95% CI) | P value |
| Age (years) | 23.7 | 23.2, 24.3 | 23.8 | 23.1, 24.6 | 0.8226 |
| Height (cm) | 170.3 | 169.2, 171.3 | 171.4 | 170.1, 172.7 | 0.1891 |
| Weight (kg) | 68.5 | 67, 70 | 72.2 | 69.9, 74.5 | 0.0063 |
| BMI | 23.5 | 23.1, 23.9 | 24.5 | 23.8, 25.1 | 0.0078 |
| Waist circumference (cm) | 79.3 | 78.1, 80.4 | 81.9 | 80.1, 83.6 | 0.0117 |
| B | |||||
| Normotensive females | WT (n = 204) | WT (95% CI) | V (n = 127) | V (95% CI) | P value |
| Age (years) | 23.5 | 22.8, 24.2 | 23.7 | 22.8, 24.7 | 0.6750 |
| Height (cm) | 165.6 | 164.7, 166.6 | 165.6 | 165.4, 167.6 | 0.2335 |
| Weight (kg) | 62.7 | 61.2, 64.2 | 66.1 | 63.9, 68.4 | 0.0085 |
| BMI | 22.8 | 22.3, 23.3 | 23.9 | 23, 24.7 | 0.0258 |
| Waist circumference (cm) | 75.7 | 74.4, 76.9 | 78.0 | 76.1, 79.8 | 0.0343 |
| C | |||||
| Normotensive males | WT (n = 108) | WT (95% CI) | V (n = 68) | V (95% CI) | P value |
| Age (years) | 24.2 | 23.2, 25.1 | 24.0 | 22.7, 25.3 | 0.8377 |
| Height (cm) | 179.1 | 177.8, 180.4 | 180.5 | 178.8, 182.3 | 0.1801 |
| Weight (Kg) | 79.5 | 77.2, 81.9 | 83.5 | 79.7, 87.4 | 0.0605 |
| BMI | 24.8 | 24.2, 25.4 | 25.6 | 24.6, 26.5 | 0.1227 |
| Waist circumference (cm) | 86.1 | 84.2, 87.9 | 89.1 | 86.2, 92.0 | 0.0642 |
The GPER P16L variant-associated rise in blood pressure is gender-specific
Females carrying the GPER P16L variant had significantly higher mean arterial pressure (P16/P16, 80 ± 1 mmHg (n = 204) vs. P16L carriers, 82 ± 1 mmHg (n = 127), P < 0.05, 95% CI BP difference 0.6, 4.0), systolic blood pressure (P16/P16, 105 ± 1 mmHg vs. P16L carriers, 108 ± 1 mmHg, P < 0.05, 95% CI BP difference 1.0, 5.1) and diastolic blood pressure (P16/P16, 66 ± 1 mmHg vs. P16L carriers 68 ± 1, P < 0.05, 95% CI BP difference 0.2, 3.6). Additionally, females carrying the GPER P16L allele had increased BMI and waist circumference (Table 1B). The difference in blood pressure associated with carrying the allele remained significant in females after adjusting for covariates (i.e. age, BMI, waist circumference) (MAP: 82 ± 1 mmHg in P16L carriers vs. 80 ± 1 in P16/P16 carriers, P < 0.05, 95% CI for BP difference 1.8, 2.2, Table 2). In contrast no statistically significant differences in any blood pressure parameter were evident in males carrying the GPER P16L variant (Figure 3).
Table 2.
Summary of anova for genotype and quantitative traits in normotensive subjects
| Whole population (507) | F value | P value |
|---|---|---|
| Dependent variable: mean arterial blood pressure | ||
| Sources of variation | ||
| GPER P16L | 2.78 | NS (0.096) |
| Age | 9.61 | 0.002 |
| BMI | 0.27 | NS (0.60) |
| Waist circumference | 10.30 | 0.001 |
| Females (331) | F value | P value |
| Dependent variable: mean arterial blood pressure | ||
| Sources of variation | ||
| GPER P16L | 5.20 | 0.02 |
| Age | 11.10 | 0.001 |
| BMI | 0.90 | NS (0.34) |
| Waist circumference | 0.15 | NS (0.70) |
| Males (176) | F value | P-value |
| Dependent variable: mean arterial blood pressure | ||
| Sources of variation | ||
| GPER P16L | 0.00 | NS (0.99) |
| Age | 0.17 | NS (0.68) |
| BMI | 2.23 | NS (0.14) |
| Waist circumference | 0.33 | NS (0.57) |
Figure 3.
