

Abstract
FTY720 (fingolimod), an FDA-approved drug for treatment of multiple sclerosis, has beneficial effects in the CNS that are not yet well understood, independent of its effects on immune cell trafficking. We show that FTY720 enters the nucleus, where it is phosphorylated by sphingosine kinase 2 (SphK2), and that nuclear FTY720-P binds and inhibits class I histone deacetylases (HDACs), enhancing specific histone acetylations. FTY720 is also phosphorylated in mice and accumulates in the brain, including the hippocampus, inhibits HDACs and enhances histone acetylation and gene expression programs associated with memory and learning, and rescues memory deficits independently of its immunosuppressive actions. Sphk2−/− mice have lower levels of hippocampal sphingosine-1-phosphate, an endogenous HDAC inhibitor, and reduced histone acetylation, and display deficits in spatial memory and impaired contextual fear extinction. Thus, sphingosine-1-phosphate and SphK2 play specific roles in memory functions and FTY720 may be a useful adjuvant therapy to facilitate extinction of aversive memories.
Fingolimod, also known as FTY720, is the first orally available therapeutic agent approved for treatment of relapsing-remitting multiple sclerosis, a disorder of the CNS1. FTY720, a synthetic analog of sphingosine, is a prototype of immunomodulatory prodrugs that is phosphorylated in vivo to FTY720-phosphate (FTY720-P) by sphingosine kinases (SphKs), especially SphK2, to a mimetic of sphingosine-1-phosphate (S1P). This bioactive sphingolipid metabolite is now emerging as an important regulator of many physiological and disease processes of the immune, central nervous and cardiovascular systems. Most of the known actions of S1P are mediated by binding to five specific G protein–coupled receptors, designated S1PR1–S1PR5. While FTY720-P is an agonist at all of the S1PRs except S1PR2, its beneficial action in multiple sclerosis is generally believed to result from the induction of irreversible downregulation and degradation of S1PR1, a receptor that is critical for lymphocyte trafficking1,2. Decreased S1PR1 surface expression on activated T cells prevents their egress from lymphoid tissues and infiltration of autoaggressive lymphocytes into the CNS. Preclinical studies suggest that FTY720 accumulates and is phosphorylated in brain3 and has beneficial effects in the CNS that are not understood yet, independent of its immune cell trafficking activity1,4.
S1P also has important intracellular actions5,6. SphK2, which is present in the nucleus of many cells5,7, produces nuclear S1P that specifically binds to HDAC1 and HDAC2, inhibits their enzymatic activities and increases histone acetylation, linking nuclear S1P to epigenetic regulation of gene expression5. Ye t it is still unknown whether nuclear SphK2 and S1P function similarly in vivo. HDAC1 and 2 belong to a large family of zinc-dependent HDACs, and HDAC inhibitors (HDACi) have long been used in psychiatry and various brain disorders and are being investigated as possible treatments for many diseases8,9. Because SphK2 is the main SphK isoenzyme that phosphorylates FTY720 in vivo and FTY720-P is a close structural analog of S1P, we wondered where in the cell FTY720 is phosphorylated and whether it also mimics the intracellular actions of S1P and inhibits HDACs to regulate histone acetylation, gene expression and brain functions.
RESULTS
FTY720-P is generated in the nucleus by SphK2 and enhances histone acetylation
FTY720 was rapidly taken up by human SH-SY5Y neuroblastoma cells. SphK2, which was predominantly found in the nucleus of these cells, as in many other types of cells, robustly phosphorylated FTY720, and hence FTY720-P accumulated over time to a greater level in the nucleus than in the cytoplasm (Fig. 1a–c). There was much less secreted FTY720-P as compared to the intracellular pools in both primary hippocampal neurons (18 ± 3 as compared to 230 ± 32 pmol) and neuroblastoma cells (Fig. 1d). Overexpression of SphK2, but not the catalytically inactive SphK2G212E, increased formation of nuclear FTY720-P by >100-fold (Fig. 1e), suggesting that nuclear SphK2 phosphorylates FTY720. The nucleus contains large amounts of sphingosine5, and overexpression of SphK2 also increased nuclear S1P (Fig. 1f). Treatment with FTY720 decreased nuclear S1P in neuroblastoma cells (Fig. 1f) and in hippocampal neurons (Fig. 1g), as expected, since FTY720 competes with the substrate sphingosine for phosphorylation by SphK2. We obtained similar results in other cell types (Supplementary Fig. 1a,b).
Figure 1.
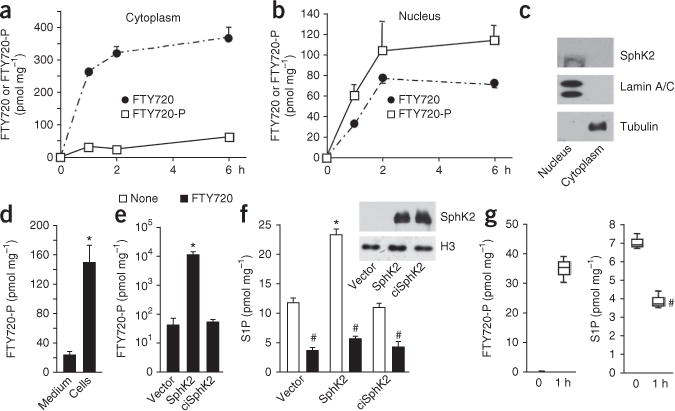
FTY720-P is produced in the nucleus by SphK2. (a–d) SH-SY5Y neuroblastoma cells were treated with 5 μM FTY720 for the indicated times (a,b) or for 6 h (d) (N = 3 independent cell cultures per group). Cytoplasmic (a) and nuclear (b) levels of FTY720 and FTY720-P were determined by liquid chromatography–electrospray injection–tandem mass spectroscopy (LC-ESI-MS/MS). (c) Equal amounts of protein were separated by SDS-PAGE and immunoblotted with SphK2-specific antibody. Antibodies against lamin A/C and tubulin were used as nuclear and cytosolic markers, respectively. (d) Total intracellular and secreted FTY720-P were determined by LC-ESI-MS/MS. *P < 0.001 as compared to medium; unpaired Student’s t-test. (e,f) SH-SY5Y neuroblastoma cells transfected with vector, SphK2 or catalytically inactive SphK2G212E (ciSphK2) constructs were treated without or with FTY720 for 6 h and nuclear levels of FTY720-P (e) and S1P (f) determined by LC-ESI-MS/MS. Equal expression was confirmed by immunoblotting. Data are expressed as mean ± s.d. *P < 0.005 as compared to vector; #P < 0.001 as compared to untreated; unpaired Student’s t-test. All western blots were performed three times. Full-length blots are presented in Supplementary Figure 10. (g) Hippocampal neurons were treated with FTY720 for 1 h and nuclear levels of FTY720-P and S1P determined by LC-ESI-MS/MS (N = 3 independent cell culture replicates per group). Box plots indicate median (center line), 25–75th percentile (box limits), and minimum and maximum (whiskers).
We next examined whether FTY720-P produced in the nucleus by SphK2 mimics the nuclear actions of S1P. Treatment of SH-SY5Y cells with FTY720 increased acetylation of Lys9 of histone H3 (H3K9), Lys5 of histone H4 (H4K5) and Lys12 of histone H2B (H2BK12) (Fig. 2a), the same residues that nuclear S1P increases5, without affecting acetylation of other lysines. Similarly, after treatment of hippocampal neurons with FTY720, nuclear FTY720-P gradually increased, concomitantly with an increase in histone H3K9 acetylation (Fig. 2b). In accord with the increase in nuclear FTY720-P (Fig. 1e and Supplementary Fig. 1a), overexpression of SphK2, but not catalytically inactive SphK2G212E, enhanced the effect of FTY720 on histone acetylation (Supplementary Fig. 1c). To exclude the possibility that these effects were due to secreted FTY720-P that acts by binding to S1PRs on the plasma membrane, we examined the effects of FTY720-P on histone acetylation in highly purified nuclei, which do not contain S1PRs. Like addition of S1P5, addition of FTY720-P to isolated nuclei increased specific histone acetylations (Fig. 2c and Supplementary Fig. 1d). Moreover, histone acetylations induced by FTY720 itself added to isolated nuclei were prevented by downregulation of SphK2 (Fig. 2d), which was associated with decreased nuclear formation of FTY720-P (326 ± 7 to 53 ± 8 pmol per mg protein). In contrast, treatment of cells with FTY720-P or S1P, which activates all of its receptors, as demonstrated by enhanced extracellular signal-regulated kinases (ERK1/2) phosphorylation, did not cause detectable changes in global histone acetylation (Fig. 2e and Supplementary Fig. 1e). Taken together, these results indicate that FTY720-P produced in the nucleus by SphK2 regulates specific histone acetylations independently of S1PRs.
Figure 2.
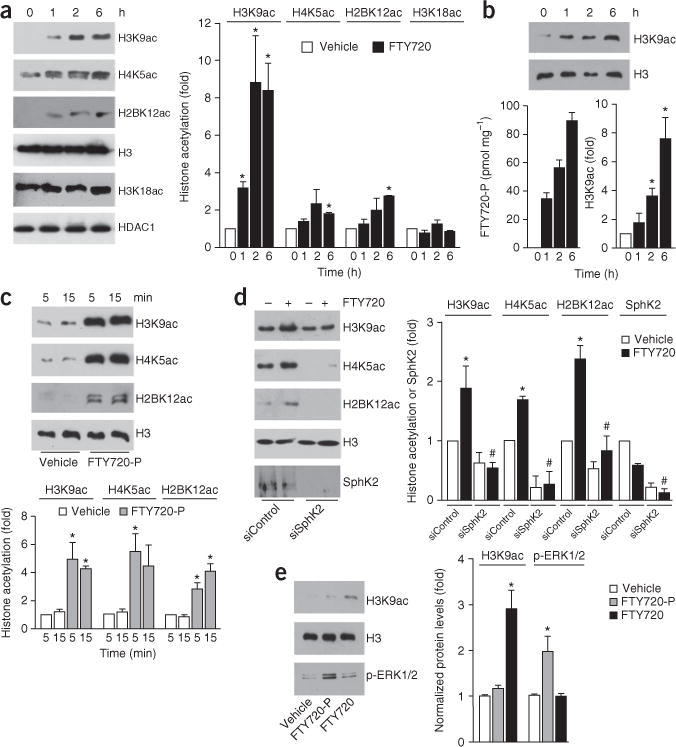
FTY720-P enhances specific histone acetylations. (a,b) SH-SY5Y neuroblastoma cells (a) or primary hippocampal neurons (b) were treated with FTY720 (5 μM) for the indicated times (N = 3 independent cell culture replicates per group). Histone acetylations in nuclear extracts were detected by immunoblotting with antibodies to specific histone acetylation sites and nuclear levels of FTY720-P were determined by LC-ESI-MS/MS (b). (c) Purified nuclei from naive SH-SY5Y neuroblastoma cells were incubated for the indicated times with vehicle or FTY720-P (1 μM) and histone acetylations determined. *P < 0.01 as compared to vehicle; unpaired Student’s t-test. (d) Purified nuclei were isolated from neuroblastoma cells transfected with siControl or siSphK2 and incubated with vehicle or FTY720 (1 μM) for 15 min. Histone acetylations were determined by immunoblotting. (e) Naive neuroblastoma cells were treated with vehicle, FTY720-P (100 nM) or FTY720 (1 μM) for 2 h and nuclear extracts analyzed by western blotting with the indicated antibodies. Quantified data are expressed as mean ± s.e.m. *P < 0.05 as compared to vehicle and #P < 0.01 as compared to siControl treated with FTY720 (unpaired Student’s t-test). H3 was used as an internal loading control. (a–e) *P < 0.05 as compared to vehicle (unpaired Student’s t-test). (d) #P < 0.01 as compared to siControl treated with FTY720 (unpaired Student’s t-test). All western blots were performed three times. Full-length blots are presented in Supplementary Figure 10.
