

Abstract
Pheochromocytoma (PHEO) is a rare but potentially lethal neuroendocrine tumor arising from catecholamine-producing chromaffin cells. Especially for metastatic PHEO, the availability of animal models is essential for developing novel therapies. For evaluating therapeutic outcome in rodent PHEO models, reliable quantification of multiple organ lesions depends on dedicated small-animal in vivo imaging, which is still challenging and only available at specialized research facilities. Here, we investigated whether whole-body fluorescence imaging and monitoring of urinary free monoamines provide suitable parameters for measuring tumor progression in a murine allograft model of PHEO. We generated an mCherry-expressing mouse PHEO cell line by lentiviral gene transfer. These cells were injected subcutaneously into nude mice to perform whole-body fluorescence imaging of tumor development. Urinary free monoamines were measured by liquid chromatography with tandem mass spectrometry. Tumor fluorescence intensity and urinary outputs of monoamines showed tumor growth–dependent increases (P < .001) over the 30 days of monitoring post-tumor engraftment. Concomitantly, systolic blood pressure was increased significantly during tumor growth. Tumor volume correlated significantly (P < .001) and strongly with tumor fluorescence intensity (rs = 0.946), and urinary outputs of dopamine (rs = 0.952), methoxytyramine (rs = 0.947), norepinephrine (rs = 0.756), and normetanephrine (rs = 0.949). Dopamine and methoxytyramine outputs allowed for detection of lesions at diameters below 2.3 mm. Our results demonstrate that mouse pheochromocytoma (MPC)-mCherry cell tumors are functionally similar to human PHEO. Both tumor fluorescence intensity and urinary outputs of free monoamines provide precise parameters of tumor progression in this sc mouse model of PHEO. This animal model will allow for testing new treatment strategies for chromaffin cell tumors.
Pheochromocytomas (PHEOs) are rare catecholamine-producing tumors mainly deriving from sympathetic chromaffin tissue of the adrenal medulla. Tumors from extra-adrenal sympathetic chromaffin tissue are referred to as extra-adrenal PHEOs or paragangliomas. Other paragangliomas deriving from parasympathetic tissue do not produce catecholamines (1–3). PHEOs are rare with an incidence of approximately eight or fewer cases per million (4, 5). Approximately 10% of PHEOs are metastatic (2). To date, a few markers, such as mutant succinate dehydrogenase subunit B and the Ki67 proliferation index (6), as well as splice isoforms of carboxypeptidase E (7), have been identified to be related to the metastatic phenotype. However, malignant PHEO is mainly diagnosed by identification of distant metastases (1, 2, 8). Generally, treatment options for metastatic PHEO include radionuclide therapy, radiofrequency ablation, or chemotherapy, which sometimes show suboptimal results, with short-term remission in some patients (2, 9). Thus, it is necessary to develop new therapeutic strategies for the treatment of metastatic PHEO.
Over the years, there has been great interest in developing PHEO cell-line models. To date, with few exceptions (10), most efforts focused on animal cell lines. Thereafter, animal models have limited utilities for evaluating therapeutic strategies. Models available with spontaneously or genetically emerging tumors (11–14) or allograft models (15–18) are the most straightforward models for evaluating therapies. The mouse pheochromocytoma (MPC) cell line was developed from PHEOs arising in neurofibromatosis type 1 (Nf1) knockout mice (14, 19) showing high-level expression of receptor tyrosine kinase RET (20), as such, involved in activation of ras-signaling and the mTOR pathway (21). Furthermore, MPC cells, instead of the frequently used PC12 cells (22, 23), resemble human chromaffin cells as they express the enzyme phenylethanolamine-N-methyltransferase converting norepinephrine to epinephrine (14, 16).
