

Abstract
Background
Legionnaires’ disease is a severe form of pneumonia caused by the environmental bacterium Legionella pneumophila. Outbreaks commonly affect people with known risk factors, but the genetic and pathogenic complexity of L. pneumophila within an outbreak is not well understood. Here, we investigate the etiology of the major Legionnaires’ disease outbreak that occurred in Edinburgh, UK, in 2012, by examining the evolutionary history, genome content, and virulence of L. pneumophila clinical isolates.
Results
Our high resolution genomic approach reveals that the outbreak was caused by multiple genetic subtypes of L. pneumophila, the majority of which had diversified from a single progenitor through mutation, recombination, and horizontal gene transfer within an environmental reservoir prior to release. In addition, we discover that some patients were infected with multiple L. pneumophila subtypes, a finding which can affect the certainty of source attribution. Importantly, variation in the complement of type IV secretion systems encoded by different genetic subtypes correlates with virulence in a Galleria mellonella model of infection, revealing variation in pathogenic potential among the outbreak source population of L. pneumophila.
Conclusions
Taken together, our study indicates previously cryptic levels of pathogen heterogeneity within a Legionnaires’ disease outbreak, a discovery that impacts on source attribution for future outbreak investigations. Furthermore, our data suggest that in addition to host immune status, pathogen diversity may be an important influence on the clinical outcome of individual outbreak infections.
Electronic supplementary material
The online version of this article (doi:10.1186/s13059-014-0504-1) contains supplementary material, which is available to authorized users.
Background
Legionella pneumophila is an ubiquitous intracellular pathogen of environmental protozoa, commonly found in freshwater reservoirs [1]. The bacterium can also cause human infections (legionellosis) and is a causative agent of the severe community-acquired pneumonia known as Legionnaires’ disease [2]. Infection may occur following exposure to contaminated aerosols, although some individuals can seroconvert without displaying any symptoms [3]. The risk of human exposure to aerosols containing L. pneumophila increases with elevated ambient temperature, vapor pressure, and bacterial density in the reservoir [4,5], while host factors such as male gender, age, smoking and underlying respiratory pathology are associated with increased risk of developing Legionnaires’ disease [6]. Studies to date have largely considered that each outbreak of legionellosis likely results from a point source of a clonal L. pneumophila population, although a recent study has reported multiple sequence types associated with a single patient [7].
The ability of L. pneumophila to establish infection within human host cells is mediated through type IV secretion systems (T4SSs), complexes of proteins homologous to conjugation systems that mediate the transfer of nucleoprotein complexes and proteins between cells [8,9]. Among L. pneumophila strains, three families of variably present T4SS have been identified; T4ASS, T4BSS and the genomic island-associated T4SS (GI-T4SS) [10-12]. T4ASS, encoded by the L. pneumophila Lvh locus, has a role in host-cell entry and intracellular replication [13], and contributes to establishment of infection at lower temperatures [14], while the Dot/Icm system encoded by a T4BSS is essential for intracellular replication [15-17]. The functions of many of the effector proteins are unclear but a large number demonstrate homology with eukaryotic proteins, which may allow L. pneumophila to modulate the host response through structural mimicry of host components [18-21].
The incidence of L. pneumophila-associated disease in Scotland, UK ranges from 15 to 40 cases per year, with approximately two-thirds of cases attributed to sporadic infections acquired during travel [22]. Between 31 May and 17 July 2012, a total of 56 confirmed and 36 suspected cases of Legionnaires’ disease were reported with an epidemiological link to the south-west region of Edinburgh in Scotland. Analysis of prevailing wind conditions preceding and during the outbreak suggested a cluster of water cooling towers in the northeast of the affected area as the likely source of aerosols containing L. pneumophila responsible for the outbreak [23]. Although the suspect cooling towers were extensively sampled for Legionella spp., cultures were not obtained from any of the environmental samples. Patients were linked to the outbreak based on the results of traditional typing methods for L. pneumophila of serogrouping, monoclonal antibody (mAb) subgrouping, and sequence-based typing (SBT) [23]. However, the low resolution of traditional typing methods limits their utility for investigating intra-clonal levels of diversity among strains from a single outbreak. A previous pilot study examined the potential application of whole genome sequencing (WGS) to Legionnaires’ disease outbreak investigation by sequencing seven L. pneumophila isolates from an outbreak in Hampshire, UK, and a possible source was inferred from the analysis which was consistent with previous assertions based on traditional epidemiological analysis [24]. In addition, a very recent WGS study of L. pneumophila isolates from Alcoy, Spain, highlighted an important role for recombination in the evolution of L. pneumophila populations leading to multiple genotypes within the same outbreak [25]. In the current study, WGS was applied to all 22 L. pneumophila clinical isolates cultured from the 2012 Edinburgh outbreak in order to investigate their genetic diversity, genome content and pathogenic potential. Unexpectedly, we discovered considerable genetic heterogeneity among the outbreak isolates which was the result of mutation, recombination and horizontal gene transfer within environmental populations prior to release. Multiple genetic subtypes were identified within individual patients, and strain-dependent differences in virulence were observed in a Galleria mellonella model of infection, consistent with variation in pathogenic potential among outbreak isolates. These data have important implications for source attribution in future Legionnaires’ disease outbreaks, and imply a putative role for L. pneumophila virulence determinants in the variable outcome of infections within an outbreak.