FTY720-P, but not FTY720, potently inhibits class I HDACs
Histone acetylation levels are regulated by the opposing activities of histone acetyltransferases (HATs) and HDACs. Because FTY720-P has no effect on HAT activity (Supplementary Fig. 2), increased acetylation of histones could be due to direct inhibition of HDACs by FTY720-P, as we previously demonstrated that nuclear S1P has no effect on HAT activity but binds to and inhibits HDAC1 and 2 (ref. 5). Indeed, FTY720-P inhibited the activities of highly purified recombinant class I HDACs (HDAC1, HDAC2, HDAC3 and HDAC8) even more potently than S1P and almost as effectively as suberoylanilide hydroxamic acid (SAHA), a commonly used inhibitor of these HDACs (Fig. 3a–d). In contrast, FTY720 had no significant effects on activity of these class I HDACs. Even though S1P inhibited HDAC1–HDAC3, it did not inhibit HDAC8 activity (Fig. 3d), and neither FTY720-P nor S1P inhibited the class II HDAC7 (Fig. 3e).
Figure 3.
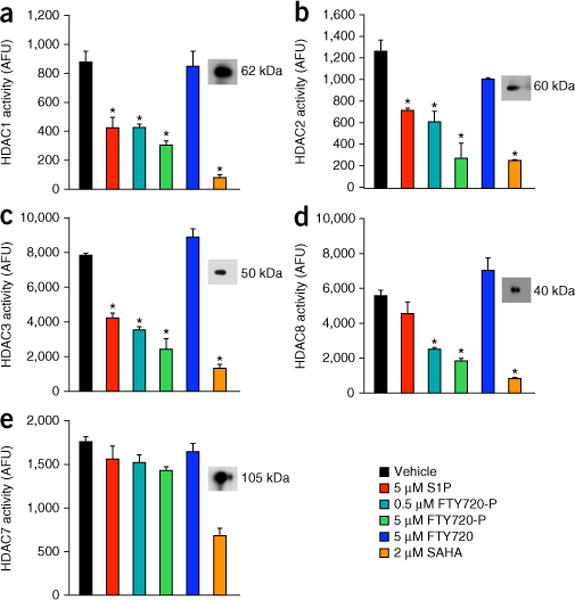
FTY720-P inhibits class I HDACs. (a–d) HDAC activities of recombinant class I HDACs (HDAC1, 2, 3 and 8) and (e) class II HDAC (HDAC7) were measured in the presence of vehicle, S1P (5 μM), FTY720-P (0.5 or 5 μM), FTY720 (5 μM) or SAHA (2 μM). Data are averages of triplicate determinations ±s.d. and expressed as arbitrary fluorescence units (AFU). *P < 0.0001 as compared to vehicle (N = 3 independent samples per group); one-way ANOVA with Bonferroni multiple-comparisons test. Insets, proteins analyzed by western blotting with HDAC-specific antibodies.
FTY720-P binds to class I HDACs
To provide further evidence that FTY720-P targets class I HDACs, we examined whether FTY720-P binds to recombinant HDACs in a similar manner to that of S1P5. FTY720-P and dihydro-S1P, as well as SAHA, completely displaced bound [32P]S1P from HDAC1 to the same extent as an excess of unlabeled S1P, indicating that they share a common or overlapping binding site (Fig. 4a). In agreement with their inability to inhibit HDAC1 (Fig. 3 and ref. 5), neither FTY720 nor sphingosine competed with binding of [32P]S1P to HDAC1, nor did lysophosphatidic acid (LPA), another bioactive lysophospholipid structurally related to S1P (Fig. 4a). Moreover, [32P]FTY720-P also specifically bound to recombinant HDAC1 and could only be displaced by excess FTY720-P, S1P, dihydro-S1P or SAHA (Fig. 4b). Displacement curves indicated that both S1P and FTY720-P bound to HDAC1 with high affinities (Supplementary Fig. 3). FTY720-P bound to HDAC1 with an apparent Kd of 6.2 nM, which is consistent with the half-maximal inhibitory concentration of ~25 nM for inhibition of HDAC1.
Figure 4.
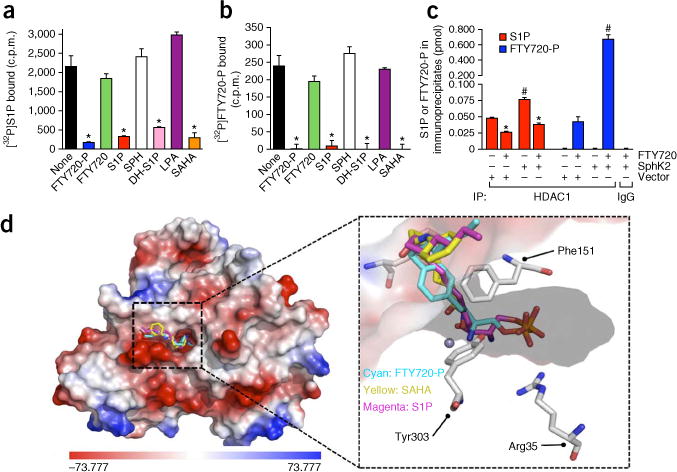
FTY720-P competes with S1P for binding to HDAC1. (a–c) Recombinant His-tagged HDAC1 immobilized to Ni-NTA resin or control Ni-NTA resin (control) was incubated with 0.1 nM [32P]S1P (a) or 0.1 nM [32P]FTY720-P (b) in the absence or presence of unlabeled 1 μM FTY720-P, FTY720, S1P, dihydro-S1P (DH-S1P), sphingosine, LPA or SAHA. Beads were washed extensively and [32P]S1P or [32P]FTY720-P bound to HDAC1 was eluted with 500 mM imidazole and radioactivity detected by scintillation counting. (c) Nuclear extracts from HeLa cells transfected with vector or SphK2 and treated without or with FTY720 (5 μM) for 4 h were immunoprecipitated with anti-HDAC1 antibody or control immunoglobulin G, and bound sphingolipids and FTY720-P determined by LC-ESI-MS/MS. Data are averages of triplicate determinations ±s.d. *P < 0.0001 as compared to none (a,b) or untreated (c) and #P < 0.0001 as compared to vector (unpaired Student’s t-test) (N = 3 independent cell cultures per group). (d) Docking of FTY720-P into the pocket of HDAC2. The low-energy conformations of FTY720-P, S1P and SAHA were calculated when docked in a model based on the crystal structure of HDAC2 (Protein Data Bank: 3MAX). Left, surface contour of the binding site with S1P, SAHA and FTY720-P. The surface contour is colored by electrostatic potential. Right, the binding of S1P (magenta), SAHA (yellow) and FTY720-P (cyan) with HDAC2. The active site residues involved in the interactions with small molecules are shown as sticks and the zinc atom as a sphere.
Next we sought to determine whether FTY720-P formed in the nucleus by SphK2 is bound to endogenous HDAC1. To this end, we treated cells with FTY720, isolated nuclei and measured FTY720-P and sphingolipids present in HDAC1 immunoprecipitates by mass spectrometry. In cells treated with FTY720, a substantial amount of FTY720-P was associated with HDAC1 immunoprecipitates, and this amount was markedly increased by SphK2 overexpression (Fig. 4c). Generation of FTY720-P in the nucleus, which decreased formation of nuclear S1P, also reduced the amount of S1P bound to HDAC1 (Fig. 4c).
Molecular docking of FTY720-P to the active site of HDAC2 on the basis of the crystal structure10 indicated that its binding mode is very similar to that of SAHA and S1P (Fig. 4d). This conserved HDAC active site consists of a tubular pocket with a zinc-binding site at the base, two aspartate-histidine charge-relay systems and a tyro-sine that stabilizes the tetrahedral oxyanion necessary for catalysis11. The hydroxyl and amino groups of FTY720-P and S1P could act similarly to the hydroxamic acid of SAHA, which chelates the zinc atom, and may explain the mechanism of class I HDAC inhibition by FTY720-P and S1P. Molecular modeling also suggests that the highly conserved arginine stabilizes the phosphate group of S1P5 and FTY720-P (Fig. 4d) and explains the low affinity of sphingosine and FTY720. Tyr303, important for catalysis, and His141 are also predicted to interact with S1P and FTY720-P (Supplementary Fig. 4). Another feature of the binding mode between FTY720-P and HDAC2 is that the phenyl ring of FTY720 could engage in π-π stacking with Phe206 and Phe151, which may increase the binding affinity. Lack of these distinctive features and the shallow binding pocket of HDAC7 may explain the lack of inhibitory effects of FTY720-P and S1P on HDAC7 (Fig. 3e). Altogether, these data indicate that FTY720-P can bind to the active site of class I HDACs and inhibit their enzymatic activity.