To date, monitoring of tumor progression in rodent PHEO models is solely based on dedicated small-animal imaging strategies only available in specialized research centers. However, conventional anatomic imaging as well as functional imaging in small animals, especially for quantifying the total burden of metastatic lesions, remains challenging and time consuming (15, 24, 25). PHEO is characterized by an overproduction of catecholamines, which is most readily assessed from increases in their plasma or urinary O-methylated metabolites (2, 26). In our current study, we investigated whether whole-body fluorescence imaging (FLI) and monitoring of urinary monoamines (catecholamines and metanephrines) provide suitable parameters for quantification of tumor progression in a murine allograft model of PHEO based on our far-red-fluorescence-tagged MPC cell line, named MPC-mCherry.
We demonstrate that whole-body FLI is a rapid, quantitative, and powerful tool to monitor tumor progression in an sc mouse model of PHEO. We also found that, to some extent, MPC-mCherry cell tumors in mice functionally recapitulate the endocrine situation in human PHEO by producing both catecholamines and O-methylated metabolites of catecholamines. Our findings, that urinary monoamines quantitatively reflect total tumor burden, may offer a perspective to substantially facilitate comprehensive preclinical diagnostic and therapeutic evaluations of multiple organ lesions of PHEO.
Materials and Methods
Cell culture
Mouse PHEO cells [MPC; clone 4/30PRR (14), passage 32] were cultured in collagen-coated flasks and maintained in RPMI 1640 including HEPES (GIBCO) in a humidified 5% CO2/95% (v/v) O2 atmosphere at 37°C. Culture medium was supplemented with 10% (v/v) heat-inactivated horse serum (GIBCO), 5% (v/v) fetal bovine serum superior (BIOCHROM) and 0.1% (v/v) gentamicin (GIBCO) and was replaced every 48–72 hours. Cells were routinely passaged every 7–10 days. Before injection into animals, cells at 70–80% confluence were detached using 0.05% (w/v) Trypsin-EDTA in magnesium- and calcium-free phosphate-buffered saline (PBS) and were adjusted to a concentration of 2 × 106 cells per 60 μL in 50% (v/v) Matrigel (BD Biosciences)/PBS.
Lentiviral vector construction and genetic modification of cancer cells
The lentiviral transfer vector p6NST70-MC is a derivative of the recently described p6NST90, harboring a multiple-cloning site with unique XbaI and HpaI sites downstream of an internal elongation factor 1 alpha promoter instead of the human ubiquitin C promoter–driven enhanced green fluorescent protein (EGFP) expression cassette (27). The open reading frame (orf) of the mCherry expression cassette was cloned into the discussed restriction sites of the multiple cloning site using NheI and PmeI restriction sites at the 5′ and 3′ end of both orf, respectively. For selection of successfully gene-modified MPC cells (passage 32; = MPC-mCherry cells; passage 0), the p6NST70-MC harbors a zeocin resistance expression cassette. Bicistronic expression for the selection marker downstream of the transgene stop codon is mediated by an internal ribosomal entry site derived from the encephalomyocarditis virus.
To generate vesicular stomatitis virus G glycoprotein pseudotyped lentivirus, the lentiviral vector DNA was cotransfected into HEK293T cells by linear polyethylenimine (Polysciences Europe) with lentiviral packaging plasmid pCD/NL-BH and vesicular stomatitis virus G glycoprotein–encoding pMD-GM plasmid. Viral supernatants were collected, concentrated by ultracentrifugation, and stored at −80°C, as described elsewhere (28, 29). Viral titers were determined by limiting dilution transduction on HT1080 cells, as described elsewhere (27, 29).
Animal experimentation
All animal procedures and experiments were carried out according to the guidelines of the German Regulations for Animal Welfare. Nude mice with rudimentary thymus (NMRI nu/nu) were purchased from the pathogen-free breeding facility of the Medical Faculty Carl Gustav-Carus Experimental Center (Technische Universität). Nude mice were preferred to ensure maximal optical tissue penetration during whole-body FLI. The protocols were approved by the local Ethical Committee for Animal Experiments. A number of 2 × 106 MPC-mCherry cells (passage 12) were transplanted subcutaneously into the right shoulder of 10-week-old male and female animals. Experimental groups consisted of 10 animals housed in a pathogen-free facility. General anesthesia was induced and maintained with inhalation of 10% (v/v) desflurane in 30/10% (v/v) oxygen/air. Imaging studies were performed every 3–4 days. Noninvasive blood pressure (BP) data and single-voided urine samples were collected weekly on different days, respectively, and in advance of any imaging study. Tumor diameters were measured using a caliper. Tumor volume V was determined according to Tomayko and Reynolds (30), assuming a triaxial ellipsoid with distinct axes a, b, and c (V = (π/6)×abc. When tumor size reached a diameter of 1.5 cm, animals were killed using CO2 inhalation and cervical dislocation.