Results and discussion
Genomic epidemiology of the 2012 Edinburgh Legionnaires’ disease outbreak
Confirmed cases of Legionnaires’ disease were defined by clinical or radiological evidence of community-acquired pneumonia in conjunction with either isolation of Legionella species from respiratory secretions, detection of L. pneumophila antigen in urine, or a positive L. pneumophila serogroup (Sg) antibody response. Based on these criteria, 56 cases of Legionnaires’ disease were confirmed, while an additional 36 cases were classed as probable (based on a positive respiratory secretion L. pneumophila Sg 1 PCR), or suspected L. pneumophila. Isolates were cultured from 15 of 92 patients, a culture success rate consistent with previous studies, and linked to the outbreak on the basis of date and location of isolation. Results of typing techniques indicated all isolates to be L. pneumophila Sg 1, mAb subgroup Knoxville and sequence type (ST)191. Viable cultures were not obtained from any of the water samples from suspected outbreak sources precluding a definitive source attribution. In order to investigate the genetic relatedness of the clinical L. pneumophila isolates from the outbreak, we sequenced the genomes of all 22 isolates which included multiple colonies from the primary isolation plates of each of 4 patients (Table 1). An additional three contemporaneous clinical isolates that were epidemiologically unrelated to the Edinburgh outbreak, four environmental isolates of ST191 obtained previously in the UK, and a single Sg 1, ST591 isolate were sequenced to provide a phylogenetic context to the outbreak isolates (Table 1). Sequence assemblies resulted in 26 to 103 contigs per genome, with N50 values ranging from 81,559 to 718,197 bp (Table S1 in Additional file 1). Sequence analysis confirmed that 21 of the 22 sequenced isolates that were linked to the outbreak were ST191. However, isolate 12_4117 from patient 3, which had previously been reported to be Sg 1, ST191, was determined to be a novel sequence type ST1418. The original sample from patient 3 was re-examined and colonies of ST191 and ST148 isolates were identified from the same clinical sample, consistent with a co-infection of the patient with multiple sequence types of L. pneumophila.
Table 1.
Origin, traditional typing, and genomic subtyping of L. pneumophila outbreak isolates
| Patient | Isolate | Date of isolation | Serogroup | mAb | ST | Genomic subtype |
|---|---|---|---|---|---|---|
| Edinburgh outbreak isolates | ||||||
| 1 | 12_4030 | 31/05/2012 | 1 | Knoxville | 191 | A |
| 12_4054 | 31/05/2012 | 1 | Knoxville | 191 | C | |
| 2 | 12_4042 | 01/06/2012 | 1 | Knoxville | 191 | A |
| 3 | 12_4117 | 02/06/2012 | 10 | NA | 1418 | NA |
| 4 | 12_4058 | 03/06/2012 | 1 | Knoxville | 191 | A |
| 5 | 12_4053 | 04/06/2012 | 1 | Knoxville | 191 | A |
| 6 | 12_4561 | 06/06/2012 | 1 | Knoxville | 191 | B |
| 7 | 12_4169 | 06/06/2012 | 1 | Knoxville | 191 | B |
| 8 | 12_4555 | 06/06/2012 | 1 | Knoxville | 191 | B |
| 9 | 12_4563 | 07/06/2012 | 1 | Knoxville | 191 | B |
| 10 | 12_4499 | 08/06/2012 | 1 | Knoxville | 191 | C |
| 12_4480 | Not recorded | 1 | Knoxville | 191 | C | |
| 11 | 12_5064 | 08/06/2012 | 1 | Knoxville | 191 | B |
| 11 | 12_4437 | 12/06/2012 | 1 | Knoxville | 191 | B |
| 12 | 12_4240 | 11/06/2012 | 1 | Knoxville | 191 | D |
| 13 | 12_4903 | 17/06/2012 | 1 | Knoxville | 191 | B |
| 14 | 12_5223 | 20/06/2012 | 1 | Knoxville | 191 | B |
| 15 | 12_5251 | 29/06/2012 | 1 | Knoxville | 191 | A |
| 12_5392 | 29/06/2012 | 1 | Knoxville | 191 | A | |
| 12_5383 | 29/06/2012 | 1 | Knoxville | 191 | A | |
| 12_5414 | 29/06/2012 | 1 | Knoxville | 191 | A | |
| 12_5415 | 29/06/2012 | 1 | Knoxville | 191 | A | |
| Contemporary non-outbreak isolates | ||||||
| NA | 12_3965 | 31/05/2012 | 1 | Benidorm | 42 | NA |
| NA | 12_4251 | 07/06/2012 | 1 | Philadelphia | 616 | NA |
| NA | 12_4904 | 21/06/2012 | 1 | Philadelphia | 37 | NA |
| Historical non-outbreak isolates | ||||||
| NA | H080160261 | 2008 | 6 | NA | 191 | NA |
| NA | H080160262 | 2008 | 6 | NA | 191 | NA |
| NA | H080160263 | 2008 | 6 | NA | 191 | NA |
| NA | H064020049 | 2006 | 1 | Allentown | 591 | NA |
| NA | H092620872 | 2009 | 6 | NA | 191 | NA |
mAb, monoclonal antibody subtype; NA, not applicable; ST, sequence type.
In order to investigate the relatedness of the outbreak isolates to the breadth of known L. pneumophila diversity, we reconstructed the phylogeny of all 30 sequenced isolates in addition to 9 additional L. pneumophila strains for which the genome sequence was publicly available (Table 2, Figure 1). The maximum-likelihood phylogeny indicated that the outbreak ST191 isolates were more closely related to each other than to non-outbreak isolates, but the non-outbreak, environmental ST191 isolates formed a sister clade in the phylogeny (Figure 1). The phylogeny also confirmed that the three L. pneumophila isolates obtained from cases of Legionnaires’ disease which were contemporaneous but had no epidemiological link to the Edinburgh outbreak (L. pneumophila strains 12_3965, 12_4251, and 12_4904) were not closely related to the outbreak isolates (Figure 1). Of note, while L. pneumophila Sg 1 has been responsible for the great majority of previously reported clinical infections [26], ST191 has not previously been reported as a common cause of legionellosis. However, it has been widely detected in environmental samples from the UK, Germany, the Netherlands, Poland, and Russia (PHE Legionella Database), highlighting its potential for future outbreaks. As stated, Legionella spp. were not cultured from any samples of the suspect water reservoirs implicated in the Edinburgh Legionnaires’ disease outbreak. In the future, culture-free sequencing techniques may be useful for identifying the existence of related genetic subtypes within suspected water sources or patient samples without the requirement for culture [27,28].
Table 2.