FTY720-P inhibits hippocampal HDACs, enhances histone acetylations, and facilitates fear extinction in SCID mice
Recent studies suggest that FTY720 also has nonimmunological actions in experimental autoimmune encephalomyelitis and multiple sclerosis1,12. FTY720-P accumulates in the brain and has beneficial effects that are not well understood in the CNS, independent of its immunosuppressive activity1,12. Therefore, we next sought to examine the effects of FTY720 administration on HDAC activity and histone acetylation in vivo. As expected1,13,14, 24 h after oral administration of FTY720 to mice, circulating lymphocytes were significantly decreased, with a depletion of 85% at a dose of 0.5 mg per kilogram body weight, correlating with the increased serum levels of FTY720-P (Supplementary Fig. 5a,b). In accord with reports of brain accumulation of FTY720-P in rats3 and humans15, FTY720-P accumulated in the brains of mice, including nuclei of hippocampal cells, in a dose-dependent manner (Supplementary Fig. 5c). Notably, FTY720 administration inhibited hippocampal HDAC activity (Supplementary Fig. 5d) and also increased histone H3K9 acetylation, even at the lowest dose of FTY720 tested (Supplementary Fig. 5e).
Chromatin remodeling, especially histone tail acetylation, has been implicated in memory formation, and pharmacological and mouse genetic approaches have demonstrated that HDACs influence memory and learning processes8,9. Because we found that FTY720 is phosphorylated in the nucleus by SphK2 and that FTY720-P inhibits HDACs, we investigated whether, like other HDAC inhibitors16–20, it might also affect learning and memory in mice. However, because the immune system has complex effects on learning and memory, and to circumvent the known effects of FTY720-P on immunosuppression and lymphocyte trafficking, we decided to test its effects in severe combined immune deficient (SCID) mice, which are deficient in both T and B cell responses and substantially impaired in acquisition of the ability to perform cognitive tasks21,22.
To examine the effects of FTY720 on learning and memory, SCID mice were administered daily oral FTY720 (1 mg/kg) or saline and exposed to a novel context, followed by electric footshock to elicit acquisition of hippocampus-dependent fear memory that was measured as percentage time freezing, defined as a lack of movement other than respiration. Saline- and FTY720-treated groups exhibited similar levels of freezing preshock, postshock and 48 h after conditioning (two-way repeated-measures ANOVA; interaction: F2,26 = 0.05, P = 0.95; time: F2,26 = 68.91, P < 0.0001; treatment: F1,26 = 0.02, P = 0.90). Hence, FTY720 treatment did not alter the response to footshock, nor did it affect the acquisition of fear memories in these mice, as both groups displayed similar high levels of freezing (Fig. 5a). These findings indicate that FTY720-treated mice did not forget the association between the context and footshock 48 h after conditioning.
Figure 5.
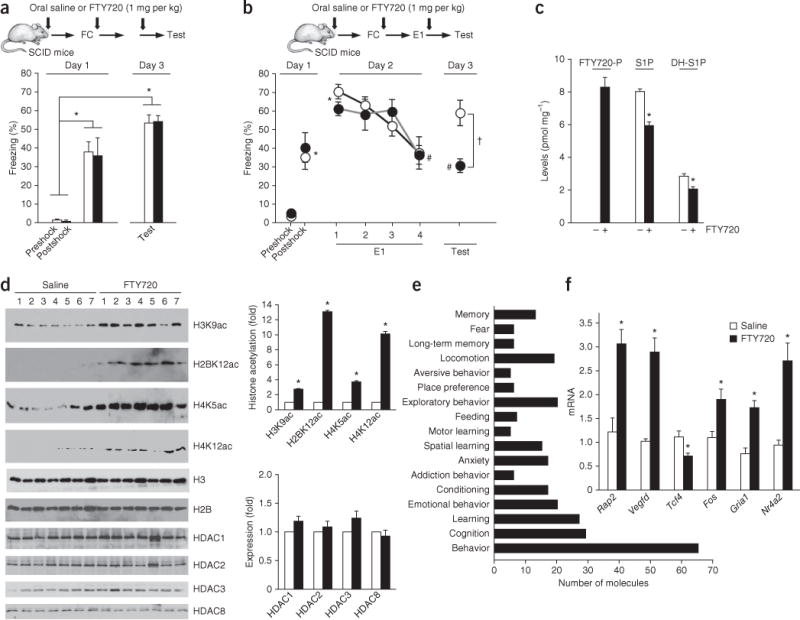
FTY720 administration-facilitated contextual fear extinction in SCID mice correlates with formation of hippocampal FTY720-P, inhibition of HDACs and enhanced histone acetylation and memory-related gene expression. (a) SCID mice were administered oral FTY720 (1 mg/kg) or saline daily by gavage 16 h before fear conditioning. Mice were fear conditioned with three footshocks and preshock and postshock freezing determined. Forty-eight hours later (day 3), freezing was evaluated in a 2.5-min test, with no extinction session. *P < 0.001 as compared to preshock (Bonferroni post hoc test). N = 9 mice per group. (b–f) SCID mice were administered oral FTY720 (1 mg/kg) or saline daily by gavage 16 h before fear conditioning. Mice were fear conditioned with three footshocks and preshock and postshock freezing determined. Twenty-four hours later (day 2), mice received a single, 10-min extinction session (E1; consecutive 2.5-min bins are indicated by 1, 2, 3, 4) and then evaluated on day 3 in a 2.5-min consolidation test. (b) Both groups exhibited significant freezing postshock and in the first 2.5 min of the extinction session as compared to preshock, *P < 0.001 (Bonferroni post hoc test); #P < 0.001 versus the first bin of the E1 extinction session (Bonferroni post hoc test). Only FTY720-treated mice displayed a significant decrease in freezing behavior in the test on day 3. †P = 0.008 between FTY720- and saline-treated mice (Bonferroni post hoc test). N = 8 mice per group. Data are presented as mean ± s.e.m. (c–f) Hippocampi were removed 1 h after the consolidation test on day 3. (c) Hippocampal nuclear FTY720-P, S1P and DH-S1P were measured by LC-ESI-MS/MS. *P < 0.001 as compared to vehicle; one-way ANOVA with Bonferroni multiple-comparisons test. (d) Histone acetylations were examined by immunoblotting with the indicated antibodies. Each lane corresponds to an individual mouse. Quantified data are expressed as mean ± s.e.m. *P < 0.0001 as compared to saline (unpaired t-test). H3 was used as an internal loading control. Full-length blots are presented in Supplementary Figure 10. (e,f) mRNA extracted for determination of gene expression by microarray analysis (e) and quantitative PCR (f). (e) Ingenuity pathway analysis of differentially expressed memory-related genes. (f) Expression of selected genes was analyzed by quantitative PCR and normalized to Gapdh. Data are mean ± s.e.m. *P < 0.05 as compared to saline (unpaired Student’s t-test).
Differential regulation of hippocampal histone acetylation has been shown to be important not only for memory acquisition but also for extinction of fear memories17,18,23. Fear extinction is a specific form of learning and is a mechanism for reducing excess fear of aversive events. Thus, we decided to test whether FTY720 treatment would affect contextual fear extinction. FTY720 (1 mg/kg) or saline was administered daily to the SCID mice, beginning 16 h before fear conditioning, followed by an extinction session and behavioral testing (Fig. 5a,b). This is a clinically relevant dose and regimen that maintains therapeutic levels of FTY720 and FTY720-P in mice and humans1,24.
During extinction training, the mice were reexposed to the conditioned stimulus (the context) without receiving the footshock again (extinction session, E1), which resulted in a decline in the freezing response. FTY720 and saline-treated SCID mice displayed comparable magnitudes of postshock freezing and freezing behavior upon exposure to the conditioning chamber 24 h after shock (two-way repeated-measures ANOVA; interaction: F2,28 = 1.25, P = 0.30; postshock time: F2,28 = 89.04, P < 0.0001; treatment: F1,28 = 0.11, P = 0.75). Moreover, both groups had similar extinction rates during the 10-min acute extinction training session (two-way repeated measures ANOVA; interaction: F3,42 = 1.40, P = 0.26; extinction time: F3,42 = 15.85, P < 0.0001; treatment: F1,42 = 0.08, P = 0.78). Remarkably, however, we observed marked differences on reexposure to the conditioning chamber 1 d later (Fig. 5b; two-way repeated measures ANOVA; interaction: F1,14 = 6.10, P = 0.03; treatment: F1,14 = 12.04, P = 0.004; day: F1,14 = 29.50, P < 0.0001). FTY720-treated mice maintained low levels of freezing behavior during the consolidation test (day 3) (P = 0.008; Bonferroni post hoc test), indicating that extinction was preserved, whereas the saline-treated SCID mice, as expected25, displayed a marked deficit in extinction memory as manifested by elevated freezing (Fig. 5b). These data reveal that FTY720 rescues extinction deficits in SCID mice.
FTY720-P also accumulated in various brain areas of SCID mice, including the hippocampus (Fig. 5c and Supplementary Fig. 6), an area of the brain important for learning and memory, at much higher levels than in serum. Hippocampal S1P and dihydro-S1P were slightly decreased after FTY720 treatment (Fig. 5c). As in C57BL/6 mice (Supplementary Fig. 5d), treatment of SCID mice with FTY720 reduced hippocampal HDAC activity by 50% without discernible differences in expression of HDAC1, 2, 3 or 8 (Fig. 5d). Nevertheless, FTY720 administration also enhanced acetylation of specific histone lysine residues in the hippocampus (Fig. 5d), particularly H3K9, H4K12 and H4K5, associated with regulation of memory processes26,27.
Additional groups of SCID mice were trained in the Morris water maze (MWM) task, a hippocampus-dependent spatial memory test. In agreement with others, we found that SCID mice performed very poorly in the MWM21,22, and treatment with FTY720 did not affect their performance (ref. 28 and Supplementary Fig. 7a–c). Like treatment with other HDAC inhibitors19,27, treatment with FTY720 did not affect exploratory behavior in a novel environment or basal anxiety-like behavior (Supplementary Fig. 7d–f), nor tone-dependent fear conditioning that is hippocampus independent, nor did it affect extinction of response to the tone conditioned stimulus (Supplementary Fig. 7g). Altogether, these data indicate that FTY720 protected the SCID mice from deficits in expression of extinction in the contextual fear model. This aspect of learning is important for the organism to reduce fear-related behavior in response to a stimulus that no longer predicts an aversive event.