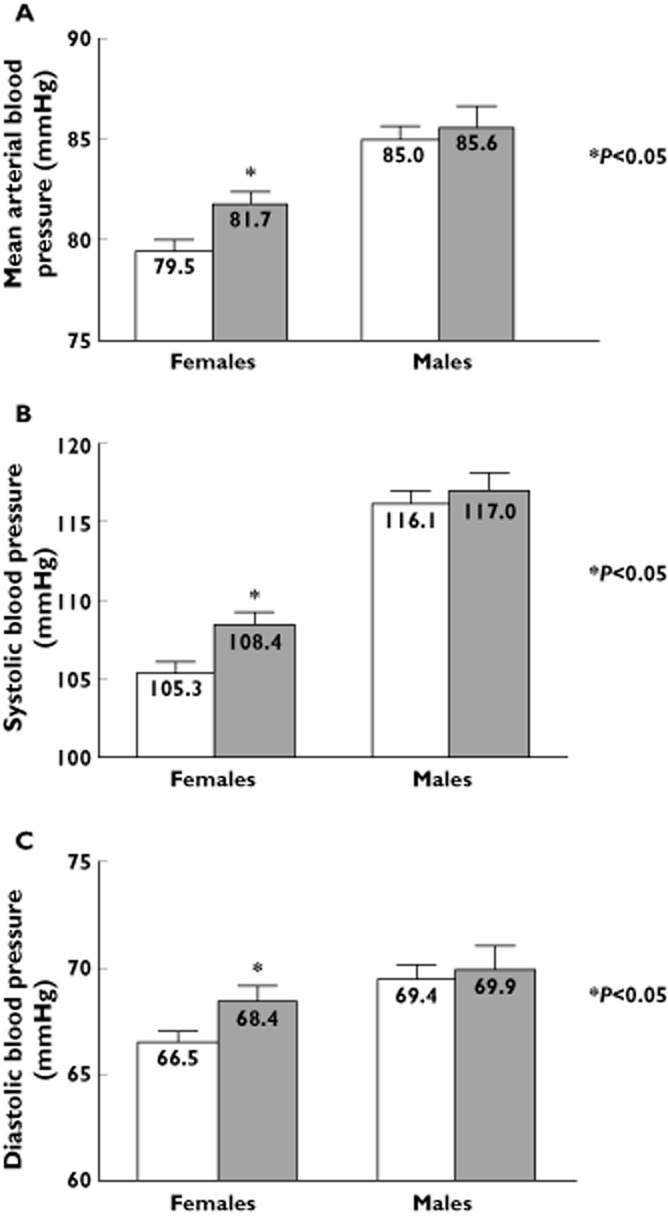
Gender-specific effect of GPER P16L expression on blood pressure in normotensive subjects. (A) Mean arterial blood pressure, (B) systolic blood pressure or (C) diastolic blood pressures were significantly higher in females, but not males, carrying the GPER P16L variant (V). * represents P < 0.05 vs. wild type.  , WT (females = 204, males = 108);
, WT (females = 204, males = 108);  , P16L (females = 127, males = 68)
, P16L (females = 127, males = 68)
The allele frequency of the GPER P16L genetic variant is significantly higher in females with hypertension
As an initial approach to the question of whether carrying the P16L GPER variant is associated with the development of hypertension, we assessed the allele frequency of the GPER P16L variant in a population of hypertensive subjects (n = 150) referred to a tertiary level care hypertension clinic primarily for management of difficult-to-treat hypertension. The hypertensive subject population was almost entirely White (98%). The allele frequency of the GPER P16L variant was statistically significantly higher in hypertensive females in comparison with hypertensive males (31% vs. 16%, P < 0.05, allele ratio 0.5, 95% CI 0.32, 0.76, P < 0.05). Further, the allele frequency of the GPER P16L variant in hypertensive women was statistically significantly higher than that found among normotensive females (31% vs. 23%, allele ratio 0.7, 95% CI 0.53, 0.97, P < 0.05). In contrast, in males with hypertension the allele frequency of the GPER P16L variant was not significantly different from in normotensive males and in fact, tended to be lower in males with hypertension (16% vs. 22%). Overall, the hypertensive patients (predictably) were older, had higher BMIs and waist circumferences. However, no differences in age, BMI or waist circumference were evident between hypertensive groups based on carriage of the P16L GPER allele (Table 3). Further, there were no differences in the baseline number of antihypertensive medications prescribed for those with or without the allele (Table 4). The percentage of use of the varying classes of antihypertensive medications in the variant vs. wild type groups is presented in Table 4.
Table 3.
Subject demographics: hypertensive patients
| A | |||||
|---|---|---|---|---|---|
| All patients | WT (n = 91) | WT (95% CI) | V (n = 59) | V (95% CI) | P value |
| Age (years) | 53.5 | 51.5, 55.6 | 55.1 | 52.57, 57.53 | 0.3487 |
| Height (cm) | 171.1 | 168.6, 173.6 | 168.5 | 165.7, 171.3 | 0.1787 |
| Weight (kg) | 98.8 | 94.5, 103.2 | 92.9 | 87.1, 98.6 | 0.0986 |
| BMI | 33.8 | 32.31, 35.29 | 33.4 | 33.2, 35.5 | 0.7222 |
| Waist circumference (cm) | 105.7 | 102.6, 108.9 | 102.4 | 98.06, 106.7 | 0.2044 |
| B | |||||
| Hypertensive females | WT (n = 27) | WT (95% CI) | V (n = 31) | V (95% CI) | P value |
| Age (years) | 53.0 | 48.57, 57.43 | 52.8 | 49.4, 56.2 | 0.9411 |
| Height (cm) | 159.7 | 157.2, 162.2 | 160.8 | 158.5, 163.1 | 0.5201 |
| Weight (kg) | 93.7 | 82.76, 104.6 | 87.9 | 80.1, 94.7 | 0.3445 |
| BMI | 36.6 | 32.77, 40.33 | 33.6 | 31.1, 36.2 | 0.1836 |
| Waist circumference (cm) | 103.0 | 96.19, 109.9 | 99.3 | 93.4, 105.3 | 0.4044 |
| C | |||||
| Hypertensive males | WT (n = 64) | WT (95% CI) | V (n = 28) | V (95% CI) | P value |
| Age (years) | 53.8 | 51.5, 56.1 | 53.0 | 48.8, 57.1 | 0.7143 |
| Height (cm) | 176.0 | 173.4, 178.6 | 177.1 | 174, 180.2 | 0.6328 |
| Weight (kg) | 101.0 | 96.7, 105.3 | 98.4 | 88.9, 107.9 | 0.5668 |
| BMI | 32.6 | 31.2, 34.0 | 33.0 | 29.3, 36.8 | 0.7977 |
| Waist circumference (cm) | 106.9 | 103.4, 110.4 | 105.9 | 99.4, 112.4 | 0.7781 |
Table 4.