Origin and characteristics of previously sequenced L. pneumophila strains included in this study
| Isolate | Isolation date | Isolation country | Sg | mAb | ST | Source | Reference |
|---|---|---|---|---|---|---|---|
| Alcoy | 1999 | Spain | 1 | ND | 578 | Clinical | [10] |
| Corby | NA | UK | 1 | Knoxville | 51 | Clinical | [11] |
| ATCC43290 | 1987 | USA | 12 | NA | 187 | Clinical | [29] |
| 130b | 1978 | USA | 1 | Benidorm | 42 | Clinical | [30] |
| Lens | NA | France | 1 | Benidorm | 15 | Clinical | [19] |
| Paris | NA | France | 1 | Philadelphia | 1 | Clinical | [19] |
| Philadelphia | 1974 | USA | 1 | Philadelphia | 36 | Clinical | [31] |
| NC_018139 | NA | France | 1 | ND | 47 | Clinical | [32] |
| NC_018140 | NA | France | 1 | ND | 734 | Environmental | [32] |
mAb, mAb, monoclonal antibody subtype; NA, not applicable; ND, not determined; Sg, serogroup; ST, sequence type.
Figure 1.
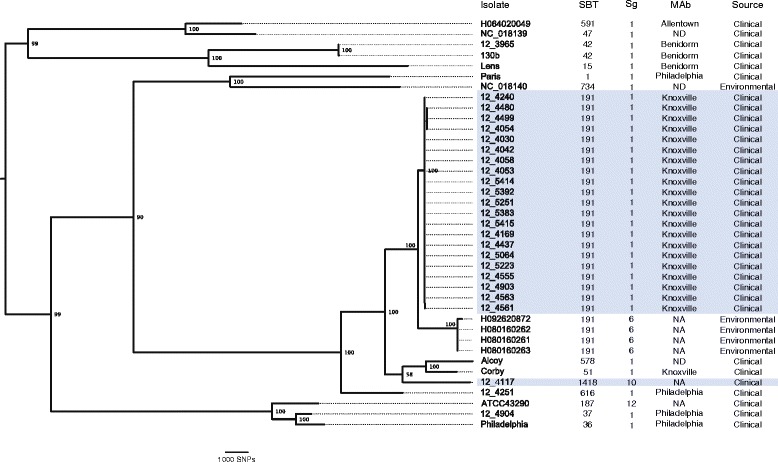
The majority of Edinburgh Legionnaires’ disease outbreak isolates belong to a single ST191 clade. Maximum-likelihood phylogeny based on the core genome of L. pneumophila outbreak and diverse reference isolates with Legionella longbeachae included as outgroup. Isolates from the Edinburgh Legionnaires’ disease outbreak are indicated in blue shading. Characteristics based on traditional typing schemes are denoted by: mAb, monoclonal antibody subgroup; NA, not applicable; ND, not determined; SBT, sequence-based typing; Sg, serogroup.
The Edinburgh Legionnaires’ disease outbreak was caused by multiple genetic subtypes of ST191 L. pneumophila
In order to elucidate the relatedness of the 21 ST191 outbreak isolates to each other, the core genome variation among the ST191 outbreak isolates only was examined. Inspection of the sequence alignment revealed the presence of three regions of high SNP density in each of three isolates (12_4480, 12_4499, 12_5054), which were suggestive of recombination events in those strains (Figure 2). Removal of these genomic regions yielded an alignment of 2,694,741 bp, with a total of 42 polymorphic sites (Figure 2). Phylogenetic reconstruction using maximum likelihood and Bayesian methods revealed four distinct subtypes (A to D) among the ST191 outbreak isolates from 15 patients (Figure 2, Table 1). Of note, for patients 10, 11, and 15 the multiple isolates obtained from each were identical, suggesting that the short incubation periods did not support extensive within-host diversification. However, isolates from patient 1 were represented by multiple genetic subtypes of ST191 (clades A and C), which were differentiated by 20 core genome SNPs (Figure 2). Of the four subtypes, three were identified in multiple patients, including subtype A in four patients, subtype B in seven patients, and subtype C in two patients, indicating their wide distribution among patients infected during the outbreak (Table 1). The short timescale between exposure to and isolation of the pathogen during the outbreak and the lack of person-to-person transmission for L. pneumophila [33,34] strongly suggest that the genetic subtypes of ST191 existed in the outbreak source prior to release and evolved from a recent progenitor within the water reservoir by a combination of gene mutation and recombination.
Figure 2.
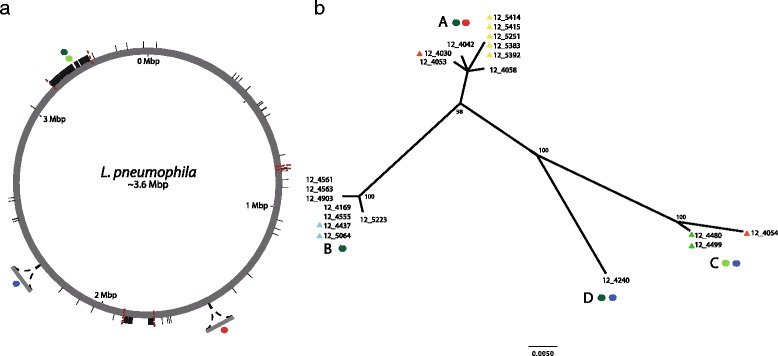
ST191 outbreak isolates are represented by multiple genetic subtypes which arose by mutation, recombination and horizontal gene transfer. (a) Genome distribution of mutations and predicted recombinant regions among ST191 outbreak isolates. Polymorphisms are mapped to the reference strain Corby. SNPs are represented by short black lines, and variant T4SSs are indicated by colored circles; Dot/Icm T4BSS (variant 1), dark green; Dot/Icm T4BSS (variant 2), light green; Lvh T4ASS (Philadelphia), red; Lvh T4ASS (novel), blue. Predicted recombinant regions are flanked by dashed red lines. (b) Maximum likelihood un-rooted radial phylogeny reconstructed using the non-recombinant core genome of ST191 outbreak isolates. For each node, maximum likelihood bootstrap values are displayed. Filled triangles indicate multiple isolates that were obtained from a single patient as follows; patient 1, yellow; patient 2, red; patient 12, green; patient 13, blue. Presence or absence of genomic regions encoding T4SS is indicated by colored filled circles as per (a).