Hippocampal memory-related gene expression
We next examined whether the effects of FTY720 on histone acetylation correlated with changes in specific hippocampal programs of gene expression. One hour after the consolidation test, we isolated hippocampal RNA and analyzed the hippocampal transcriptome. This revealed that 475 genes (216 upregulated and 259 downregulated) were differentially expressed in FTY720-treated SCID mice as compared to the control saline-treated group. Bioinformatic analysis indicated that many of the differentially expressed genes were specifically linked to learning-regulated genes (Supplementary Table 1), and 280 have been implicated in processes important for spatial and motor learning, cognition and memory (Fig. 5e). Functional over-representation analysis with both Ingenuity and TOPPGene identified gene networks related to neuroplasticity, associative learning and behavior. Quantitative PCR analysis verified a significant increase in expression of Vegfd (also known as Figf), recently shown to be involved in cognitive function29, and decreased expression of the transcription factor Tcf4 (Fig. 5f), a schizophrenia risk gene30 whose upregulation in mice correlates with reduced cognitive perform-ance31. Expression of several HDAC-regulated memory-associated genes, including Fos (cFos), Gria1 (also known as Glur1) and nuclear receptor subfamily 4, group A, member 2 (Nr4a2)19,32, was also increased in the hippocampus of FTY720-treated mice (Fig. 5f). Our attention was drawn to VEGFD because it is also a cFos-induced growth factor33, and cFos, an immediate-early gene and transcription factor, is regulated by SphK2 and nuclear S1P5. In accord with its effect on histone acetylation (Supplementary Fig. 5e), Vegfd expression was increased 6- to 7-fold 24 h after treatment of C57BL/6 mice with FTY720 (Supplementary Fig. 5f). Expression of brain-derived neurotrophic factor (Bdnf) was also significantly increased (P < 0.01), albeit to a lesser extent (Supplementary Fig. 5g). Owing to its short half-life, Fos mRNA expression was transiently increased much earlier, whereas Vegfd expression remained sustained (Supplementary Fig. 5h,i).
We next sought to determine whether the increased pattern of specific gene expression correlated with increased synaptic plasticity. To this end, we assessed the effect of FTY720 on long-term potentiation (LTP) of the Schaffer collateral input to CA1, which forms excitatory synapses on pyramidal cells in the stratum radiatum, by electrophysiological recordings. LTP was induced using 100 Hz trains patterned as theta bursts, a stimulation protocol known to induce long-lasting, transcription-dependent LTP23,34. As in previous results with HDAC inhibitors19,23,26,35,36, we found that pretreat-ment of hippocampal slices with FTY720, which was phosphorylated to FTY720-P, did not affect baseline stimulus–response curves or paired-pulse facilitation (Fig. 6a,b,d). In contrast, like other HDAC inhibitors19,23,26,35,36, FTY720 treatment significantly facilitated LTP (Fig. 6c). Hence, treatment with FTY720 enhances synaptic plasticity but does not affect basal synaptic transmission.
Figure 6.
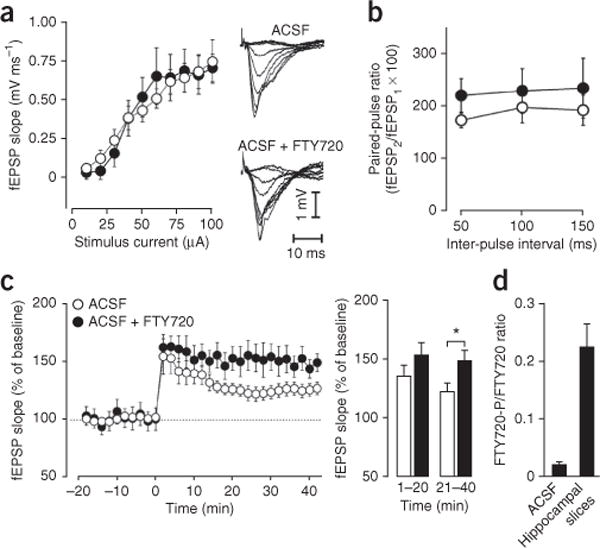
Treatment with FTY720 enhances LTP, but does not alter basal synaptic transmission of Schaffer-collateral synapses. Electrophysiological recordings of CA1 region of hippocampal slices incubated in artificial cerebrospinal fluid (ACSF) without (N = 8 mice) or with (N = 6 mice) 10 μM FTY720. (a) Input-output curves of field excitatory postsynaptic potentials (fEPSPs) evoked by graded stimulus currents were not significantly altered by FTY720 (Mann-Whitney U statistic; Z = 0.227, P = 0.821). Data are mean ± s.e.m. Insets, fEPSP responses for representative slices in ACSF in the absence or presence of FTY720. (b) Paired-pulse facilitation was not significantly altered by FTY720 (mixed-model ANOVA, with treatment as the between-subjects variable and interpulse interval as a repeated measure; F1,9 = 1.279; P = 0.287). Data are presented as mean ± s.e.m. of the facilitation of the second response relative to the first response. (c) FTY720 treatment enhances LTP induced by 100-Hz stimulation applied as theta bursts. Left, mean ± s.e.m. fEPSP slopes before and after stimulation applied at time 0. Right, mean fEPSP slope amplitude was significantly elevated in FTY720-treated slices as compared to ACSF slices at 21–40 min (mixed-model ANOVA, with treatment as the between-subjects variable and time after conditioning as the repeated measure; F1,12 = 5.223; *P = 0.041). (d) At the end of the electrophysiological recording, FTY720 and FTY720-P levels in ACSF and hippocampal slices were measured by LC-ESI-MS/MS. Data are expressed as the ratio of FTY720-P to FTY720 and are mean ± s.d.
Sphk2−/− mice have reduced hippocampal histone acetylation and learning deficits
Because SphK2, not SphK1, is the main isoform in the brain and is highly expressed in hippocampus37,38, we used Sphk2−/− mice to examine the function of SphK2-produced S1P in the regulation of hippocampal HDAC activity and histone acetylation in learning and memory models. Sphk2−/− mice had significantly less S1P and dihydro-S1P in the hippocampus than wild type (WT) (Fig. 7a). In contrast to its effect in the colon39, ablation of SphK2 was not accompanied by compensatory upregulation of SphK1 in the hippocampus (Fig. 7a). Deletion of SphK2 also significantly decreased hippocampal histone acetylation on specific lysine residues (H3K9, H4K5, H4K12) (Fig. 7b) that are linked to memory impairment26,27. There were, however, no discernible differences in brain anatomy, suggesting that absence of SphK2 is not detrimental to brain development.
Figure 7.
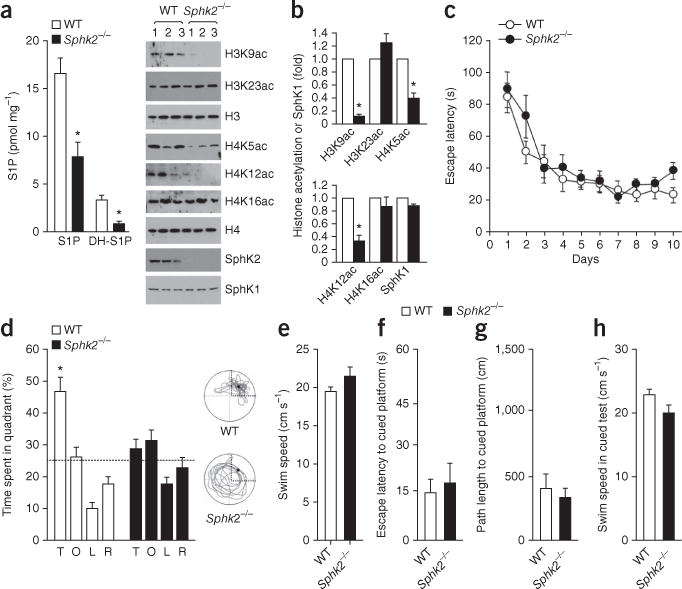
SphK2 knockout mice have reduced hippocampal histone acetylations and learning deficits. (a,b) Hippocampi were removed and nuclei isolated from 3-month-old Sphk2−/− and WT littermates and S1P and DH-S1P levels determined by LC-ESI-MS/MS (a). *P < 0.01 as compared to WT (unpaired t-test). (b) Nuclear SphK2 and histone acetylations and SphK1 in cytoplasm were examined by immunoblotting with the indicated antibodies. Quantified data are expressed as mean ± s.e.m. *P < 0.01 as compared to WT (unpaired t-test). H3 and H4 were used as internal loading controls. (c–h) Mice were tested on the Morris water maze. Data are mean ± s.e.m. (c) Sphk2−/− and WT mice had similar acquisition performance in a fixed-platform water maze task. Escape latencies for WT and Sphk2−/− mice differed significantly from day 1, starting from days 2 and 3, respectively (P < 0.001, Bonferroni post hoc test). (d) In a 120-s probe trial 24 h after day 10 of fixed platform training, percentage time spent in each quadrant was measured: T, target quadrant; L, left quadrant; O, opposite quadrant; R, right quadrant. WT but not Sphk2−/− mice spent significantly more time in the target quadrant (*P < 0.01, Bonferroni post hoc test). Right, representative swim paths of mice during probe test. There were no significant differences in swim speed in the probe trial (e) or in escape latencies (f), path lengths (g) and swim speed (h) when the platform was made visible in the cued test.
Next we evaluted these mice on the MWM. In the fixed-platform test, mice learn the location of a hidden platform using distal visual cues. Both WT and Sphk2−/− mice found the platform faster as training proceeded, with no impairment in Sphk2−/− mice (two-way, repeated-measures ANOVA; interaction: F9,171 = 0.96, P = 0.48; day: F9,171 = 21.58, P < 0.0001; genotype: F1,171 = 1.22, P = 0.28) (Fig. 7c). After 10 d of fixed-platform training, we conducted a probe trial in which we removed the hidden platform and measured the time spent in each quadrant of the water maze. Sphk2−/− mice did not show a spatial preference for the target quadrant, whereas the WT littermates spent significantly more time in the target quadrant (Fig. 7d) (two-way, repeated-measures ANOVA; genotype × quadrant interaction: F3,57 = 5.96; P = 0.0013; Bonferroni post hoc test: P < 0.001 for WT versus knockout mice in the target quadrant), suggesting that Sphk2−/− mice have spatial memory deficits in the MWM task. Both genotypes displayed similar swim speeds (Fig. 7e) and identical performance in the visible-platform test (Fig. 7f–h), indicating that gross sensorimotor and/or motivational deficits are unlikely to account for the poor performance of Sphk2−/− mice during the probe trial.