Fluorescence imaging
All imaging studies were performed using the In-Vivo Xtreme System (BRUKER) equipped with a 400W xenon illuminator and a back-illuminated 4 MP CCD detector. For quantification of mCherry in vitro, cells were serially diluted from 2 × 106 to 6.25 × 104 cells in PBS in a 96-well plate and imaged for 1–5 seconds with a 550-nm excitation band-pass filter and a 600-nm emission long-pass filter. Fluorescence microscopy of paraformaldehyde-fixed cells was performed using a laser-scanning microscope IX83 (OLYMPUS). Nuclei were stained with Hoechst 33258 (1:10 000; stock solution: 1 mg mL−1 in dimethylsulfoxide).
For detection of mCherry in vivo, continuously anesthetized animals were imaged for 1–5 seconds with a 600-nm excitation band-pass filter and a 700-nm emission long pass filter. All images were analyzed using MI 7.1.1 software (Bruker). Fluorescence intensity (total photon counts) was determined as the sum of the background-subtracted pixel values within a region of interest of constant size and shape for each animal. Local background was defined as the median of each region of interest perimeter intensity.
Determination of urinary free monoamines
Each animal was allowed to roam in an empty, clean, conventional cage. After spontaneous urination, the animal was removed and the voided urine was aspirated with a pipet tip, avoiding contamination with feces. Samples were aliquoted and stored at −66°C. Urinary free metanephrines and catecholamines were determined simultaneously by liquid chromatography tandem mass spectrometry (LC-MS/MS), as described elsewhere (26). Urinary creatinine excretion was determined using the picric acid method of Jaffe in a DxC 800 analytical system (Beckman Coulter) and was used for volume corrections.
Blood pressure measurement
Blood pressure measurements were performed using the noninvasive CODA 2 system for mice and rats (Emka Technologies). Awake animals were allowed to acclimatize to the holder for 10 minutes. Blood pressure was determined by averaging at least five consecutive measurements in a 20-minute session.
Statistical analysis
Statistical analysis was performed using GraphPad Prism version 5.02 for Windows (Graphpad Software, www.graphpad.com). Experimental groups consisted of randomly selected animals; n represents the number of animals or cell culture dishes investigated. Data are presented as arithmetic means ± SEM (for n < 10) or mean ± SD (for n ≥ 10). In vitro growth data were fitted with the logistic equation:
In vivo tumor growth data were fitted with the exponential equation:
Comparison of fits was done using the Extra sum-of-squares F-test. Significance of differences was tested by analysis of variance using Sidak's post hoc test. Differences were considered significant at values of P < .05. Significance of relationships was analyzed using Spearman's linear correlation test and displayed as Spearman's correlation coefficient, rs.
Results
Generation of a stably transduced mCherry-expressing MPC cell line
In this study, consecutive expression of mCherry in MPC cells was used for observing tumor progression in a murine sc allograft model of PHEO. For stable genetic modification of MPC cells, we generated the lentiviral transfer vector p6NST70-MC harboring an mCherry expression cassette driven by the elongation factor 1 alpha promoter. We named this new cell line MPC-mCherry.