Antihypertensive drugs and drug class distribution
| Antihypertensive class (n (%)) | Wild type (CC) (n = 91) | Variant (CT/TT) (n = 59) | ||
|---|---|---|---|---|
| Females (n = 27) | Males (n = 64) | Females (n = 31) | Males (n = 28) | |
| β-adrenoceptor antagonists | 6 (22%) | 11 (17%) | 4 (13%) | 4 (14%) |
| Calcium channel blockers | 10 (37%) | 28 (44%) | 6 (19%) | 8 (29%) |
| Diuretics | 8 (30%) | 19 (30%) | 12 (39%) | 6 (21%) |
| Angiotensin converting enzyme inhibitor (ACE-I) | 12 (44%) | 20 (31%) | 15 (48%) | 15 (54%) |
| Angiotensin II receptor blockers (ARB) | 7 (26%) | 21 (33%) | 10 (32%) | 6 (21%) |
| Potassium sparing diuretic | 4 (15%) | 2 (3%) | 3 (10%) | 1 (4%) |
| Number of antihypertensive treatments (95% CI) | 2.0 (1.6, 2.4) | 1.7 (1.4, 2.0) | 1.5 (1.2, 1.9) | 1.7 (1.3, 2.1) |
Discussion
The importance of the G protein coupled receptor, GPER, in mediating the vascular effects of oestradiol has been increasingly appreciated 27,28. However, the importance of GPER in regulation of cardiovascular function in humans is undefined. Utilizing a genetic variant approach and examining the impact of a relatively common missense GPER variant, GPER P16L, the current studies demonstrate that i) the gene product of this single nucleotide polymorphism is hypofunctional when expressed in vascular smooth muscle cells, ii) females, but not males, carrying this P16L variant have higher blood pressure and iii) females with hypertension are more likely to carry a GPER P16L allele. In total, these studies suggest that genetic down-regulation of GPER activity parallels an increase in blood pressure and risk of hypertension in women, but not men. These data in aggregate support the hypothesis that GPER regulation is important in the regulation of cardiovascular function in women.
G protein coupled receptors and their linked transmembrane signalling proteins are important regulators of both vascular reactivity and vascular growth. Based on this premise, these proteins have been the focus of candidate gene approaches to elucidate their roles in cardiovascular disease generally and in hypertension 29. GPCR/G protein/associated signalling proteins whose genetic variants have been linked to increased blood pressure and/or hypertension include the β1- and β2-adrenoceptors 30, G-proteins (GNB3) 29, regulators of G protein signalling (RGS2) 31,32 G protein receptor kinases (GRK4) 29 and adenylyl cyclases (ADCY6) 33. However, the associations between blood pressure and many of these candidate gene variations have been inconsistent 34.
The current studies suggest that the P16L variant of the GPCR, GPER, has significant effects on cardiovascular regulation evident both in vitro in vascular smooth muscle cells and in vivo with regard to blood pressure and risk of hypertension in women. Previous studies by our laboratory and others have suggested an important role of GPER in regulation of ERK activation and vascular cell growth and death 8. Our current studies demonstrate that in rat aortic vascular smooth muscle cells (which lose expression of native GPER when maintained in culture) the expression of the GPER P16L variant vs. WT GPER results in attenuated GPER-mediated ERK activation and GPER-mediated apoptosis. Notably, in these studies, we compared the effects of multiple doses of each construct (achieving a range of GPER protein expression levels) and normalizing the effect of these doses based on the extent of GPER protein expression achieved. We have previously utilized this approach in our delineation of the impact of expression of a common adenylyl cyclase 6 (ADCY6) genetic variant 33. The importance of using this approach is to establish that the differences seen with expression of the GPER P16L are truly due to qualitative differences in the functionality of the genetic variant P16L vs. WT receptor rather than simply quantitative differences in expression of GPER as mediated by two different adenoviral cDNA constructs.
GPER activation in acute studies mediates vasodilation and reduces blood pressure 35. Thus, we hypothesized that impaired GPER function as mediated by a hypofunctional GPER variant would be reflected by increased blood pressure. An increase in blood pressure was detected in those women carrying the GPER P16L hypofunctional variant. The effect on blood pressure of carrying this allele is almost 2 mmHg, an impact equivalent to or greater than that of several of the genetic variants of proteins more clearly linked to blood pressure regulation in humans 36. The potential impact of expression of the GPER P16L genetic variant on blood pressure regulation was supported by the increased allele frequency of this genetic variant in patients referred to a tertiary care hypertension clinic. The enrichment in allele frequency of almost 1.4 times for carrying the genetic variant in females with hypertension is well within the range of reported ratios of other genetic variants of proteins clearly associated with BP regulation, including aldosterone synthase 37 catechol-O-methyl transferase 38, GNB3 39. Beyond the impact of carrying the GPER P16L as a risk factor for the development of hypertension, these studies support the concept that GPER regulation has a significant impact on cardiovascular function.