We then evaluated the mice in a contextual fear conditioning task that included assessment of extinction. There were no significant differences in acquisition of fear memories between Sphk2−/− and WT mice (Fig. 8a and Supplementary Fig. 8a), and magnitudes of postshock freezing and freezing behaviors were comparable upon reexposure to the conditioning chamber 48 h (Supplementary Fig. 8a) or 96 h (Fig. 8a) after shock (two-way, repeated-measures ANOVA; interaction: F2,34 = 2.36, P = 0.11; time: F2,34 = 151, P < 0.0001; genotype: F1,34 = 1.83, P = 0.19). Both genotypes displayed significant increases in freezing behavior (P < 0.001, Bonferroni post hoc) as compared with preshock freezing levels, indicating that memory for the context and footshock even 96 h after conditioning was not disrupted by the gene deletion. Moreover, both genotypes had similar extinction rates during the 10-min extinction training session, E1, when reexposed to the novel context without a shock (Supplementary Fig. 8b). However, after repeated reexposure to the conditioned context on subsequent days (24-h intervals) without receiving the footshock again (extinction trials E2–E4), WT and Sphk2−/− mice displayed significant differences in extinction of contextual fear memory (Fig. 8b) (two-way ANOVA; genotype × day interaction: F3,48 = 1.40, P = 0.25; genotype: F1,48 = 8.06, P = 0.01; day: F3,48 = 19.60, P < 0.0001). While freezing behavior in the WT group declined during further extinction training (P < 0.05 for days 3–5, Bonferroni post hoc test), Sphk2−/− mice showed elevated freezing throughout the extinction sessions (Fig. 8b). Of note, impaired expression of extinction exhibited by Sphk2−/− mice was not rescued by FTY720 administration (two-way, repeated measures ANOVA; treatment × day interaction: F3,54 = 2.51, P = 0.07; treatment: F1,54 = 0.13, P = 0.72; day: F3,54 = 27.66, P < 0.0001). This finding is consistent with the notion that SphK2 is the main isoform in the brain that phosphorylates FTY720 to its active form (ref. 1 and Fig. 8c). The impairment of fear extinction of the Sphk2−/− mice was not due to decreased initial fear responses or locomotor activity, because reaction to shock during the training session (Fig. 8a and Supplementary Fig. 8a), as well as exploratory and basal anxietylike behaviors, were virtually identical between the two genotypes (Supplementary Fig. 9a–d). Moreover, freezing in response to tone-conditioned stimulus also did not differ between the Sphk2−/− and WT mice (Supplementary Fig. 9e).
Figure 8.
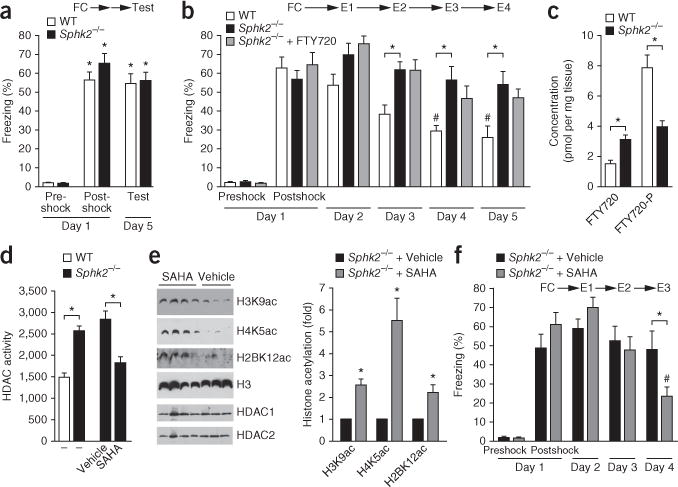
SAHA administration to SphK2 knockout mice reversed the increased HDAC activity, reinstated hippocampal histone acetylations and rescued their extinction deficit in contextual fear conditioning. (a) Deletion of SphK2 has no effect on contextual fear conditioning. Top, experimental design. Sphk2−/− and WT mice were fear conditioned (FC) with three footshocks. Preshock, postshock and 96-h (day 5) freezing were evaluated, with no extinction session. *P < 0.001 as compared to preshock. N = 9 mice per group. (b) Sphk2−/− mice display deficits in contextual fear extinction that are not rescued by FTY720. WT and Sphk2−/− mice were treated daily with saline or FTY720 (1 mg/kg) as indicated. Mice were fear conditioned with three footshocks on day 1 and received daily 10-min extinction sessions in the conditioning chamber without footshocks on days 2–5. Fear extinction consolidation on the indicated days was determined during the first 2.5 min of the extinction session. #P < 0.05 versus E1 (day 2). *P < 0.05 between WT and Sphk2−/− mice (Bonferroni post hoc test). N = 8 WT mice, 10 Sphk2−/− mice. (c) Accumulation of FTY720 and FTY720-P in hippocampus from the WT and Sphk2−/− mice was measured by LC-ESI-MS/MS. *P < 0.05 between WT and Sphk2−/− mice. N = 5 mice. (d–f) SAHA rescues histone acetylation and extinction of fear memories in Sphk2−/− mice. Sphk2−/− mice were injected intraperitoneally with vehicle or SAHA (25 mg/kg) daily starting 10 d before behavior testing26. (f) Mice were then exposed to contextual fear conditioning and tested 24 h later. Extinction of contextual fear was performed on consecutive days, as described above, and fear extinction on the indicated days was determined during the first 2.5 min of the extinction session. #P < 0.05 versus E1 (day 2). *P < 0.05 versus Sphk2−/− mice treated with vehicle (Bonferroni post hoc test). N = 7 vehicle-treated and 9 SAHA-treated mice. Data represent means ± s.e.m. Hippocampi were isolated 1 h after the last consolidation test and HDAC activity in hippocampal nuclear extracts determined with a fluorometric assay (d; *P < 0.001, unpaired Student’s t-test) and histone acetylations examined by immunoblotting with the indicated antibodies (e; *P < 0.05 as compared to vehicle, unpaired t-test). Quantified data are expressed as mean ± s.e.m. H3 was used as an internal loading control.
Because SphK2 knockout mice showed a deficit in extinction of contextual fear memories that correlated with lack of inhibition of HDACs as a result of decreased levels of nuclear S1P, the only known endogenous inhibitor of HDAC5, and decreased histone acetylations, we examined whether treatment of these mice with the potent HDAC inhibitor SAHA would rescue the memory deficit. Indeed, SAHA administered to SphK2 knockout mice reversed the increased HDAC activity (Fig. 8d) and reinstated hippocampal histone acetylations (Fig. 8e). Notably, SAHA treatment facilitated expression of fear extinction in Sphk2−/− mice (Fig. 8f) (two-way repeated measures ANOVA: treatment × day interaction: F2,28 = 6.75, P = 0.004), and SAHA-treated Sphk2−/− mice displayed a significant decrease in freezing on day 4 (P < 0.05; Bonferroni post hoc test) as compared to those treated with vehicle. These data reveal that SAHA can rescue extinction deficits in Sphk2−/− mice.
DISCUSSION
Our study has uncovered a new mechanism of action of the prod-rug FTY720 and revealed that FTY720 enters the nucleus, where it is phosphorylated by SphK2. In turn, FTY720-P that accumulates there binds and inhibits class I histone deacetylases (HDACs) and consequently enhances specific histone acetylations independently of S1PRs. We demonstrated this with recombinant HDACs, in neuronal cell culture, and in vivo. In mice, this enhances acetylation of histone lysine residues linked to epigenetic regulation of learning and memory genes and facilitates fear extinction independently of its established effect on lymphocyte trafficking.
Another noteworthy aspect of the actions of FTY720 is that, in spite of the facilitation of contextual fear extinction in SCID mice, it had no effect on spatial memory performance in the MWM, which depends on both visual and motor functions. This could be a consequence of different requirements and structures underlying these forms of learning. Moreover, mice may use multiple behavioral tactics to escape from the water, and some of these strategies may be comparably efficient but distinct in their requirement for hippocampal function35.
Systemic or intrahippocampal administration of HDACi facilitates fear extinction in mice16–18,23,40, increases synaptic plasticity, enhances long-term memory19,20 and improves memory function in aging mice27 and in mouse models of neurodegenerative disorders9,41. Nevertheless, several HDAC inhibitors also enhance acquisition of conditioned fear memories, and some of these compounds are potentially toxic or brain impermeant and cannot be administered to humans. In contrast, we have demonstrated that FTY720, which readily penetrates the CNS of rodents3 and humans15, is converted to FTY720-P, inhibits HDACs in the hippocampus, enhances LTP in hippocampal neurons and facilitates extinction of aversive memories without enhancing fear memory acquisition. Owing to these distinctive features, FTY720 might be more effective than other HDAC inhibitors as an adjuvant therapy for eliminating aversive memories. Enhancing extinction of fear memory is of great interest for treatment of anxiety disorders, such as post-traumatic stress disorder42. It is possible that development of similar analogs of sphingosine and FTY720 that can be phosphorylated by SphK2 to a mimetic of S1P that retains its nuclear actions but lacks immunomodulatory effects on S1PRs might be useful for extinguishing fear memories.
Our studies suggested that the enhancement of contextual fear extinction by FTY720 did not globally alter gene expression but involved epigenetic regulation of transcription of certain genes that are essential for behavior and long-term synaptic plasticity and memory. Especially intriguing is the upregulation of the growth factors VEGFD and BDNF. VEGFD controls maintenance of dendrite arborization in the adult mouse hippocampus in an autocrine manner and is required for cognitive function and memory formation29. Thus, the large increase in hippocampal expression of VEGFD in mice might contribute to memory enhancement upon FTY720 administration. Like HDACi16, and in agreement with other findings43, FTY720 also enhanced expression of BDNF, a neurotrophin involved in synaptic plasticity processes that are required for long-term memory16,44. Although in cortical neurons FTY720-P mediates increased BDNF by ERK1/2 signaling downstream of S1PR activation43, it is not known whether the increased BDNF expression in a mouse model of Rett syndrome after 4 weeks of FTY720 administration involves S1PRs43 or, as we suggest here, is due to its intracellular actions. Of relevance, in animals that successfully extinguished fear, endogenous BDNF was elevated only in the hippocampus, and infusion of BDNF into hippocampus reduced fear even in the absence of extinction training but did not disrupt performance or the fear memory itself44. These results might be related to the impairment of extinction in both mice and humans by a BDNF polymorphism45.
Expression of the orphan nuclear receptor Nr4a2, a HDAC- and CREB-dependent gene that has been implicated in long-term memory19, was also increased following the memory-enhancing effect of FTY720. In this regard, long-term memory enhancement by hippocampus-specific HDAC3 deletion or inhibition is abolished by intrahippocampal delivery of Nr4a2 short interfering RNA32, suggesting that negative regulation of memory formation by HDAC3 requires Nr4a2. Moreover, blocking hippocampal Nr4a2 transcriptional activity impairs long-term memory but does not affect short-term memory, and it prevents memory enhancement by HDACi46. Thus, Nr4a target genes may contribute to memory enhancement by FTY720. Notably, a recent study reported that a selective inhibitor of class I HDACs epigenetically primes the expression of neuroplasticity-related genes (for example, Fos) to overcome the resilience of remote fear memories to successful extinction23.