As demonstrated by fluorescence microscopy, mCherry was located throughout the cytoplasm and around the nucleus of MPC-mCherry cells (Figure 1B). To examine other effects of the genetic modification, we compared the biological properties of MPC-mCherry cells with the nontransfected parental MPC cells in vitro. Both cell lines were found to exhibit identical cell morphology (Figure 1, A and B). Logistic growth characteristics (Figure 1 C) did not differ significantly (P = .551, F = 0.705 [3;90]) between MPC cells (ymax = 71.3, y0 = 1.65, k = 0.457) and MPC-mCherry cells (ymax = 65.0, y0 = 1.21, k = 0.520). mCherry expression was quantified by measuring the fluorescence intensity of serially diluted MPC-mCherry cells at excitation/emission wavelengths of 550/600 nm (Figure 1E). We observed a proportional relation (y = 9.93 × 10−4x; R2 = 0.999) of cell number and fluorescence intensity (rs = 1; P = .003) confirming a stable genomic integration of the mCherry expression cassette.
Figure 1.
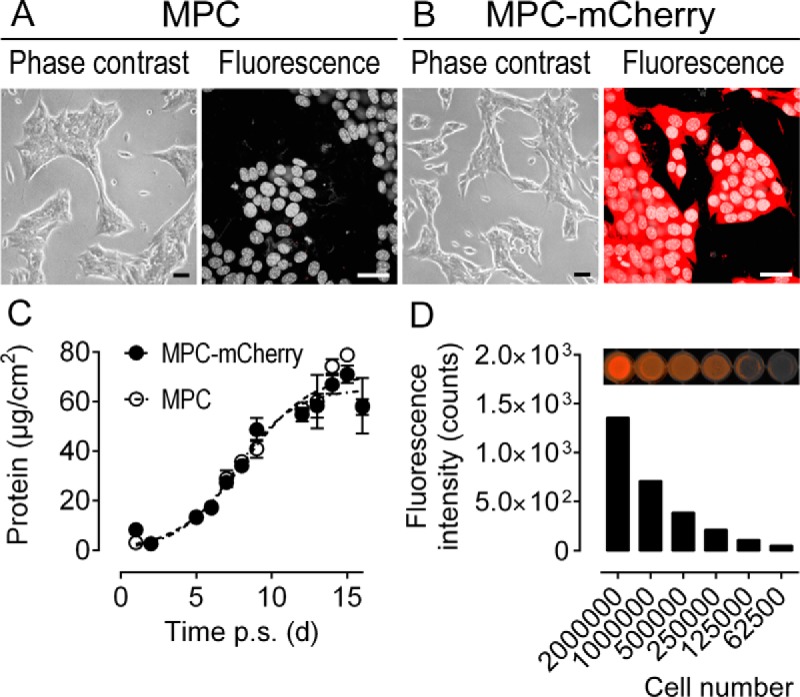
Biological properties of MPC-mCherry cells compared with nontransduced parent MPC cells in vitro; A and B, Morphological appearance and fluorescence under microscopy; nuclei stained with Hoechst 33258; scale bars, 30 μm. C, Growth of 2 × 105 MPC and MPC-mCherry cells during 16 days post subculture quantified as protein of adherent cells per cm2, presented as means ± SEM, n = 4. D, Positive proportional relation of MPC-mCherry cell number and corresponding fluorescence intensities at λEx/Em = 550/600 nm.
Fluorescence imaging and progression of MPC-mCherry-derived tumors
To study tumor progression in vivo, we injected 2 × 106 MPC-mCherry cells subcutaneously into the shoulders of nude mice with rudimentary thymus (NMRI nu/nu). To support the engraftment, we used Matrigel in PBS as the injection vehicle. Tumor progression was monitored at regular intervals over 4 weeks. Tumor fluorescence intensity was measured by performing whole-body FLI at excitation/emission wavelengths of 600/700 nm allowing for deep tissue penetration at the optimal signal-to-background ratio, as determined by multispectral analysis (data not shown). As a reference parameter, tumor volume was determined with calipers (range from 2–463 mm3). To exclude any possible sex effects on tumor progression, male and female animals were monitored specifically using the identical experimental setup.