The finding that the haemodynamic impact of carrying the GPER P16L variant is restricted to females is notable. GPER was initially characterized as mediating the effects of oestradiol 40. Hence an effect of expression of a hypofunctional GPER leading to increased blood pressure solely in females might be expected. However, it should be noted that in the normotensive population blood pressure tended to be higher in males as well as females carrying the P16L GPER genetic variant. Thus whether the inability to determine a significant increase in blood pressure in males carrying the GPER P16L variant reflected either a lesser importance of GPER in regulating blood pressure in males or a type II error for the ability to determine a change in blood pressure in males comparable with that seen in females cannot be conclusively determined.
It may be reasonable to query why this genetic variant has not been previously suggested as a candidate gene based on prior genome-wide association studies in hypertension. However, as far as we have been able to determine, none of the arrays used in the hypertension studies listed in the catalogue of published genome-wide association studies (GWAS) carried the SNP of interest (rs11544331) (https://www.genome.gov/page.cfm?pageid=26525384#searchForm) 41–50. In this context, our current studies emphasize that the full range of potential candidate genes in hypertension may have been incompletely characterized in past GWAS efforts and as well emphasizes the importance of gender-specific considerations in the identification of hypertension candidate genes.
The increase in blood pressure in those carrying the GPER genetic variant paralleled an increase in BMI and visceral fat (as assessed by waist circumference) in the normotensive patient population. It is of note that this genetic variant of GPER has not been identified to date as a potential candidate gene for obesity in association studies. However it is notable that genetic variants of other GPCRs as well as G proteins have been linked with obesity, such as associations between GNB3 C825T variant and waist circumference in North American aboriginal people 51,52. Some polymorphisms affecting genes encoding GPCRs, such as B3ADR encoding the β3-adrenoceptor, also have been associated with indices of obesity 53, although less consistently.
Important limitations to these studies should be noted. The population of hypertensive patients studied was one that had been referred to a tertiary care level subspecialty clinic generally reserved for difficult-to-treat hypertension. Thus whether the increased odds ratio seen in the current study was specific for more difficult-to-treat patients with hypertension or was more generalized to patients with milder forms of hypertension has yet to be determined. Further, although we have demonstrated that carrying a GPER variant which is hypofunctional, when expressed in vascular smooth muscle cells, parallels increased blood pressure and a greater frequency of carriage in hard-to-treat hypertensive females, the basis for the causal relationship between the attenuation in GPER-mediated vascular effects and increased blood pressure remains conjecture. GPER activation has been described to have a range of metabolic and CNS effects 54. Further, the increase in blood pressure in those females carrying the variant GPER allele was also associated with increased BMI (a well established risk factor for the development of hypertension). However, even when adjusted for BMI, the increase in blood pressure in females carrying the P16L GPER genetic variant remained significant. Notwithstanding, impairment of GPER responses at non-vascular target sites, which might also underlie the association with increased BMI, could also contribute to the increase in blood pressure seen in females carrying the GPER P16L allele.
In summary, our studies demonstrate that expression of a common GPER genetic variant parallels impaired GPER-mediated function, increased blood pressure and an increased risk of hypertension. Overall, they support an important role of GPER in regulation of cardiovascular function in women.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare RDF, RG and RAH had support from Heart and Stroke Foundation of Canada for the submitted work. There are no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
References
- Prossnitz ER, Barton M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol. 2011;7:715–726. doi: 10.1038/nrendo.2011.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeci C, Thompson DA, Ring HZ, Francke U, Weigel RJ. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics. 1997;45:607–617. doi: 10.1006/geno.1997.4972. [DOI] [PubMed] [Google Scholar]