Another related observation in our study was that Sphk2−/− mice, which had decreased levels of S1P in the hippocampus, displayed reduced histone acetylation and had impaired spatial memory and contextual fear extinction. The lack of inhibition of HDACs associated with decreased levels of nuclear S1P in Sphk2−/− mice could be overcome by treatment with a potent inhibitor of HDACs, which also reinstated hippocampal histone acetylation and the contextual fear extinction deficits. However, a caveat of these studies is that they do not conclusively demonstrate that these deficits are due to the loss of SphK2. Although Sphk2−/− mice showed impaired fear extinction, memory acquisition was not altered. Extinction is an active mnemonic process that has some similarity with other steps of memory formation, yet increasing evidence now suggests that distinct pathways are involved in acquisition and extinction of fear memories41,47–49. Our data suggest that the SphK2-S1P-HDAC axis is important in epigenetic regulation of expression of genes mediating extinction of aversive memories and that targeting specific hippocampal HDACs with compounds such as FTY720 deserves consideration as an adjuvant therapy for post-traumatic stress disorder and other anxiety disorders.
ONLINE METHODS
Cell culture and transfection
Hippocampal neurons were cultured from embryonic day 18 C57BL/6 mouse embryos as described50. Briefly, the hippocampus was dissected free from the rest of the brain, minced, and incubated for 30 min at 37 °C with trypsin and DNase in Neurobasal medium (Invitrogen) supplemented with B-27 additives (Invitrogen), l-glutamine (0.5 mM), glutamate (25 μM) and an antibiotic mixture. Tissue was triturated, resuspended in medium, filtered twice through 70-μm-pore nylon mesh, then plated in Neurobasal medium. The cultures were almost exclusively neurons as assessed by neuronal nuclear (NeuN) or microtubule-associated protein 2 (MAP2) immunostaining; glial contamination at time of use in experiments was less than 2%. For western blot analyses, 2 × 106 cells were plated per well in six-well plates coated with poly-l-lysine. Human neuroblastoma SH-SY5Y and HeLa cells (ATCC) were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% (vol/vol) serum. Cells were transfected with vector, SphK2 or catalytically inactive SphK2G212E constructs or with ON-TARGETplus SMARTpool siRNA against SphK2 (5′-CAAGGCAGCUCUACACUCA-3′; 5′-GAGACGGGCUGCUCCAUGA-3′; 5′-GCUCCUCCAUGGCGAGUUU-3′; 5′-CCACUGCCCUCACCUGUCU-3′) and control siRNA from Dharmacon as previously described5.
Nuclea extracts
Cells were washed with cold PBS and resuspended in buffer containing 10 mM HEPES (pH 7.8), 10 mM KCl, 0.1 mM EDTA, 1 mM Na3VO4, 1 mM DTT, 1:500 protease inhibitors (Sigma), and incubated on ice for 15 min. NP-40 was added to 0.75% (vol/vol) and cells were vortexed for 10 s. Nuclei and supernatant (“cytoplasm”) were separated by centrifugation at 1,000g for 3 min at 4 °C. Nuclei were resuspended in buffer containing 20 mM HEPES (pH 7.8), 0.4 M NaCl, 1 mM EDTA, 1 mM Na3VO4, 1 mM DTT and 1:500 protease inhibitors and incubated on ice for 15 min. Nuclear extracts were cleared by centrifugation at 14,000g for 5 min at 4 °C.
HDAC activity measurements
HDAC activity of purified recombinant His6-tagged HDAC1–HDAC3, HDAC8 and HDAC7 purified from Sf9 cells was determined with a fluorometric HDAC activity assay as described5. Reaction mixtures containing Boc-Lys(Ac)-AMC as substrate were incubated at 37 °C for 30 min, lysine developer was added and the mixture was then incubated for 30 min at 37 °C. Fluorescence was measured with excitation at 360 nm and emission at 460 nm. No-enzyme controls and inhibitor controls were included.
S1P and FTY720-P binding assays
Recombinant His6-tagged HDAC1 was incubated with [32P]S1P or [32P]FTY720-P (0.1 nM, 6.8 μCi/pmol) in buffer containing 50 mM Tris (pH 7.5), 137 mM NaCl, 1 mM MgCl2, 2.7 mM KCl, 15 mM NaF and 0.5 mM NaV3O4 for 25 min at 30 °C. His6-tagged protein was then immobilized on Ni-NTA resin and washed three times with the same buffer to remove unbound proteins, and bound proteins were eluted with 500 mM imidazole. S1P or FTY720-P associated with the eluted proteins was quantified with a LS6500 scintillation counter (Beckman). Where indicated, unlabeled S1P, DH-S1P, sphingosine, FTY720-P, FTY720, LPA or SAHA (Enzo) were added 10 min before addition of the labeled compounds.
HAT activity
HAT activity in nuclear extracts was determined using a colorimetric assay kit (Abcam) in which free CoA produced serves as a coenzyme for NADH production that is detected spectrophotometrically (440 nm) upon reacting with a soluble tetrazolium dye, as described previously5.
Mass spectrometry measurements
Lipids were extracted. Phosphorylated and unphosphorylated sphingoid bases, FTY720 and FTY720-P were quantified by liquid chromatography–electrospray ionization–tandem mass spectrometry (LC-ESI-MS/MS, 4000 QTRAP, AB Sciex) as described39.
Immunoblotting
Equal amounts of protein were separated by SDS-PAGE, trans-blotted to nitrocellulose and incubated with primary antibodies. The antibodies used were as follows: rabbit polyclonal antibodies to histone H4 (07-108), H2B (07-371), H3K23ac (07-355), H3K18ac (07-354) and H4K16ac (07-329) (Millipore, 1:1,000 dilution); histone H3 (ab24834), H3K9ac (ab10812), H4K5ac (ab51997) and H2BK12ac (ab61228) (Abcam, 1:1,000 dilution); H4K12ac (2591), lamin a/c (2032), tubulin (2145), p-ERK1/2 (4372), HDAC3 (3949) and HDAC7 (2882) (Cell Signaling, 1:1,000 dilution); HDAC1 (sc-7872), HDAC2 (sc-7899) and HDAC8 (sc-11405) (Santa Cruz Biotechnology, 1:1,000 dilution); V5 (R960-25, Invitrogen, 1:5,000 dilution). Immunopositive bands were visualized by enhanced chemiluminescence using secondary antibodies conjugated with horseradish peroxidase (goat anti-rabbit (111-035-045, 1:5,000) and goat anti-mouse (115-035-166, 1:10,000), Jackson ImmunoResearch Laboratories) and Super-Signal West Pico chemiluminescent substrate (Pierce). Blots were not stripped and reprobed. Optical densities of bands associated with proteins of interest were quantified using AlphaEaseFC software (Alpha Innotech) and normalized to the optical densities of their respective H3 bands.
Mice
Male SCID mice (CB17-Prkdcscid/J) were purchased from the Jackson Laboratory. C57BL/6 wild-type and Sphk2−/− mice were from R. Proia (NIH). Three-month-old male mice with littermate controls to assure the same genetic background were used for all experiments. Animal procedures were approved by the Institutional Animal Care and Use Committee at Virginia Commonwealth University.
FTY720 administration
Mice were treated daily by oral administration of 1 mg/kg FTY720 in saline, unless indicated otherwise, by gavage. FTY720 was administered 16 h before fear conditioning and behavioral assessments.
SAHA administration
Suberoylanilide hydroxamic acid (SAHA, vorinostat) was dissolved in DMSO at a concentration of 50 mg/ml and then diluted to 5 mg/ml in saline just before injection. Mice received intraperitoneal injections daily with SAHA (25 mg/kg) or vehicle starting 10 d before memory tests and were alternated daily between left and right sides of the abdomen, always 16 h before testing as described26.
Contextual fear extinction test
To measure associative learning, contextual fear conditioning was used as described previously with minor modifications51. The training consisted of a single exposure to the novel experimental chamber (47.5 × 41 × 22 cm) for 2.5 min followed by three electric foot shocks (0.70 mA; 30 s ITI (intertrial interval)). Baseline freezing behavior was measured in the 2.5 min before the shock was administered and postshock freezing evaluated for 30 s after the third shock. Mice were then returned to their home cages. Context-dependent freezing, a conditioned fear–related response, was assessed 24 h later in the first 2.5-min bin. Mice were assessed for extinction by giving them a 10-min exposure to the conditioned context without footshock, which results in a decline of the time spent freezing. On subsequent days, mice were evaluated in a 2.5-min consolidation test to determine whether freezing behavior was still extinguished. ANY-maze video tracking system and software (Stoelting) was used to track the mice and analyze immobility.
Tone-paired conditioned fear test and extinction
Mice were assessed in tone-paired conditioned fear as previously described52. Mice were placed in an olfactory-paired, transparent, Plexiglas experimental chamber (47.5 × 41 × 22 cm) with the shock floor in place. After a 3-min acclimation period, a 20-s tone (80 dB) was presented that coterminated with a scrambled 2-s (0.7 mA, alternating current) electric foot shock. SCID mice received five tone-shock pairings. Mice were returned to their home cage 1 min later. On successive days, mice underwent extinction training in a different experimental chamber that was paired with a new olfactory cue and lacked shock grids. During extinction sessions, mice were placed in the novel chamber for a 180-s acclimation period, presented with the tone for 200 s, and removed 60 s later from the apparatus and returned to their respective home cages. In the conditioning session, percentage of time spent freezing was assessed 180 s before tone-shock pairings (pre-shock) and 60 s after tone-shock pairings (postshock). In each extinction session, the percentage of time spent freezing during the 200-s tone was determined.
Exploratory behavior and basal anxiety tests
Mice were placed in a plastic arena (47.5 × 41 × 22 cm). The exploratory behavior of the animals, distance traveled during the first 3 min of the test and thigmotaxia time, defined as time spent less than 5 cm away from the wall of the apparatus, were determined using ANY-maze video tracking and software. Light/dark testing used a small (36 × 10 × 34 cm) enclosed, dark box with a passageway (6 × 6 cm) leading to a larger (36 × 21 × 34 cm), light box. Before testing, mice were acclimated in the testing room for 1 h. Mice were then placed in the light side of the box and allowed to freely explore the apparatus for 5 min. Time spent in the light and dark sides was measured by ANY-maze software. The marble-burying test was carried out in a polycarbonate cage (33 × 21 × 19 cm) filled to a depth of 5 cm with pine wood bedding. Before testing, 20 clear, glass marbles (10 mm diameter) were arranged in an evenly spaced, grid-like fashion across the surface of the bedding and the cages were placed in a lit, sound-attenuated chamber. Mice were placed in the cage, which was then covered with a transparent, Plexiglas lid with air holes, and assessed for 20 min. The number of marbles buried (defined as 50% or more of the marbles covered by bedding) was counted by a trained observer.