As the ST191 isolate sequences in the current study did not facilitate determination of the mutation rate of the outbreak isolates (Figure S1 in Additional file 1), we estimated the lower limit of the time to the most recent common ancestor (tMRCA) of the ST191 outbreak isolates using a previous estimate of the evolutionary rate for L. pneumophila [25] as a prior in Bayesian phylogenetic analysis. This indicated that the MRCA likely existed many months prior to the outbreak (Table S2 in Additional file 1). Consistent with this, a previous study demonstrated that L. pneumophila can persist in cooling towers for periods of at least 5 years, a time-frame which could easily account for the identified genetic diversity among the ST191 isolates from the Edinburgh outbreak [35].
Finally, the identification of multiple L. pneumophila strains in patients 1 and 3 raises questions concerning our capacity to confidently link infections to a single environmental source in Legionnaires’ disease outbreak situations. Of note, a previous study demonstrated the presence of L. pneumophila with indistinguishable pulsed field gel electrophoresis profiles in multiple cooling towers within a 1 km radius, suggesting that cross-contamination of water cooling towers may lead to the existence of closely related subtypes in distinct reservoirs, thereby complicating source attribution [35].
Outbreak strains differ in content of genes encoding T4SSs
Considering the existence of multiple genetic subtypes of the outbreak population of ST191 L. pneumophila, we also examined the variation in the accessory genome of ST191 isolates. Several regions of difference were identified among the isolates examined, including three genetic elements encoding T4SSs (Figure 2). For example, all nine isolates in clade A (Figure 2) contained a 39,441 bp Lvh T4ASS which shared 100% nucleotide identity with a genetic element in the genome of the Philadelphia 1 strain, derived from the original Legionnaire’s disease outbreak in Philadelphia in 1976 [31]. Of note, a recently sequenced genome of a Sg 6 isolate from Thunder Bay, Canada, contained the same genetic element with one SNP (among 39,441 bp) [36], demonstrating a remarkably high level of nucleotide conservation for the genetic element encoding this T4SS in clinical isolates obtained on two continents almost four decades apart. Isolates belonging to clades C and D had a novel 45 kb region not identified in the other outbreak isolates which contained 46 predicted coding sequences, including homologs of lvrA, lvrB, lvrC, and virB4 (Table S3 in Additional file 1), suggesting a putative role as a novel Lvh T4ASS (Figure S2 in Additional file 1). In addition, all isolates contained a copy of the T4BSS encoding the dot/icm system, which has previously been demonstrated to have an essential role in pathogenesis, but a high density of polymorphic sites at the dotA/icmVWX locus differentiated the Dot/Icm T4BSS into 2 distinct molecular variants associated with 18 isolates in clades A, B, and D (variant 1), and 4 isolates in clade C (variant 2), respectively (Figure 2). Finally, although indistinguishable in the core genome, the two isolates from patient 13 of genetic subtype B differed by the presence a 55 kb element including genes encoding resistance to heavy metals, and a 2.7 kb region encoding two hypothetical proteins. In summary, the data indicate considerable variation in accessory genome content among isolates from a single outbreak. Taken together, our genome analysis revealed that L. pneumophila persisting within the outbreak source diversified through a combination of mutation and gene flow, including recombination and horizontal gene transfer, leading to a heterogeneous population responsible for the Edinburgh Legionnaires’ disease outbreak.
Variation in T4SS gene content among outbreak isolates correlates with virulence in a G. mellonella infection model
The T4SSs play a central role in the capacity of L. pneumophila to infect free-living amoeba and survive within vacuoles in human alveolar macrophages. Mouse models have traditionally been applied to examine the role of specific Legionella spp. determinants in pathogenesis but an infection model of the G. mellonella (waxmoth) larvae has been developed recently, providing an effective model of L. pneumophila human infection which allows analysis of T4SS-dependent virulence [37]. We used the G. mellonella infection model to examine the virulence of all L. pneumophila clinical isolates obtained in the 2012 Edinburgh outbreak, in addition to a reference strain L. pneumophila Paris of known virulence [37]. Considerable strain-dependent variation in Galleria host survival was observed after infection (Figure 3), and a significant difference in killing capacity was identified between groups of isolates with unique combinations of T4SSs. In particular, strains with the novel putative T4SS resulted in more rapid killing of Galleria larvae than strains without it (P = 0.04; Figure 3). There was no significant difference between isolates containing different variants of the Dot/Icm T4SS (data not shown). Analysis of clinical data for each patient for which there was comparable information (n = 13) was carried out. The small number of patients infected with the more virulent subtype containing the novel Lvh T4SS was not sufficiently powered to facilitate a robust statistical analysis, and there was no statistically significant difference in clinical disease indicators between patients infected with strains containing the novel Lvh T4SS (n = 3), and those infected with strains lacking the Lvh T4SS (n = 10). However, patients with the novel T4SS required more clinical care intervention, including higher intensive care unit (ICU) admission, a higher proportion requiring mechanical ventilation, and fewer ICU-free days. Taken together, we have identified heterogeneity in virulence among closely related L. pneumophila isolates from the same Legionnaires’ disease outbreak that may influence the outcome of infection.
Figure 3.
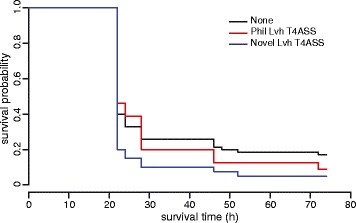
The virulence of L. pneumophila outbreak isolates correlates with T4SS content. G. mellonella survival curves representing the mean for isolates grouped according to the combination of T4SS, including presence of the Lvh T4ASS Philadelphia (red), Lvh T4ASS novel (blue), and isolates without T4ASS Philadelphia or T4ASS novel (black). Larvae infected with isolates which encoded the Lvh T4ASS novel had a lower survivability compared with those with infected other isolates (P = 0.04).