Progression of the sc tumors is shown in Figure 2, A–C. During the first 7 days after cell injection, the initial fluorescence intensity at the injection site decreased, probably due to cell death or dispersal. The remaining MPC-mCherry cells gave rise to a solid sc tumor, which did not metastasize during the period of observation. Monitoring tumor progression by FLI (Figure 2B) revealed that the exponential tumor growth characteristics between male animals (y0 = 365083, k = 0.132, R2 = 0.604) and female animals (y0 = 756 451, k = 0.110, R2 = 0.544) did not differ significantly (P = .165, F = 1.825 [2;133]). Monitoring tumor progression using volume measurement by caliper also showed that the exponential tumor growth characteristics between male animals (y0 = 2.66, k = 0.169, R2 = 0.728) and female animals (y0 = 6.41; k = 0.137; R2 = 0.795) did not differ significantly (P = .249, F = 1.405 [2;132]). Analysis of covariance between tumor volume (mm3) and tumor fluorescence intensity (total photon counts) revealed a significant positive correlation (rs = 0.946, P < .001). Taken together, whole-body FLI enabled us to noninvasively monitor the tumor cells in nonmetastatic MPC-mCherry cell-derived sc tumors in vivo.
Figure 2.

Progression of sc tumors in male and female nude mice after sc injection of 2 × 106 MPC-mCherry cells. A, Overlays of x-ray and fluorescence images (λEx/Em = 600/700 nm) of a representative animal at 1 h to 29 d post injection. B, Tumor fluorescence intensities in vivo over time. C, Tumor volumes over time; data are presented as mean ± SD, (male animals, n = 10; female animals, n = 10). D, Correlation analysis between tumor volume and tumor fluorescence intensity; n = 20; confidence interval, 95%; number of XY pairs, 136; p.i., post injection; px, pixel.
Physiologic parameters during progression of MPC-mCherry cell-derived tumors
To investigate whether urinary free monoamines reflect the total tumor burden in mice bearing MPC-mCherry definable tumors, we regularly measured concentrations of urinary free catecholamines and their corresponding metanephrines by LC-MS/MS. In addition, BP was monitored at regular intervals. To examine any possible sex effects, we systematically compared male and female animals separately.
In all animals, we found steadily increasing urinary concentrations of the several monoamines during 27 days of tumor progression compared with basal concentrations (−8 and −2 days) before cell injection (Figure 3, A–E). In this effect, we did not observe any significant differences between male and female animals. We observed the earliest alterations in urinary monoamine concentrations at 6 days after cell injections. The animals showed a 1.7-fold increase in dopamine (DA) from 0.68–1.18 μmol/mmol creatinine and a 2.1-fold increase in its corresponding metabolite 3-methoxytyramine (MTY) from 0.35–0.76 μmol/mmol creatinine (Figure 4). In contrast, during this time period, norepinephrine (NE) decreased from 0.60–0.30 μmol/mmol creatinine and its corresponding metabolite normetanephrine (NMN) decreased from 1.20–0.70 μmol/mmol creatinine. At this time the average tumor volume was approximately 12.3 mm3 (tumor diameter of approximately 2.3 mm) and the fluorescence intensity at the cell injection site reached the minimum level for detection (Figure 2, A and B).
Figure 3.

Physiologic parameters during tumor progression in male and female nude mice after sc injection of 2 × 106 MPC-mCherry cells. A–D, Urinary concentration of free monoamines −10 and −2 d before, and 6, 13, 20, and 27 d after cell injection; presented as mean ± SD (male animals, n = 10, female animals, n = 10). E, Changes in the urinary concentration pattern of free monoamines (net chart) presented as mean (male + female animals, n = 20). F, Systolic BP −10 and −4 d before and 3, 10, 17, 24, 31 d after cell injection presented as mean (male animals, n = 10; female animals, n = 10) and mean ± SD (male + female animals, n = 20); DA, dopamine; MTY, 3-methoxytyramine; NE, norepinephrine; NMN, normetanephrine; EPI, epinephrine; MN, metanephrine; p.i., post injection. **, P < 0.01; ***, P < 0.001.
Figure 4.