- Lin BC, Suzawa M, Blind RD, Tobias SC, Bulun SE, Scanlan TS, Ingraham HA. Stimulating the GPR30 estrogen receptor with a novel tamoxifen analogue activates SF-1 and promotes endometrial cell proliferation. Cancer Res. 2009;69:5415–5423. doi: 10.1158/0008-5472.CAN-08-1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He YY, Cai B, Yang YX, Liu XL, Wan XP. Estrogenic G protein-coupled receptor 30 signaling is involved in regulation of endometrial carcinoma by promoting proliferation, invasion potential, and interleukin-6 secretion via the MEK/ERK mitogen-activated protein kinase pathway. Cancer Sci. 2009;100:1051–1061. doi: 10.1111/j.1349-7006.2009.01148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey DP, Lappano R, Albanito L, Madeo A, Maggiolini M, Picard D. Estrogenic GPR30 signalling induces proliferation and migration of breast cancer cells through CTGF. EMBO J. 2009;28:523–532. doi: 10.1038/emboj.2008.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariazi EA, Brailoiu E, Yerrum S, Shupp HA, Slifker MJ, Cunliffe HE, Black MA, Donato AL, Arterburn JB, Oprea TI, Prossnitz ER, Dun NJ, Jordan VC. The G protein–coupled receptor GPR30 inhibits proliferation of estrogen receptor–positive breast cancer cells. Cancer Res. 2010;70:1184–1194. doi: 10.1158/0008-5472.CAN-09-3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan QK, Lam HM, Ng CF, Lee AY, Chan ES, Ng HK, Ho SM, Lau KM. Activation of GPR30 inhibits the growth of prostate cancer cells through sustained activation of Erk1/2, c-jun/c-fos-dependent upregulation of p21, and induction of G(2) cell-cycle arrest. Cell Death Differ. 2010;17:1511–1523. doi: 10.1038/cdd.2010.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Gros R, Limbird LE, Chorazyczewski J, Feldman RD. Estradiol-mediated ERK phosphorylation and apoptosis in vascular smooth muscle cells requires GPR 30. Am J Physiol Cell Physiol. 2009;297:C1178–C1187. doi: 10.1152/ajpcell.00185.2009. [DOI] [PubMed] [Google Scholar]
- Gros R, Ding Q, Liu B, Chorazyczewski J, Feldman RD. Aldosterone mediates its rapid effects in vascular endothelial cells through GPER activation. Am J Physiol Cell Physiol. 2013;304:C532–C540. doi: 10.1152/ajpcell.00203.2012. [DOI] [PubMed] [Google Scholar]
- Broughton BR, Miller AA, Sobey CG. Endothelium-dependent relaxation by G protein-coupled receptor 30 agonists in rat carotid arteries. Am J Physiol Heart Circ Physiol. 2010;298:H1055–1061. doi: 10.1152/ajpheart.00878.2009. [DOI] [PubMed] [Google Scholar]
- Haas E, Bhattacharya I, Brailoiu E, Damjanović M, Brailoiu GC, Gao X, Mueller-Guerre L, Marjon NA, Gut A, Minotti R, Meyer MR, Amann K, Ammann E, Perez-Dominguez A, Genoni M, Clegg DJ, Dun NJ, Resta TC, Prossnitz ER, Barton M. Regulatory role of G protein–coupled estrogen receptor for vascular function and obesity. Circ Res. 2009;104:288–291. doi: 10.1161/CIRCRESAHA.108.190892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey SH, Cohen JA, Brosnihan KB, Gallagher PE, Chappell MC. Chronic treatment with the G protein-coupled receptor 30 agonist G-1 decreases blood pressure in ovariectomized mRen2.Lewis rats. Endocrinology. 2009;150:3753–3758. doi: 10.1210/en.2008-1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessup JA, Lindsey SH, Wang H, Chappell MC, Groban L. Attenuation of salt-induced cardiac remodeling and diastolic dysfunction by the GPER agonist G-1 in female mRen2.Lewis rats. PLoS ONE. 2010;5:e15433. doi: 10.1371/journal.pone.0015433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey SH, Yamaleyeva LM, Brosnihan KB, Gallagher PE, Chappell MC. Estrogen receptor GPR30 reduces oxidative stress and proteinuria in the salt-sensitive female mRen2.Lewis rat. Hypertension. 2011;58:665–671. doi: 10.1161/HYPERTENSIONAHA.111.175174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mårtensson UEA, Salehi SA, Windahl S, Gomez MF, Swärd K, Daszkiewicz-Nilsson J, Wendt A, Andersson N, Hellstrand P, Grände P-O, Owman C, Rosen CJ, Adamo ML, Lundquist I, Rorsman P, Nilsson B-O, Ohlsson C, Olde B, Leeb-Lundberg LMF. Deletion of the G protein-coupled receptor 30 impairs glucose tolerance, reduces bone growth, increases blood pressure, and eliminates estradiol-stimulated insulin release in female mice. Endocrinology. 2009;150:687–698. doi: 10.1210/en.2008-0623. [DOI] [PubMed] [Google Scholar]
- Langer G, Bader B, Meoli L, Isensee J, Delbeck M, Noppinger PR, Otto C. A critical review of fundamental controversies in the field of GPR30 research. Steroids. 2010;75:603–610. doi: 10.1016/j.steroids.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Giess M, Lattrich C, Springwald A, Goerse R, Ortmann O, Treeck O. GPR30 gene polymorphisms are associated with progesterone receptor status and histopathological characteristics of breast cancer patients. J Steroid Biochem Mol Biol. 2010;118:7–12. doi: 10.1016/j.jsbmb.2009.09.001. [DOI] [PubMed] [Google Scholar]