Conclusions
The application of genomics offers great potential for enhanced understanding of the biology of infectious disease outbreaks. Here, the high resolution of WGS revealed previously unappreciated levels of genetic and pathogenic complexity underlying a major Legionnaires’ disease outbreak. In particular, our data suggest that the genetic diversity of L. pneumophila environmental populations associated with an outbreak may make source attribution challenging, particularly in the light of potential cross-contamination of neighboring water coolers [35]. Intra- and inter-species horizontal gene transfer is common among Legionella spp., and variation in T4SS content between strains has been reported previously [10,13,30,32,38]. However, differences in T4SS content and associated virulence have not previously been described among strains from a single outbreak. While susceptibility to infection is strongly influenced by known host-associated risk factors, our data suggest that variation in content of virulence genes among outbreak isolates may also influence the clinical outcome of individual infections.
Materials and methods
L. pneumophila culture and DNA extraction
L. pneumophila was incubated on buffered charcoal yeast extract (BCYE) agar at 37°C for 48 h in a humid environment. A sweep of L. pneumophila was taken from the agar culture using a sterile loop, suspended in 2 ml phosphate-buffered saline (PBS), and pelleted by centrifugation for 10 minutes at 5,000 × g. Genomic DNA extraction was performed according to the standard isolation of DNA from Gram-negative bacteria protocol using the QIAcube platform (QIAGEN, Benelux B.V., Netherlands Netherlands).
DNA sequencing, assembly and alignment
Genomic DNA libraries were prepared using the Illumina TruSeq kit, and sequenced using either 150 bp paired-end runs on an Illumina MiSeq, or 100 bp paired-end runs on an Illumina HiSeq 2000. A sequence project has been created at the European Nucleotide Archive with project accession PRJEB6631. The raw FASTQ files were examined for Illumina adaptor sequences using cutadapt v.1.2 [39] to facilitate removal of adaptor contamination. The 5' and 3' ends of reads were trimmed to remove low quality scoring bases (Q <30) using Sickle v.1.2 [40]. Reads containing low frequency sequencing errors were corrected with Quake v.0.3 using a k-mer size of 15 [41]. Processed sequence reads for each isolate were assembled de novo using the de Bruijn graph-based assembler Velvet v.1.1 [42]. Optimal k-mer length and coverage cutoff parameters were calculated using the VelvetOptimiser v.2.2.5 script [43]. Assembled contigs, along with the genome sequences of representative clinical and environmental isolates from public databases, were aligned using progressiveMauve with default parameters [44]. Locally collinear blocks not common to all sequences or <1,000 bp in length were removed, resulting in a gap-free core genome alignment.
Mapping of Illumina sequence reads
Sequence reads were mapped to the genome sequence of L. pneumophila strain Corby (accession number NC_009494.2) using the Burrows-Wheeler Aligner v.0.6.2 [45] with the Smith-Waterman algorithm disabled. Base calls were made at sites that were covered by at least five sequencing reads, and a core genome alignment was produced, with the core genome defined as nucleotide positions with a base call in all isolates.
Recombination detection
To assess the level of recombination among the sequences in the alignment, the gap-free genome alignments from progressiveMauve were used as input for BratNextGen [46]. One hundred iterations of recombination learning were performed, until parameters had converged.
Phylogenetic reconstruction and dating analysis
Core genome alignments were used as input for maximum likelihood phylogenetic reconstruction in RAxML v.8 [47]). A GTR model of nucleotide substitution was applied with the gamma model of rate heterogeneity. Support for nodes was assessed using 1,000 bootstrap replicates, and species tree was rooted by using the sequence of a Legionella longbeachae isolate as outgroup. Core genome alignments of outbreak ST191 isolates were used as input for BEAST v.1.8.0 in order to estimate the date for the most recent common ancestor [48]. A HKY model of nucleotide substitution was applied with a gamma model of rate heterogeneity plus invariant sites. Three demographic models were investigated (constant, exponential, and Bayesian skyline), using an uncorrelated lognormal clock with the previously reported rate of evolution for L. pneumophila as a prior (1.39 × 10-7, 95% HPD intervals of 5.41 × 10-8 to 2.30 × 10-7) [25]. For each demographic model, 3 independent chains were run for 1 × 108 generations, with sampling every 10,000 generations and 10% discarded as burn-in.
Genome annotation and identification of variable gene content
Variable gene content among the outbreak isolates was examined using a combination of genome annotation and alignment. Predicted protein and RNA coding sequences in the assembled contigs for the sequenced isolates were annotated using the prokka pipeline v.1.5.2 [49], and a custom BLAST database of Legionella sequences. The Gram-negative option was specified to predict signal sequences appropriate for L. pneumophila. The annotated assembled contigs were aligned using Mugsy v.1r.2.2 [50] and gene presence or absence determined with mugsy-annotator v.0.5 [51].
G. mellonella larvae infection model
Single colonies of L. pneumophila were incubated in ACES [N-(2-acetamido)-2-aminoethanesulfonic acid] yeast extract broth at 37°C for 21 h [30]. The OD600 of liquid cultures was adjusted to 0.5 using PBS. Groups of 10 G. mellonella larvae were injected with 10 μl of either PBS or 1.6 ± 0.4 × 107 CFU bacteria. The larvae were incubated at 37°C for 74 h and checked periodically for death. Kaplan-Meier survival curves were produced using the Survival package for R v.3.0.2 [52]. Differences between survival probability distributions were assessed using a log-rank test.
Patient clinical information
Clinical information on all patients infected in the outbreak was extracted from public health interviews and ‘travel diaries’ to ascertain place of residence and work, date of symptom onset and co-morbidities. Following the outbreak, data on all confirmed and probable cases were extracted from patients’ medical records. These data included age, gender, co-morbidities, hospital length of stay and hospital outcome. Documented cardiovascular disease (including a history of ischemic or valvular heart disease or heart failure) respiratory disease (asthma, chronic obstructive pulmonary disease or lung fibrosis), chronic kidney disease, chronic liver disease or immunosuppression (systemic steroids or immunosuppressant therapy) was recorded. Where patients were admitted to critical care, data on demographics, acute physiology, therapeutic interventions and outcome were extracted using the Scottish Intensive Care Society Audit Group’s data collection program Wardwatcher. CURB65 score was also calculated to assess severity of pneumonia at presentation. CURB65 is a clinical prediction score validated for predicting mortality in community-acquired pneumonia [53].