Morris water maze test
The water maze consisted of a circular steel pool (1.8 m diameter, 0.6 m height) filled with opaque water (17–22 °C). A white platform (10 cm diameter) was submerged 1 cm below the water’s surface. Black geometric shapes on the walls surrounding the maze served as visual cues. Videomax-one (Columbus Instruments) was used to track the swim paths of each subject. Fixed-platform training was conducted as previously described53. Before platform training, the mice received a single, 5-min acclimation session in which the platform was not present in the water maze. The mice were then given a daily acquisition session for 5 d (SCID) or 10 d (WT and Sphk2−/−) to locate the submerged platform that remained in a fixed location. Testing sessions consisted of four 120-s trials per day, with an inter-trial interval of approximately 10 min. Four different points along the perimeter of the maze served as starting points for each trial. Once a mouse located the platform, it was allowed to remain there for 30 s. If a mouse failed to locate the platform within 120 s, it was manually guided to the platform and removed 30 s later. For each trial, escape latency (time (s) to find the hidden platform), path length (cm) to the platform location and swim speed (path length/escape latency) were determined. The mean escape latency, path length and swim speed of the four daily trials were analyzed.
Memory retention for the platform location was assessed 24 h after the final day of fixed platform training during a 120-s probe trial, in which the platform was removed from the water maze. Escape latency, path length and swim speed to the former platform location were determined. The percentage of time spent in the target quadrant (where the platform had been located), as well as each of the other three quadrants, was assessed.
Mice were then tested in the cued platform version of the water maze task to evaluate whether noncognitive factors, including sensorimotor or motivational deficits, contributed to the impaired water maze performance. In the cued task, the location of the platform was made visible by placing a black rubber stopper, which extended approximately 2 cm above the surface of the water, on top of the submerged platform53. Mice were trained in the cued task for 3 d (2 trials per day). The mice were then tested 24 h later and the mean escape latencies, path lengths and swim speeds of the two trials were analyzed.
Isolation of hippocampus and nuclear fractions
Brain regions of interest were dissected from fresh brains immediately after rapid decapitation as previously described54. The hippocampus was dissected from the surface of the brain after removing the cortex. Hippocampi were homogenized in buffer containing 10 mM HEPES pH 7.8, 10 mM KCl, 0.1 mM EDTA, 1 mM Na3VO4, 1 mM DTT and protease inhibitor cocktail (Sigma) and incubated on ice for 15 min. NP-40 was added to a final concentration of 0.75% (vol/vol), and the tissue suspension was vortexed for 10 s and then incubated on ice for 2 min. Nuclear and cytoplasmic fractions were separated by centrifugation at 1,000g for 3 min at 4 °C. Nuclei were resuspended in high salt buffer containing 20 mM HEPES pH 7.8, 0.4 M NaCl, 1 mM EDTA, 1 mM Na3VO4, 1 mM DTT and protease inhibitors, and nuclear proteins were extracted as described above.
Electrophysiological analysis
Mice were anesthetized with 4% isoflurane for 4 min and the brain rapidly removed. Horizontal 400-μm slices were cut into artificial cerebrospinal fluid (ACSF; 2–4 °C) containing (in mM) NaCl 124, KCl 3, MgSO4 1, NaHCO3 25, NaH2PO4 1.25, CaCl2 2, glucose 10 (pH 7.4), saturated with 95% O2/5% CO2. Slices were held in oxygenated dishes containing ACSF in the absence or presence of 10 μM FTY720 for 2 h before electrophysiological recording. During this equilibration period and subsequent recording, bathing solutions were held at 32 °C. For recording, a slice was transferred to a submersion-type recording chamber perfused at a rate of 2 ml/min with the solution used for equilibration. A bipolar stimulating electrode and a micropipette recording electrode (filled with ACSF, resistance 4–6 MΩ) were positioned in CA1 stratum radiatum, separated by approximately 0.5 mm. Constant current pulses (0.2 ms duration) were used to evoke field excitatory postsynaptic potentials (fEPSPs). Signals were amplified (bandpass 0.1–5,000 Hz), digitized at 20 KHz and analyzed offline using pClamp v9.2 software (Molecular Devices, LLC, Sunnyvale, CA, USA). Paired-pulse facilitation (PPF) was assessed using paired stimulus presentations (interpulse intervals of 50, 100 and 150 ms), at current intensities subthreshold for target cell discharge. For long-term potentiation (LTP) experiments, a stimulating intensity that evoked fEPSPs of 50% maximum, as based on input-output testing, was delivered at a rate of one pulse every 30 s and used to obtain a 30-min period of baseline recording. LTP was then induced using three trains of theta-burst high frequency stimulation (HFS), consisting of 10 bursts of four pulses at 100 Hz, with 200 ms separating the onset of each burst. Each train was separated by 20 s. Following HFS, fEPSPs were acquired for 60 min using stimulus parameters identical to those of the baseline recording. For LTP baseline and post-HFS data, mean fEPSP slopes were aggregated into 2-min epochs for graphical and statistical analyses.
Quantitative real-time PCR
Hippocampi were dissected, total RNA was isolated with TRIzol (Invitrogen) and reverse transcribed with the High Capacity cDNA Reverse Transcription Kit and premixed primer probe sets from Applied Biosystems, and cDNA was amplified with the ABI 7900HT as previously described5.
Microarray analysis
Total RNA was isolated from individual hippocampi using Stat-60 (Tel-Test) reagent and a Tekmar homogenizer. RNA quality and quantity was assessed by 260 nm/280 nm absorbance ratios and RNA quality indicator (RQI) values calculated by an Experion analyzer (Bio-Rad). All samples had RQI > 9.0. Total RNA (100 ng) was used as the template for synthesis and amplification of bioti-nylated aRNA using the GeneChip 3′ IVT Express Kit (Affymetrix). Labeled aRNA was fragmented, hybridized to a total of eight GeneChip Mouse Genome 430A 2.0 microarrays (Affymetrix), stained and scanned as described previously55.
Before statistical analysis, microarray quality was evaluated using a standard battery of quality control metrics55. All arrays had greater than 60% probesets called as present using Affymetrix Expression Console Software MAS 5.0 expression calls. The effect of FTY720 on hippocampal transcript abundance was measured using the S-score algorithm as described56. The S-score uses a probe-level analysis to determine statistical significance of probe-set differences between individual Affymetrix microarrays, with results output as a standard normal distribution having a mean of 0 (no change) and s.d. of 1. A positive S-score indicated upregulation with FTY720 treatment and a negative S-score indicated downregulation. Biological reproducibility of gene expression differences identified by S-scores was determined by one-class statistical analysis of microarrays (SAM), a rank based permutation method using a 5% false discovery rate (FDR) threshold. Transcripts with average \S\ < 1.5 were filtered, and only genes passing this statistical filtering scheme were used in subsequent bioinformatics analyses.
Functional biological enrichment analysis of FTY720 responsive genes was determined by using ToppGene Suite57 and also by Ingenuity Pathway Analysis (Ingenuity Systems). Genes were analyzed for over-representation in annotation categories including Gene Ontology terms using an FDR of 5% to account for multiple testing. GeneWeaver, a web-based repository that allows for integration of distinct empirically derived gene lists58, was used to investigate the intersection of our results with independent gene lists obtained from relevant published experiments.
Statistical analysis
Statistical analysis was performed using unpaired two-tailed Student’s t-test for comparison of two groups and analysis of variance (ANOVA) for analyzing experiments consisting of three or more groups (GraphPad Prism). In all behavioral assays, subjects were randomly assigned to a group and the experiments were blind with respect to group assignments. Data distribution was assumed to be normal but was not formally tested. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in previous publications51–53. Significant ANOVA results were followed by post hoc tests for multiple comparisons. To analyze the effect on gene expression, P-values were calculated using an unpaired homoscedastic t-test, where n is taken to be the number of independent experiments (at least three in all cases). In all cases, homoscedasticity was first confirmed using an F-test. P < 0.05 was considered significant. Electrophysiological results were evaluated using mixed-model ANOVAs, with treatment as a between-subjects variable and time after HFS or interpulse interval as repeated measures.
A Supplementary Methods Checklist is available.
Supplementary Material
Acknowledgments
This work was supported by US National Institutes of Health (NIH) grant R37GM043880 to S.S. Behavioral studies were supported by 5P01DA009789 to A.H.L. and R21AG042745 to L.E.W. LTP studies were supported by R01NS057758 to T.M.R. The Lipidomics core was supported in part by NIH grant P30CA16059 to the Massey Cancer Center. Modeling studies were supported by National Natural Science Foundation of China grant 91029704 to C.L. We thank R. Proia (US National Institutes of Health) for providing the Sphk2−/− mice, B.L. Mason for technical assistance and S. Lima for discussions.
Footnotes
Accession codes. GEO: microarray data, GSE57015.
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
N.C.H. designed, performed research and analyzed; L.E.W. performed behavior studies and analyzed data; S.M. and S.S. developed the concept, created figures and wrote the manuscript. J.C.A., D.A. and M.O. performed research; J.L. and C.L. performed molecular modeling; A.H.L. and M.F.M. contributed to data interpretation; P.E.K. provided reagents; T.M.R. carried out LTP studies.
COMPETING FINANCIAl INTERESTS
The authors declare no competing financial interests.