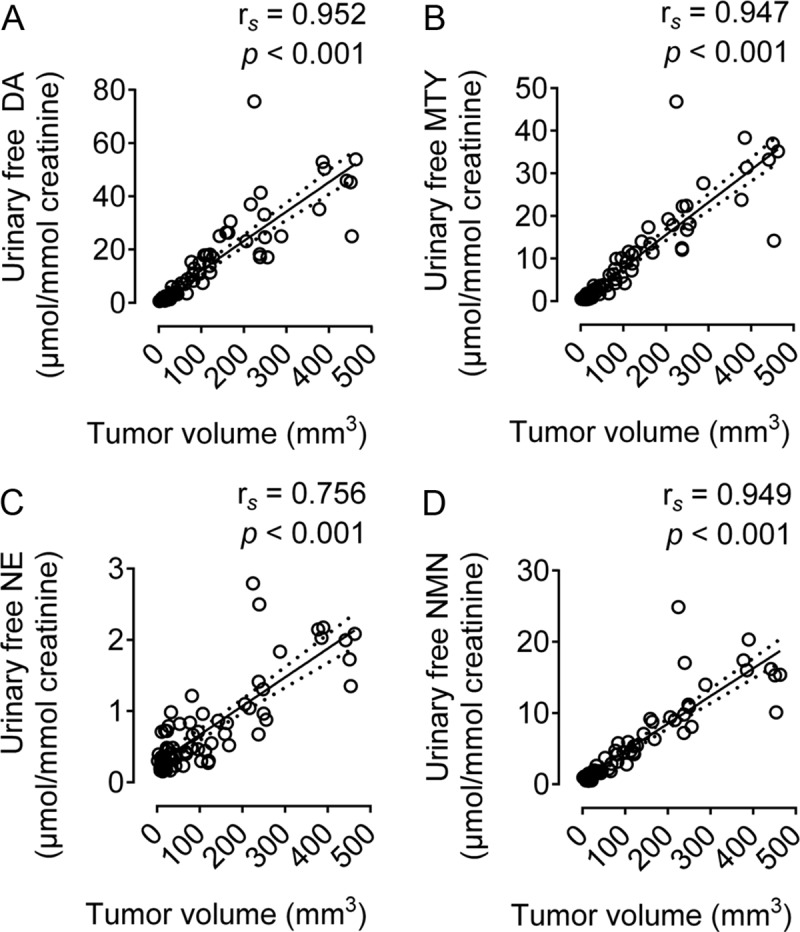
Analysis of covariance between urinary free monoamines and tumor volume. DA, dopamine; MTY, 3-methoxytyramine; NE, norepinephrine; EPI, epinephrine; NMN, normetanephrine. n = 20, confidence interval, 95%, number of XY pairs, 78.
Within 27 days, we observed dramatic increases in urinary monoamine outputs of DA, MTY, and NMN. The animals showed a significant 50-fold increase in DA from 0.68–35.18 μmol/mmol creatinine, a significant 70-fold increase in MTY from 0.35–24.26 μmol/mmol creatinine, a nonsignificant 3-fold increase in NE from 0.55–1.56 μmol/mmol creatinine, and a significant 11-fold increase in NMN from 1.21–13.03 μmol/mmol creatinine. Throughout the experiment, epinephrine (EPI) and metanephrine (MN) remained at basal levels of 0.15–0.30 μmol/mmol creatinine, respectively (Figure 3E). Tumor volume (mm3) showed significant positive correlations with urinary outputs of DA (rs = 0.952, P < .001), MTY (rs = 0.947, P < .001), NE (rs = 0.756, P < .001), and NMN (rs = 0.949, P < .001).
In addition to the altered catecholamine pattern, we observed increasing systolic BP (Figure 3F). With this effect, we did not observe any significant differences between male and female animals. Changes in systolic BP were found to be significant (P < 0.01) after 24 days' post-cell injections among a mixed population of both male (n = 10) and female (n = 10) animals.
In conclusion, MPC-mCherry cell-derived sc tumors altered the physiologic catecholamine pattern and BP in nude mice. Among all monoamines investigated, urinary concentrations of free DA, MTY, NE, and NMN allowed for biochemical monitoring of tumor progression in vivo.