Ethics and data permissions
This study was undertaken as part of the further analysis of the factors underlying an outbreak of Legionnaires’ disease [23] and under the auspices of the Incident Management Team. Advice was sought from the relevant research ethics committee and it was confirmed that these outbreak-related investigations did not require research ethics approval. All tissue samples were handled in line with the requirements of the Lothian Bioresource. The authors who reviewed the clinical and tissue data were the relevant members of the Incident Management Team and data were anonymized at the earliest opportunity to minimize the risk of disclosure. The NHS Lothian Caldicott Guardian oversaw the governance, advised on the data flows, reviewed the collected clinical data for disclosivity and approved the final manuscript.
Data availability
The Illumina sequences generated and used in this study are deposited and available in the European Nucleotide Archive [54], along with the draft genome assembly contigs under project accession number PRJEB6631.
Acknowledgements
We are grateful to T Harrison and C Buchrieser for provision of L. pneumophila reference strains, and H Brown for advice on statistics. We also thank C Buchrieser for critical analysis of the manuscript. We thank the Microbiology scientific staff, at the Department of Laboratory Medicine, Royal Infirmary of Edinburgh, the Scottish Haemophilus, Legionella, Meningococcus, and Pneumococcus Reference Laboratory, and Edinburgh Genomics for technical assistance. We acknowledge institute strategic grant funding from the Biotechnology and Biological Sciences Research Council (UK), in addition to a doctoral training grant from the Medical Research Council (UK).
Abbreviations
- bp
base pair
- ICU
intensive care unit
- mAb
monoclonal antibody
- PBS
phosphate-buffered saline
- SBT
sequence-based typing
- Sg
serogroup
- SNP
single nucleotide polymorphism
- ST
sequence type
- T4SS
type IV secretion system
- tMRCA
time to most recent common ancestor
- WGS
whole genome sequencing
Additional file
Output from Path-O-Gen representing a regression plot of root-tip distances against isolation time for outbreak isolates indicating a lack of temporal signal in the outbreak isolate sequences. Figure S2. Schematic representation of the genomic region encoding a novel putative T4ASS in some ST191 isolates. Arrows represent predicted coding sequences (CDSs) identified using the prokka annotation pipeline, colored as follows: blue, functional homology to previously identified T4SS proteins; green, homology to integrase family proteins. Unlabeled CDSs encode predicted hypothetical proteins. Table S1. Sequence assembly metrics for each L. pneumophila isolate sequenced. Table S2. Estimated time to the most recent common ancestor for the outbreak ST191 isolates. Table S3. Nearest homologs for ORFs identified in the genomic island encoding the novel putative T4SS among Edinburgh outbreak L. pneumophila.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Author contributions
JRF, PRM, and GFE designed the study. PRM, CWVDB, MG, MW and MJW carried out research. JRF, PRM, JMS, and MJW analyzed data. DSJL, MH, and MW provided research tools and reagents. JRF and PRM wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Paul R McAdam, Email: paul.mcadam@unimelb.edu.au.
Charles W Vander Broek, Email: Charles.Vander-Broek@roslin.ed.ac.uk.
Diane SJ Lindsay, Email: diane.lindsay@nhs.net.
Melissa J Ward, Email: Melissa.Ward@ed.ac.uk.
Mary F Hanson, Email: Mary.Hanson@nhslothian.scot.nhs.uk.
Michael Gillies, Email: Michael.Gillies@ed.ac.uk.
Mick Watson, Email: mick.watson@roslin.ed.ac.uk.
Joanne M Stevens, Email: Jo.Stevens@roslin.ed.ac.uk.
Giles F Edwards, Email: gilesedwards@btinternet.com.
J Ross Fitzgerald, Email: Ross.Fitzgerald@ed.ac.uk.
References
- 1.Rowbotham TJ. Preliminary report on the pathogenicity of Legionella pneumophila for freshwater and soil amoebae. J Clin Microbiol. 1980;33:1179–1183. doi: 10.1136/jcp.33.12.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McDade JE, Shepard CC, Fraser DW, Tsai TR, Redus MA, Dowdle WR. Legionnaires’ disease: isolation of a bacterium and demonstration of its role in other respiratory disease. N Engl J Med. 1977;297:1197–1203. doi: 10.1056/NEJM197712012972202. [DOI] [PubMed] [Google Scholar]
- 3.Rudbeck M, Molbak K, Uldum SA. Dynamics of Legionella antibody levels during 1 year in a healthy population. Epidemiol Infect. 2009;137:1013–1018. doi: 10.1017/S0950268808001684. [DOI] [PubMed] [Google Scholar]
- 4.Brown CM, Nuorti PJ, Breiman RF, Hathcock AL, Fields BS, Lipman HB, Llewellyn GC, Hofmann J, Cetron M. A community outbreak of Legionnaires’ disease linked to hospital cooling towers: an epidemiological method to calculate dose of exposure. Int J Epidemiol. 1999;28:353–359. doi: 10.1093/ije/28.2.353. [DOI] [PubMed] [Google Scholar]
- 5.Conza L, Casati S, Limoni C, Gaia V. Meteorological factors and risk of community-acquired Legionnaires’ disease in Switzerland: an epidemiological study. BMJ Open. 2013;3:e002428. doi: 10.1136/bmjopen-2012-002428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marston BJ, Lipman HB, Breiman RF. Surveillance for Legionnaires’ disease. Risk factors for morbidity and mortality. Arch Intern Med. 1994;154:2417–2422. doi: 10.1001/archinte.1994.00420210049006. [DOI] [PubMed] [Google Scholar]