References
- 1.Brinkmann V, et al. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov. 2010;9:883–897. doi: 10.1038/nrd3248. [DOI] [PubMed] [Google Scholar]
- 2.Cyster JG, Schwab SR. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol. 2012;30:69–94. doi: 10.1146/annurev-immunol-020711-075011. [DOI] [PubMed] [Google Scholar]
- 3.Foster CA, et al. Brain penetration of the oral immunomodulatory drug FTY720 and its phosphorylation in the central nervous system during experimental autoimmune encephalomyelitis: consequences for mode of action in multiple sclerosis. J Pharmacol Exp Ther. 2007;323:469–475. doi: 10.1124/jpet.107.127183. [DOI] [PubMed] [Google Scholar]
- 4.Hla T, Brinkmann V. Sphingosine 1-phosphate (S1P): physiology and the effects of S1P receptor modulation. Neurology. 2011;76:S3–S8. doi: 10.1212/WNL.0b013e31820d5ec1. [DOI] [PubMed] [Google Scholar]
- 5.Hait NC, et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science. 2009;325:1254–1257. doi: 10.1126/science.1176709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alvarez SE, et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature. 2010;465:1084–1088. doi: 10.1038/nature09128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Igarashi N, et al. Sphingosine kinase 2 is a nuclear protein and inhibits DNA synthesis. J Biol Chem. 2003;278:46832–46839. doi: 10.1074/jbc.M306577200. [DOI] [PubMed] [Google Scholar]
- 8.Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov. 2008;7:854–868. doi: 10.1038/nrd2681. [DOI] [PubMed] [Google Scholar]
- 9.Fischer A, Sananbenesi F, Mungenast A, Tsai LH. Targeting the correct HDAC(s) to treat cognitive disorders. Trends Pharmacol Sci. 2010;31:605–617. doi: 10.1016/j.tips.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 10.Bressi JC, et al. Exploration of the HDAC2 foot pocket: Synthesis and SAR of substituted N-(2-aminophenyl)benzamides. Bioorg Med Chem Lett. 2010;20:3142–3145. doi: 10.1016/j.bmcl.2010.03.091. [DOI] [PubMed] [Google Scholar]
- 11.Finnin MS, et al. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401:188–193. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 12.Choi JW, et al. FTY720 (fngolimod) effcacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc Natl Acad Sci USA. 2011;108:751–756. doi: 10.1073/pnas.1014154108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mandala S, et al. Alteration of lymphocyte traffcking by sphingosine-1-phosphate receptor agonists. Science. 2002;296:346–349. doi: 10.1126/science.1070238. [DOI] [PubMed] [Google Scholar]
- 14.Allende ML, et al. Mice defcient in sphingosine kinase 1 are rendered lymphopenic by FTY720. J Biol Chem. 2004;279:52487–52492. doi: 10.1074/jbc.M406512200. [DOI] [PubMed] [Google Scholar]
- 15.Briard E, et al. BZM055, an iodinated radiotracer candidate for PET and SPECT imaging of myelin and FTY720 brain distribution. ChemMedChem. 2011;6:667–677. doi: 10.1002/cmdc.201000477. [DOI] [PubMed] [Google Scholar]
- 16.Bredy TW, et al. Histone modifcations around individual BDNF gene promoters in prefrontal cortex are associated with extinction of conditioned fear. Learn Mem. 2007;14:268–276. doi: 10.1101/lm.500907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lattal KM, Barrett RM, Wood MA. Systemic or intrahippocampal delivery of histone deacetylase inhibitors facilitates fear extinction. Behav Neurosci. 2007;121:1125–1131. doi: 10.1037/0735-7044.121.5.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bredy TW, Barad M. The histone deacetylase inhibitor valproic acid enhances acquisition, extinction, and reconsolidation of conditioned fear. Learn Mem. 2008;15:39–45. doi: 10.1101/lm.801108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vecsey CG, et al. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J Neurosci. 2007;27:6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stefanko DP, Barrett RM, Ly AR, Reolon GK, Wood MA. Modulation of long-term memory for object recognition via HDAC inhibition. Proc Natl Acad Sci USA. 2009;106:9447–9452. doi: 10.1073/pnas.0903964106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kipnis J, Cohen H, Cardon M, Ziv Y, Schwartz M. T cell defciency leads to cognitive dysfunction: implications for therapeutic vaccination for schizophrenia and other psychiatric conditions. Proc Natl Acad Sci USA. 2004;101:8180–8185. doi: 10.1073/pnas.0402268101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brynskikh A, Warren T, Zhu J, Kipnis J. Adaptive immunity affects learning behavior in mice. Brain Behav Immun. 2008;22:861–869. doi: 10.1016/j.bbi.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 23.Gräff J, et al. Epigenetic priming of memory updating during reconsolidation to attenuate remote fear memories. Cell. 2014;156:261–276. doi: 10.1016/j.cell.2013.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kappos L, et al. A placebo-controlled trial of oral fngolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362:387–401. doi: 10.1056/NEJMoa0909494. [DOI] [PubMed] [Google Scholar]
- 25.Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun. 2011;25:181–213. doi: 10.1016/j.bbi.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 26.Guan JS, et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peleg S, et al. Altered histone acetylation is associated with age-dependent memory impairment in mice. Science. 2010;328:753–756. doi: 10.1126/science.1186088. [DOI] [PubMed] [Google Scholar]
- 28.Derecki NC, et al. Regulation of learning and memory by meningeal immunity: a key role for IL-4. J Exp Med. 2010;207:1067–1080. doi: 10.1084/jem.20091419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mauceri D, Freitag HE, Oliveira AM, Bengtson CP, Bading H. Nuclear calcium-VEGFD signaling controls maintenance of dendrite arborization necessary for memory formation. Neuron. 2011;71:117–130. doi: 10.1016/j.neuron.2011.04.022. [DOI] [PubMed] [Google Scholar]
- 30.Stefansson H, et al. Common variants conferring risk of schizophrenia. Nature. 2009;460:744–747. doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brzózka MM, Radyushkin K, Wichert SP, Ehrenreich H, Rossner MJ. Cognitive and sensorimotor gating impairments in transgenic mice overexpressing the schizophrenia susceptibility gene Tcf4 in the brain. Biol Psychiatry. 2010;68:33–40. doi: 10.1016/j.biopsych.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 32.McQuown SC, et al. HDAC3 is a critical negative regulator of long-term memory formation. J Neurosci. 2011;31:764–774. doi: 10.1523/JNEUROSCI.5052-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orlandini M, Marconcini L, Ferruzzi R, Oliviero S. Identifcation of a c-fos-induced gene that is related to the platelet-derived growth factor/vascular endothelial growth factor family. Proc Natl Acad Sci USA. 1996;93:11675–11680. doi: 10.1073/pnas.93.21.11675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patterson SL, et al. Some forms of cAMP-mediated long-lasting potentiation are associated with release of BDNF and nuclear translocation of phospho-MAP kinase. Neuron. 2001;32:123–140. doi: 10.1016/s0896-6273(01)00443-3. [DOI] [PubMed] [Google Scholar]
- 35.Alarcón JM, et al. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42:947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 36.Levenson JM, et al. Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem. 2004;279:40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- 37.Liu H, et al. Molecular cloning and functional characterization of a novel mammalian sphingosine kinase type 2 isoform. J Biol Chem. 2000;275:19513–19520. doi: 10.1074/jbc.M002759200. [DOI] [PubMed] [Google Scholar]
- 38.Blondeau N, et al. Distribution of sphingosine kinase activity and mRNA in rodent brain. J Neurochem. 2007;103:509–517. doi: 10.1111/j.1471-4159.2007.04755.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liang J, et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal infammation, and development of colitis-associated cancer. Cancer Cell. 2013;23:107–120. doi: 10.1016/j.ccr.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fujita Y, et al. Vorinostat, a histone deacetylase inhibitor, facilitates fear extinction and enhances expression of the hippocampal NR2B-containing NMDA receptor gene. J Psychiatr Res. 2012;46:635–643. doi: 10.1016/j.jpsychires.2012.01.026. [DOI] [PubMed] [Google Scholar]
- 41.Day JJ, Sweatt JD. Epigenetic mechanisms in cognition. Neuron. 2011;70:813–829. doi: 10.1016/j.neuron.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zovkic IB, Sweatt JD. Epigenetic mechanisms in learned fear: implications for PTSD. Neuropsychopharmacology. 2013;38:77–93. doi: 10.1038/npp.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deogracias R, et al. Fingolimod, a sphingosine-1 phosphate receptor modulator, increases BDNF levels and improves symptoms of a mouse model of Rett syndrome. Proc Natl Acad Sci USA. 2012;109:14230–14235. doi: 10.1073/pnas.1206093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peters J, Dieppa-Perea LM, Melendez LM, Quirk GJ. Induction of fear extinction with hippocampal-infralimbic BDNF. Science. 2010;328:1288–1290. doi: 10.1126/science.1186909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soliman F, et al. A genetic variant BDNF polymorphism alters extinction learning in both mouse and human. Science. 2010;327:863–866. doi: 10.1126/science.1181886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hawk JD, et al. NR4A nuclear receptors support memory enhancement by histone deacetylase inhibitors. J Clin Invest. 2012;122:3593–3602. doi: 10.1172/JCI64145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee YS, Silva AJ. The molecular and cellular biology of enhanced cognition. Nat Rev Neurosci. 2009;10:126–140. doi: 10.1038/nrn2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maren S. Seeking a spotless mind: extinction, deconsolidation, and erasure of fear memory. Neuron. 2011;70:830–845. doi: 10.1016/j.neuron.2011.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tronson NC, Corcoran KA, Jovasevic V, Radulovic J. Fear conditioning and extinction: emotional states encoded by distinct signaling pathways. Trends Neurosci. 2012;35:145–155. doi: 10.1016/j.tins.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beaudoin GM, III, et al. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat Protoc. 2012;7:1741–1754. doi: 10.1038/nprot.2012.099. [DOI] [PubMed] [Google Scholar]
- 51.Fitting S, et al. Synaptic dysfunction in the hippocampus accompanies learning and memory defcits in human immunodefciency virus type-1 Tat transgenic mice. Biol Psychiatry. 2013;73:443–453. doi: 10.1016/j.biopsych.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Plendl W, Wotjak CT. Dissociation of within- and between-session extinction of conditioned fear. J Neurosci. 2010;30:4990–4998. doi: 10.1523/JNEUROSCI.6038-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Varvel SA, Hamm RJ, Martin BR, Lichtman AH. Differential effects of delta 9-THC on spatial reference and working memory in mice. Psychopharmacology (Berl) 2001;157:142–150. doi: 10.1007/s002130100780. [DOI] [PubMed] [Google Scholar]
- 54.Lazenka MF, Selley DE, Sim-Selley LJ. DeltaFosB induction correlates inversely with CB receptor desensitization in a brain region-dependent manner following repeated delta-THC administration. Neuropharmacology. 2014;77:224–233. doi: 10.1016/j.neuropharm.2013.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kerns RT, et al. Ethanol-responsive brain region expression networks: implications for behavioral responses to acute ethanol in DBA/2J versus C57BL/6J mice. J Neurosci. 2005;25:2255–2266. doi: 10.1523/JNEUROSCI.4372-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang L, Wang L, Ravindranathan A, Miles MF. A new algorithm for analysis of oligonucleotide arrays: application to expression profling in mouse brain regions. J Mol Biol. 2002;317:225–235. doi: 10.1006/jmbi.2001.5350. [DOI] [PubMed] [Google Scholar]
- 57.Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009;37:W305–W311. doi: 10.1093/nar/gkp427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baker EJ, Jay JJ, Bubier JA, Langston MA, Chesler EJ. GeneWeaver: a web-based system for integrative functional genomics. Nucleic Acids Res. 2012;40:D1067–D1076. doi: 10.1093/nar/gkr968. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.