Discussion
Our study characterized a sophisticated animal model developed for evaluating novel therapeutic strategies for the treatment of PHEO. We employed the murine MPC cell line for developing an allograft mouse model of PHEO. MPC cells are biochemically and molecularly similar to human PHEOs, and are a suitable tool for testing new therapies for PHEO (18). However, this model may not be suitable for investigating metastatic spread, generally arising from a primary tumor. However, in nude mice, iv and ip, but potentially not sc injections of MPC cells, can be used to generate multiple organ lesions (31). The difficulty thereafter is to monitor disease progression. We have therefore first employed MPC cells to develop an sc nonmetastatic mouse model of PHEO in which the location of one single lesion is known and its size and morphology can easily be determined and matched to other parameters such as fluorescence intensity and urinary monoamine concentrations to validate their use for monitoring disease progression.
In our study, the use of Matrigel increased the reproducibility of tumor cell engraftment as well as the homogeneity in tumor shapes among the animals and, thereby improved tumor volume measurements by caliper. Precise and complete determination of tumor volume was a prerequisite for studying intensities of tumor fluorescence and concentrations of urinary free monoamines as surrogate markers of disease progression in this murine model of PHEO.
To monitor tumor progression using FLI, we generated an mCherry-expressing MPC cell line by lentiviral transduction, named MPC-mCherry. Genetic manipulation neither altered the morphology nor the growth characteristics compared with the parental MPC cell line. Genetic properties of this particular MPC-mCherry clone remain to be investigated. MPC-mCherry cells consecutively express red fluorescent mCherry protein driven by the elongation factor alpha promoter, enabling easy optical detection of the cells both in vitro and in vivo. To investigate whether the intensity of tumor fluorescence is a suitable parameter to monitor disease progression in nude mice, we performed whole-body FLI in the far-red portion of the spectrum, detecting only the tumor cells in MPC-mCherry cell-derived sc tumors in vivo. FLI allows for semiquantitative measurements of tumor progression, metastasis, and treatment response (32), and unlike other in vivo imaging strategies, it does not require the addition of a substrate, an imaging probe, or a radiotracer. Thus, FLI is preferred for multiple examinations at a minimum stress level for animals. Fluorescent proteins emitting in the far-red portion of the spectrum allow for biological interrogations at deep optical tissue penetration (33, 34). Among these proteins, mCherry, owing to its spectral class (excitation maximum, 587 nm; emission maximum, 610 nm), brightness, and superior photostability, is the best general purpose agent for whole-body optical imaging (35, 36). These optical properties allow for detection of especially small changes in number of tumor cells observed during the first 7 days after cell injection. However, these effects on the number of tumor cells may be due to cell death, diffusion, invasion of neutrophils, or newly built blood vessels and connective tissue. The strongly positive correlation between tumor size and fluorescence intensity suggests a minor influence of light-scattering artifacts when the total fluorescence intensity of the tumor is used as a quantitative parameter.
Unlike caliper measurements, FLI eliminates most contributions of the nontumor cell-derived volume, such as capsule, edema, necrotic areas, blood vessels, or immune cell infiltration, inside the heterogeneous structure of a solid tumor. Taken together, FLI of MPC-mCherry cell-derived tumors is considered to provide a powerful and rapid method for evaluating tumor progression during preclinical therapeutic investigations in nude mice.
In our study, we used nude mice to ensure maximal optical tissue penetration during whole-body FLI. In addition, the immunodeficiency of athymic mice will allow for extending the animal model to the engraftment of primary human PHEO cells or newly developed human PHEO cell lines in the future. Referring to the latter, a recent study reported about a human PHEO precursor cell line established form a primary human tumor (10). Our findings that sc MPC-mCherry cell-derived tumors did not metastasize in nude mice verify earlier studies (31). This outcome may be due to the fact that MPC cells were generated from adrenal tumors of BL-6 heterozygous neurofibromin-1 knockout mice, which generally exhibit a low metastatic potential (14). Despite their immunodeficiency, nude mice are capable of generating a nonspecific immune response against cells from different strains, for example by natural killer (NK) cell activation (37). This effect may also contribute to the initial tumor cell death at the injection site observed during the first 7 days after injection.