- 7.Coscolla M, Fernandez C, Colomina J, Sanchez-Buso L, Gonzalez-Candelas F. Mixed infection by Legionella pneumophila in outbreak patients. Int J Med Microbiol. 2014;304:307–313. doi: 10.1016/j.ijmm.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 8.Fronzes R, Christie PJ, Waksman G. The structural biology of type IV secretion systems. Nat Rev Microbiol. 2009;7:703–714. doi: 10.1038/nrmicro2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christie PJ, Vogel JP. Bacterial type IV secretion: conjugation systems adapted to deliver effector molecules to host cells. Trends Microbiol. 2000;8:354–360. doi: 10.1016/S0966-842X(00)01792-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.D'Auria G, Jimenez-Hernandez N, Peris-Bondia F, Moya A, Latorre A. Legionella pneumophila pangenome reveals strain-specific virulence factors. BMC Genomics. 2010;11:181. doi: 10.1186/1471-2164-11-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Glockner G, Albert-Weissenberger C, Weinmann E, Jacobi S, Schunder E, Steinert M, Hacker J, Heuner K. Identification and characterization of a new conjugation/type IVA secretion system (trb/tra) of Legionella pneumophila Corby localized on two mobile genomic islands. Int J Med Microbiol. 2008;298:411–428. doi: 10.1016/j.ijmm.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 12.Juhas M, Crook DW, Dimopoulou ID, Lunter G, Harding RM, Ferguson DJ, Hood DW. Novel type IV secretion system involved in propagation of genomic islands. J Bacteriol. 2007;189:761–771. doi: 10.1128/JB.01327-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bandyopadhyay P, Liu S, Gabbai CB, Venitelli Z, Steinman HM. Environmental mimics and the Lvh type IVA secretion system contribute to virulence-related phenotypes of Legionella pneumophila. Infect Immun. 2007;75:723–735. doi: 10.1128/IAI.00956-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ridenour DA, Cirillo SLG, Feng S, Samrakandi MM, Cirillo JD. Identification of a gene that affects the efficiency of host cell infection by Legionella pneumophila in a temperature-dependent fashion. Infect Immun. 2003;71:6256–6263. doi: 10.1128/IAI.71.11.6256-6263.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo Z-Q, Isberg RR. Multiple substrates of the Legionella pneumophila Dot/Icm system identified by interbacterial protein transfer. Proc Natl Acad Sci U S A. 2004;101:841–846. doi: 10.1073/pnas.0304916101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagai H, Kagan JC, Zhu X, Kahn RA, Roy CR. A bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science. 2002;295:679–682. doi: 10.1126/science.1067025. [DOI] [PubMed] [Google Scholar]
- 17.Zusman T, Yerushalmi G, Segal G. Functional similarities between the icm/dot pathogenesis systems of Coxiella burnetii and Legionella pneumophila. Infect Immun. 2003;71:3714–3723. doi: 10.1128/IAI.71.7.3714-3723.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bruggemann H, Cazalet C, Buchrieser C. Adaptation of Legionella pneumophila to the host environment: role of protein secretion, effectors and eukaryotic-like proteins. Curr Opin Microbiol. 2006;9:86–94. doi: 10.1016/j.mib.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 19.Cazalet C, Rusniok C, Bruggemann H, Zidane N, Magnier A, Ma L, Tichit M, Jarraud S, Bouchier C, Vandenesch F, Kunst F, Etienne J, Glaser P, Buchrieser C. Evidence in the Legionella pneumophila genome for exploitation of host cell functions and high genome plasticity. Nat Genet. 2004;36:1165–1173. doi: 10.1038/ng1447. [DOI] [PubMed] [Google Scholar]
- 20.Gomez-Valero L, Rusniok C, Cazalet C, Buchrieser C. Comparative and functional genomics of legionella identified eukaryotic like proteins as key players in host-pathogen interactions. Front Microbiol. 2011;2:208. doi: 10.3389/fmicb.2011.00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lomma M, Gomez Valero L, Rusniok C, Buchrieser C. Legionella pneumophila - host interactions: insights gained from comparative genomics and cell biology. Genome Dyn. 2009;6:170–186. doi: 10.1159/000235770. [DOI] [PubMed] [Google Scholar]
- 22.Potts A, Sinka K, Love J, Donaghy M, McDonald E, Johnston F, Blatchford O, Lindsay D, Brown A, Edwards G. Legionellosis in Scotland in 2009 and 2010. HPS Weekly Rep. 2011;45:389–394. [Google Scholar]
- 23.McCormick D, Thorn S, Milne D, Evans C, Stevenson J, Llano M, Donaghy M, Team IM. Public health response to an outbreak of Legionnaires’ disease in Edinburgh, United Kingdom, June 2012. Euro Surveill. 2012;17:20216. doi: 10.2807/ese.17.28.20216-en. [DOI] [PubMed] [Google Scholar]
- 24.Reuter S, Harrison TG, Koser CU, Ellington MJ, Smith GP, Parkhill J, Peacock SJ, Bentley SD, Torok ME. A pilot study of rapid whole-genome sequencing for the investigation of a Legionella outbreak. BMJ Open. 2013;3:e002175. doi: 10.1136/bmjopen-2012-002175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanchez-Buso L, Comas I, Jorques G, Gonzalez-Candelas F. Recombination drives genome evolution in outbreak-related Legionella pneumophila isolates. Nat Genet. 2014;ᅟ:ᅟ. doi: 10.1038/ng.3114. [DOI] [PubMed] [Google Scholar]
- 26.Doleans A, Aurell H, Reyrolle M, Lina G, Freney J, Vandenesch F, Etienne J, Jarraud S. Clinical and environmental distributions of Legionella strains in France are different. J Clin Microbiol. 2004;42:458–460. doi: 10.1128/JCM.42.1.458-460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loman NJ, Constantinidou C, Christner M, Rohde H, Chan JZ, Quick J, Weir JC, Quince C, Smith GP, Betley JR, Aepfelbacher M, Pallen MJ. A culture-independent sequence-based metagenomics approach to the investigation of an outbreak of Shiga-toxigenic Escherichia coli O104:H4. JAMA. 2013;309:1502–1510. doi: 10.1001/jama.2013.3231. [DOI] [PubMed] [Google Scholar]