- Li S, Sims S, Jiao Y, Chow LH, Pickering JG. Evidence from a novel human cell clone that adult vascular smooth muscle cells can convert reversibly between noncontractile and contractile phenotypes. Circ Res. 1999;85:338–348. doi: 10.1161/01.res.85.4.338. [DOI] [PubMed] [Google Scholar]
- Welshons WV, Wolf MF, Murphy CS, Jordan VC. Estrogenic activity of phenol red. Mol Cell Endocrinol. 1988;57:169–178. doi: 10.1016/0303-7207(88)90072-x. [DOI] [PubMed] [Google Scholar]
- Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, Parker MA, Tkachenko SE, Savchuck NP, Sklar LA, Oprea TI, Prossnitz ER. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol. 2006;2:207–212. doi: 10.1038/nchembio775. [DOI] [PubMed] [Google Scholar]
- Gros R, Ding Q, Sklar LA, Prossnitz EE, Arterburn JB, Chorazyczewski J, Feldman RD. GPR30 expression is required for the mineralocorticoid receptor-independent rapid vascular effects of aldosterone. Hypertension. 2011;57:442–451. doi: 10.1161/HYPERTENSIONAHA.110.161653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmelgarn BR, McAllister FA, Myers MG, McKay DW, Bolli P, Abbott C, Schiffrin EL, Grover S, Honos G, Lebel M, Mann K, Wilson T, Penner B, Tremblay G, Tobe SW, Feldman RD. The 2005 Canadian Hypertension Education Program recommendations for the management of hypertension: part 1: blood pressure measurement, diagnosis and assessment of risk. Can J Cardiol. 2005;21:645–656. [PubMed] [Google Scholar]
- Cao H, van der Veer E, Ban MR, Hanley AJG, Zinman B, Harris SB, Young TK, Pickering JG, Hegele RA. Promoter polymorphism in PCK1 (phosphoenolpyruvate carboxykinase gene) associated with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2004;89:898–903. doi: 10.1210/jc.2003-031361. [DOI] [PubMed] [Google Scholar]
- Hegele RA, Cao H, Huff MW, Anderson CM. LMNA R482Q mutation in partial lipodystrophy associated with reduced plasma leptin concentration. J Clin Endocrinol Metab. 2000;85:3089–3093. doi: 10.1210/jcem.85.9.6768. [DOI] [PubMed] [Google Scholar]
- Larson MG. Analysis of variance. Circulation. 2008;117:115–121. doi: 10.1161/CIRCULATIONAHA.107.654335. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, McGrath JC, Catterall WA, Spedding M, Peters JA, Harmar AJ CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2013/14. Br J Pharmacol. 2013a;170:1449–1867. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschamps AM, Murphy E. Activation of a novel estrogen receptor, GPER, is cardioprotective in male and female rats. Am J Physiol Heart Circ Physiol. 2009;297:H1806–1813. doi: 10.1152/ajpheart.00283.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman RD, Gros R. Unraveling the mechanisms underlying the rapid vascular effects of steroids: sorting out the receptors and the pathways. Br J Pharmacol. 2011;163:1163–1169. doi: 10.1111/j.1476-5381.2011.01366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson MD, Cole DE, Jose PA. Pharmacogenomics of G protein-coupled receptor signaling: insights from health and disease. Methods Mol Biol (Clifton, N.J.) 2008;448:77–107. doi: 10.1007/978-1-59745-205-2_6. [DOI] [PubMed] [Google Scholar]
- Kirstein SL, Insel PA. Autonomic nervous system pharmacogenomics: a progress report. Pharmacol Rev. 2004;56:31–52. doi: 10.1124/pr.56.1.2. [DOI] [PubMed] [Google Scholar]
- Riddle EL, Rana BK, Murthy KK, Rao F, Eskin E, O'Connor DT, Insel PA. Polymorphisms and haplotypes of the regulator of G protein signaling-2 gene in normotensives and hypertensives. Hypertension. 2006;47:415–420. doi: 10.1161/01.HYP.0000200714.81990.61. [DOI] [PubMed] [Google Scholar]
- Feldman RD, Gros R. Regulator of G-protein signaling-2 as a candidate gene: the road to hypertension or just another roadside marker? Hypertension. 2006;47:337–338. doi: 10.1161/01.HYP.0000200748.73303.a6. [DOI] [PubMed] [Google Scholar]
- Hodges GJ, Gros R, Hegele RA, Van Uum S, Shoemaker JK, Feldman RD. Increased blood pressure and hyperdynamic cardiovascular responses in carriers of a common hyperfunctional variant of adenylyl cyclase 6. J Pharmacol Exp Ther. 2010;335:451–457. doi: 10.1124/jpet.110.172700. [DOI] [PubMed] [Google Scholar]
- Kitsios GD, Zintzaras E. Synopsis and data synthesis of genetic association studies in hypertension for the adrenergic receptor family genes: the CUMAGAS-HYPERT database. Am J Hypertens. 2010;23:305–313. doi: 10.1038/ajh.2009.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MR, Baretella O, Prossnitz ER, Barton M. Dilation of epicardial coronary arteries by the G protein-coupled estrogen receptor agonists G-1 and ICI 182,780. Pharmacology. 2010;86:58–64. doi: 10.1159/000315497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munroe PB, Barnes MR, Caulfield MJ. Advances in blood pressure genomics. Circ Res. 2013;112:1365–1379. doi: 10.1161/CIRCRESAHA.112.300387. [DOI] [PubMed] [Google Scholar]