- 28.Seth-Smith HM, Harris SR, Skilton RJ, Radebe FM, Golparian D, Shipitsyna E, Duy PT, Scott P, Cutcliffe LT, O'Neill C, Parmar S, Pitt R, Baker S, Ison CA, Marsh P, Jalal H, Lewis DA, Unemo M, Clarke IN, Parkhill J, Thomson NR. Whole-genome sequences of Chlamydia trachomatis directly from clinical samples without culture. Genome Res. 2013;23:855–866. doi: 10.1101/gr.150037.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amaro F, Gilbert JA, Owens S, Trimble W, Shuman HA. Whole-genome sequence of the human pathogen Legionella pneumophila serogroup 12 strain 570-CO-H. J Bacteriol. 2012;194:1613–1614. doi: 10.1128/JB.06626-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schroeder GN, Petty NK, Mousnier A, Harding CR, Vogrin AJ, Wee B, Fry NK, Harrison TG, Newton HJ, Thomson NR, Beatson SA, Dougan G, Hartland EL, Frankel G. Legionella pneumophila strain 130b possesses a unique combination of type IV secretion systems and novel Dot/Icm secretion system effector proteins. J Bacteriol. 2010;192:6001–6016. doi: 10.1128/JB.00778-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chien M, Morozova I, Shi S, Sheng H, Chen J, Gomez SM, Asamani G, Hill K, Nuara J, Feder M, Rineer J, Greenberg JJ, Steshenko V, Park SH, Zhao B, Teplitskaya E, Edwards JR, Pampou S, Georghiou A, Chou IC, Iannuccilli W, Ulz ME, Kim DH, Geringer-Sameth A, Goldsberry C, Morozov P, Fischer SG, Segal G, Qu X, Rzhetsky A, et al. The genomic sequence of the accidental pathogen Legionella pneumophila. Science. 2004;305:1966–1968. doi: 10.1126/science.1099776. [DOI] [PubMed] [Google Scholar]
- 32.Gomez-Valero L, Rusniok C, Jarraud S, Vacherie B, Rouy Z, Barbe V, Medigue C, Etienne J, Buchrieser C. Extensive recombination events and horizontal gene transfer shaped the Legionella pneumophila genomes. BMC Genomics. 2011;12:536. doi: 10.1186/1471-2164-12-536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bryant JM, Grogono DM, Greaves D, Foweraker J, Roddick I, Inns T, Reacher M, Haworth CS, Curran MD, Harris SR, Peacock SJ, Parkhill J, Floto RA. Whole-genome sequencing to identify transmission of Mycobacterium abscessus between patients with cystic fibrosis: a retrospective cohort study. Lancet. 2013;381:1551–1560. doi: 10.1016/S0140-6736(13)60632-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lieberman TD, Michel JB, Aingaran M, Potter-Bynoe G, Roux D, Davis MR, Jr, Skurnik D, Leiby N, LiPuma JJ, Goldberg JB, McAdam AJ, Priebe GP, Kishony R. Parallel bacterial evolution within multiple patients identifies candidate pathogenicity genes. Nat Genet. 2011;43:1275–1280. doi: 10.1038/ng.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanchez I, Garcia-Nunez M, Ragull S, Sopena N, Pedro-Botet ML, Rey-Joly C, Sabria M, Esteve M. Genotypic variability and persistence of Legionella pneumophila PFGE patterns in 34 cooling towers from two different areas. Environ Microbiol. 2008;10:395–399. doi: 10.1111/j.1462-2920.2007.01460.x. [DOI] [PubMed] [Google Scholar]
- 36.Khan MA, Knox N, Prashar A, Alexander D, Abdel-Nour M, Duncan C, Tang P, Amatullah H, Dos Santos CC, Tijet N, Low DE, Pourcel C, Van Domselaar G, Terebiznik M, Ensminger AW, Guyard C. Comparative genomics reveal that host-innate immune responses influence the clinical prevalence of Legionella pneumophila serogroups. PLoS One. 2013;8:e67298. doi: 10.1371/journal.pone.0067298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harding CR, Schroeder GN, Reynolds S, Kosta A, Collins JW, Mousnier A, Frankel G. Legionella pneumophila pathogenesis in the Galleria mellonella infection model. Infect Immun. 2012;80:2780–2790. doi: 10.1128/IAI.00510-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gomez-Valero L, Buchrieser C. Genome dynamics in Legionella: the basis of versatility and adaptation to intracellular replication. Cold Spring Harb Perspect Med. 2013;3:a009993. doi: 10.1101/cshperspect.a009993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal. 2011;17:10–12. [Google Scholar]
- 40.Sickle. [https://github.com/najoshi/sickle]
- 41.Kelley DR, Schatz MC, Salzberg SL. Quake: quality-aware detection and correction of sequencing errors. Genome Biol. 2010;11:R116. doi: 10.1186/gb-2010-11-11-r116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Velvetoptimiser. [http://bioinformatics.net.au/software.velvetoptimiser.shtml]
- 44.Darling AE, Mau B, Perna NT. ProgressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marttinen P, Hanage WP, Croucher NJ, Connor TR, Harris SR, Bentley SD, Corander J. Detection of recombination events in bacterial genomes from large population samples. Nucleic Acids Res. 2012;40:e6. doi: 10.1093/nar/gkr928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stamatakis A, Ludwig T, Meier H. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics. 2005;21:456–463. doi: 10.1093/bioinformatics/bti191. [DOI] [PubMed] [Google Scholar]
- 48.Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 50.Angiuoli SV, Salzberg SL. Mugsy: fast multiple alignment of closely related whole genomes. Bioinformatics. 2011;27:334–342. doi: 10.1093/bioinformatics/btq665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Angiuoli SV, Dunning Hotopp JC, Salzberg SL, Tettelin H. Improving pan-genome annotation using whole genome multiple alignment. BMC Bioinformatics. 2011;12:272. doi: 10.1186/1471-2105-12-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Therneau T (2014). A Package for Survival Analysis in S. R package version 2.37-7, http://CRAN.R-project.org/package=survival
- 53.Lim WS, van der Eerden MM, Laing R, Boersma WG, Karalus N, Town GI, Lewis SA, Macfarlane JT. Defining community acquired pneumonia severity on presentation to hospital: an international derivation and validation study. Thorax. 2003;58:377–382. doi: 10.1136/thorax.58.5.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.European Nucleotide Archive. [http://www.ebi.ac.uk/ena]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The Illumina sequences generated and used in this study are deposited and available in the European Nucleotide Archive [54], along with the draft genome assembly contigs under project accession number PRJEB6631.