- Li XM, Ling Y, Lu DR, Lu ZQ, Yi QL, Liu Y, Chen HY, Gao X. Association of the aldosterone synthase gene -344T>C polymorphism with essential hypertension and glucose homeostasis: a case-control study in a Han Chinese population. Clin Exp Pharmacol Physiol. 2011;38:598–604. doi: 10.1111/j.1440-1681.2011.05555.x. [DOI] [PubMed] [Google Scholar]
- Htun NC, Miyaki K, Song Y, Ikeda S, Shimbo T, Muramatsu M. Association of the catechol-O-methyl transferase gene Val158Met polymorphism with blood pressure and prevalence of hypertension: interaction with dietary energy intake. Am J Hypertens. 2011;24:1022–1026. doi: 10.1038/ajh.2011.93. [DOI] [PubMed] [Google Scholar]
- Bagos PG, Elefsinioti AL, Nikolopoulos GK, Hamodrakas SJ. The GNB3 C825T polymorphism and essential hypertension: a meta-analysis of 34 studies including 14,094 cases and 17,760 controls. J Hypertens. 2007;25:487–500. doi: 10.1097/HJH.0b013e328011db24. [DOI] [PubMed] [Google Scholar]
- Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science (New York, N.Y.) 2005;307:1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- Yang HC, Liang YJ, Chen JW, Chiang KM, Chung CM, Ho HY, Ting CT, Lin TH, Sheu SH, Tsai WC, Chen JH, Leu HB, Yin WH, Chiu TY, Chern CL, Lin SJ, Tomlinson B, Guo Y, Sham PC, Cherny SS, Lam TH, Thomas GN, Pan WH. Identification of IGF1, SLC4A4, WWOX, and SFMBT1 as hypertension susceptibility genes in Han Chinese with a genome-wide gene-based association study. PLoS ONE. 2012;7:e32907. doi: 10.1371/journal.pone.0032907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Tomlinson B, Chu T, Fang YJ, Gui H, Tang CS, Yip BH, Cherny SS, Hur YM, Sham PC, Lam TH, Thomas NG. A genome-wide linkage and association scan reveals novel loci for hypertension and blood pressure traits. PLoS ONE. 2012;7:e31489. doi: 10.1371/journal.pone.0031489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, Chasman DI, Smith AV, Tobin MD, Verwoert GC, Hwang SJ, Pihur V, Vollenweider P, O'Reilly PF, Amin N, Bragg-Gresham JL, Teumer A, Glazer NL, Launer L, Zhao JH, Aulchenko Y, Heath S, Sober S, Parsa A, Luan J, Arora P, Dehghan A, Zhang F, Lucas G, Hicks AA, Jackson AU, Peden JF, Tanaka T, Wild SH, Rudan I, Igl W, Milaneschi Y, Parker AN, Fava C, Chambers JC, Fox ER, Kumari M, Go MJ, van der Harst P, Kao WH, Sjogren M, Vinay DG, Alexander M, Tabara Y, Shaw-Hawkins S, Whincup PH, Liu Y, Shi G, Kuusisto J, Tayo B, Seielstad M, Sim X, Nguyen KD, Lehtimaki T, Matullo G, Wu Y, Gaunt TR, Onland-Moret NC, Cooper MN, Platou CG, Org E, Hardy R, Dahgam S, Palmen J, Vitart V, Braund PS, Kuznetsova T, Uiterwaal CS, Adeyemo A, Palmas W, Campbell H, Ludwig B, Tomaszewski M, Tzoulaki I, Palmer ND, Aspelund T, Garcia M, Chang YP, O'Connell JR, Steinle NI, Grobbee DE, Arking DE, Kardia SL, Morrison AC, Hernandez D, Najjar S, McArdle WL, Hadley D, Brown MJ, Connell JM, Hingorani AD, Day IN, Lawlor DA, Beilby JP, Lawrence RW, Clarke R, Hopewell JC, Ongen H, Dreisbach AW, Li Y, Young JH, Bis JC, Kahonen M, Viikari J, Adair LS, Lee NR, Chen MH, Olden M, Pattaro C, Bolton JA, Kottgen A, Bergmann S, Mooser V, Chaturvedi N, Frayling TM, Islam M, Jafar TH, Erdmann J, Kulkarni SR, Bornstein SR, Grassler J, Groop L, Voight BF, Kettunen J, Howard P, Taylor A, Guarrera S, Ricceri F, Emilsson V, Plump A, Barroso I, Khaw KT, Weder AB, Hunt SC, Sun YV, Bergman RN, Collins FS, Bonnycastle LL, Scott LJ, Stringham HM, Peltonen L, Perola M, Vartiainen E, Brand SM, Staessen JA, Wang TJ, Burton PR, Soler Artigas M, Dong Y, Snieder H, Wang X, Zhu H, Lohman KK, Rudock ME, Heckbert SR, Smith NL, Wiggins KL, Doumatey A, Shriner D, Veldre G, Viigimaa M, Kinra S, Prabhakaran D, Tripathy V, Langefeld CD, Rosengren A, Thelle DS, Corsi AM, Singleton A, Forrester T, Hilton G, McKenzie CA, Salako T, Iwai N, Kita Y, Ogihara T, Ohkubo T, Okamura T, Ueshima H, Umemura S, Eyheramendy S, Meitinger T, Wichmann HE, Cho YS, Kim HL, Lee JY, Scott J, Sehmi JS, Zhang W, Hedblad B, Nilsson P, Smith GD, Wong A, Narisu N, Stancakova A, Raffel LJ, Yao J, Kathiresan S, O'Donnell CJ, Schwartz SM, Ikram MA, Longstreth WT, Jr, Mosley TH, Seshadri S, Shrine NR, Wain LV, Morken MA, Swift AJ, Laitinen J, Prokopenko I, Zitting P, Cooper JA, Humphries SE, Danesh J, Rasheed A, Goel A, Hamsten A, Watkins H, Bakker SJ, van Gilst WH, Janipalli CS, Mani KR, Yajnik CS, Hofman A, Mattace-Raso FU, Oostra BA, Demirkan A, Isaacs A, Rivadeneira F, Lakatta EG, Orru M, Scuteri A, Ala-Korpela M, Kangas AJ, Lyytikainen LP, Soininen P, Tukiainen T, Wurtz P, Ong RT, Dorr M, Kroemer HK, Volker U, Volzke H, Galan P, Hercberg S, Lathrop M, Zelenika D, Deloukas P, Mangino M, Spector TD, Zhai G, Meschia JF, Nalls MA, Sharma P, Terzic J, Kumar MV, Denniff M, Zukowska-Szczechowska E, Wagenknecht LE, Fowkes FG, Charchar FJ, Schwarz PE, Hayward C, Guo X, Rotimi C, Bots ML, Brand E, Samani NJ, Polasek O, Talmud PJ, Nyberg F, Kuh D, Laan M, Hveem K, Palmer LJ, van der Schouw YT, Casas JP, Mohlke KL, Vineis P, Raitakari O, Ganesh SK, Wong TY, Tai ES, Cooper RS, Laakso M, Rao DC, Harris TB, Morris RW, Dominiczak AF, Kivimaki M, Marmot MG, Miki T, Saleheen D, Chandak GR, Coresh J, Navis G, Salomaa V, Han BG, Zhu X, Kooner JS, Melander O, Ridker PM, Bandinelli S, Gyllensten UB, Wright AF, Wilson JF, Ferrucci L, Farrall M, Tuomilehto J, Pramstaller PP, Elosua R, Soranzo N, Sijbrands EJ, Altshuler D, Loos RJ, Shuldiner AR, Gieger C, Meneton P, Uitterlinden AG, Wareham NJ, Gudnason V, Rotter JI, Rettig R, Uda M, Strachan DP, Witteman JC, Hartikainen AL, Beckmann JS, Boerwinkle E, Vasan RS, Boehnke M, Larson MG, Jarvelin MR, Psaty BM, Abecasis GR, Chakravarti A, Elliott P, van Duijn CM, Newton-Cheh C, Levy D, Caulfield MJ, Johnson T. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478:103–109. doi: 10.1038/nature10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavin TP, Feng T, Schnell A, Zhu X, Elston RC. Two-marker association tests yield new disease associations for coronary artery disease and hypertension. Hum Genet. 2011;130:725–733. doi: 10.1007/s00439-011-1009-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan S, Melander O, Johnson T, Di Blasio AM, Lee WK, Gentilini D, Hastie CE, Menni C, Monti MC, Delles C, Laing S, Corso B, Navis G, Kwakernaak AJ, van der Harst P, Bochud M, Maillard M, Burnier M, Hedner T, Kjeldsen S, Wahlstrand B, Sjogren M, Fava C, Montagnana M, Danese E, Torffvit O, Hedblad B, Snieder H, Connell JM, Brown M, Samani NJ, Farrall M, Cesana G, Mancia G, Signorini S, Grassi G, Eyheramendy S, Wichmann HE, Laan M, Strachan DP, Sever P, Shields DC, Stanton A, Vollenweider P, Teumer A, Volzke H, Rettig R, Newton-Cheh C, Arora P, Zhang F, Soranzo N, Spector TD, Lucas G, Kathiresan S, Siscovick DS, Luan J, Loos RJ, Wareham NJ, Penninx BW, Nolte IM, McBride M, Miller WH, Nicklin SA, Baker AH, Graham D, McDonald RA, Pell JP, Sattar N, Welsh P, Munroe P, Caulfield MJ, Zanchetti A, Dominiczak AF. Genome-wide association study of blood pressure extremes identifies variant near UMOD associated with hypertension. PLoS Genet. 2010;6:e1001177. doi: 10.1371/journal.pgen.1001177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiura Y, Tabara Y, Kokubo Y, Okamura T, Miki T, Tomoike H, Iwai N. A genome-wide association study of hypertension-related phenotypes in a Japanese population. Circ J. 2010;74:2353–2359. doi: 10.1253/circj.cj-10-0353. [DOI] [PubMed] [Google Scholar]
- Adeyemo A, Gerry N, Chen G, Herbert A, Doumatey A, Huang H, Zhou J, Lashley K, Chen Y, Christman M, Rotimi C. A genome-wide association study of hypertension and blood pressure in African Americans. PLoS Genet. 2009;5:e1000564. doi: 10.1371/journal.pgen.1000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, Dehghan A, Glazer NL, Morrison AC, Johnson AD, Aspelund T, Aulchenko Y, Lumley T, Kottgen A, Vasan RS, Rivadeneira F, Eiriksdottir G, Guo X, Arking DE, Mitchell GF, Mattace-Raso FU, Smith AV, Taylor K, Scharpf RB, Hwang SJ, Sijbrands EJ, Bis J, Harris TB, Ganesh SK, O'Donnell CJ, Hofman A, Rotter JI, Coresh J, Benjamin EJ, Uitterlinden AG, Heiss G, Fox CS, Witteman JC, Boerwinkle E, Wang TJ, Gudnason V, Larson MG, Chakravarti A, Psaty BM, van Duijn CM. Genome-wide association study of blood pressure and hypertension. Nat Genet. 2009;41:677–687. doi: 10.1038/ng.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Org E, Eyheramendy S, Juhanson P, Gieger C, Lichtner P, Klopp N, Veldre G, Doring A, Viigimaa M, Sober S, Tomberg K, Eckstein G, Kelgo P, Rebane T, Shaw-Hawkins S, Howard P, Onipinla A, Dobson RJ, Newhouse SJ, Brown M, Dominiczak A, Connell J, Samani N, Farrall M, Caulfield MJ, Munroe PB, Illig T, Wichmann HE, Meitinger T, Laan M. Genome-wide scan identifies CDH13 as a novel susceptibility locus contributing to blood pressure determination in two European populations. Hum Mol Genet. 2009;18:2288–2296. doi: 10.1093/hmg/ddp135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegele RA, Anderson C, Young TK, Connelly PW. G-protein beta3 subunit gene splice variant and body fat distribution in Nunavut Inuit. Genome Res. 1999;9:972–977. doi: 10.1101/gr.9.10.972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollex RL, Hanley AJ, Zinman B, Harris SB, Hegele RA. Clinical and genetic associations with hypertriglyceridemic waist in a Canadian aboriginal population. Int J Obes (Lond) 2006;30:484–491. doi: 10.1038/sj.ijo.0803152. [DOI] [PubMed] [Google Scholar]
- Rosmond R. Association studies of genetic polymorphisms in central obesity: a critical review. Int J Obes Relat Metab Disord. 2003;27:1141–1151. doi: 10.1038/sj.ijo.0802397. [DOI] [PubMed] [Google Scholar]
- Fuente-Martin E, Garcia-Caceres C, Morselli E, Clegg DJ, Chowen JA, Finan B, Brinton RD, Tschop MH. Estrogen, astrocytes and the neuroendocrine control of metabolism. Rev Endocr Metab Disord. 2013;14:331–338. doi: 10.1007/s11154-013-9263-7. [DOI] [PMC free article] [PubMed] [Google Scholar]