We also investigated whether tumor-induced alterations of physiologic parameters reflect the total tumor burden of PHEO in mice. For this, we monitored the concentrations of urinary free monoamines as well as BP at regular intervals during 4 weeks of tumor progression. Among all monoamines investigated, our analyses revealed that the most dramatic increases involved urinary levels of DA and its corresponding metabolite MTY. These results suggest that MPC-mCherry cell-derived sc tumors mainly produce DA. Most importantly, measuring urinary free DA and/or MTY allowed for the most sensitive biochemical detection of tumors at diameters below 2.3 mm in vivo, although possessing similar tumor detection limits compared with anatomic and functional small-animal imaging (magnetic resonance imaging, computed tomography, positron emission tomography, single-photon emission computed tomography) in vivo. Therefore, our investigations suggest that measuring urinary free DA and/or MTY allows for easy, noninvasive quantification of catecholamine-producing lesions.
In contrast with the increase in urinary DA, we found relatively small increases in urinary NE but larger increases in its corresponding metabolite, NMN. These findings suggest that much of the NE produced within tumors is metabolized to NMN. We did not observe tumor-induced increases in urinary free EPI or its corresponding metabolite, MN. Although MPC cells are known to express phenyl-N-methyltransferase, they also only produce small amounts of EPI from NE (14), explaining why we did not observe significant increases in urinary EPI and MN. However, adaptation of peripheral catecholamine metabolism, such as in the liver, cannot be excluded as a factor influencing the observed metabolite pattern.
Systolic BP was found to be significantly increased during tumor growth, demonstrating the physiologic influence of tumor-related NE release. Nevertheless, increases in BP were not severe, although reflecting the pattern of increases in urinary outputs of catecholamines (38).
In summary, we demonstrated that the MPC-mCherry cell-derived sc allograft mouse model exhibits functional similarities to the endocrine situation in human PHEO, characterized by increased urinary outputs of catecholamines and BP. In particular, measurement of tumor progression by whole-body FLI and urinary free DA and/or MTY by LC-MS/MS allows for noninvasive quantification of tumor progression. The introduced sc PHEO model provides a useful tool for comprehensively evaluating therapeutic strategies for PHEO, but may not be a predictor of organ metastatic lesion responses to treatment. However, FLI and measurements of urinary free monoamines may offer perspectives to substantially facilitate comprehensive preclinical diagnostic and therapeutic evaluations also in other mouse models, which present tumors in the liver, lung, kidneys, bones, and muscles. Diagnosis and monitoring of tumor progression and therapy response in human patients with metastatic PHEO may also benefit from this strategy.
Acknowledgments
This work was supported by The Deutsche Forschungsgemeinschaft (Grants ZI-1362/2–1 [to C.G.Z. and G.E.] and BE-2607/1–1 [to R.B. and J.P.]). MPC 4/30PRR cells were kindly provided by Professor Arthur Tischler, Dr James Powers, and Professor Karel Pacak.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- BP
- blood pressure
- DA
- dopamine
- EGFP
- enhanced green fluorescent protein
- EPI
- epinephrine
- FLI
- fluorescence imaging
- LC-MS/MS MC
- liquid chromatography tandem mass spectrometry
- MN
- metanephrine
- MTY
- metabolite 3-methoxytyramine
- NE
- norepinephrine
- NK
- natural killer
- NMN
- normetanephrine
- NMRI nu/nu
- nude mice with rudimentary thymus
- PBS
- phosphate-buffered saline
- PHEO
- pheochromocytomas.
References
- 1. Harari A, Inabnet WB., 3rd Malignant pheochromocytoma: A review. Am J Surg. 2011; 201(5):700–708. [DOI] [PubMed] [Google Scholar]
- 2. Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet. 2005;366(9486):665–675. [DOI] [PubMed] [Google Scholar]
- 3. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. [DOI] [PubMed] [Google Scholar]
- 4. Elder EE, Elder G, Larsson C. Pheochromocytoma and functional paraganglioma syndrome: No longer the 10% tumor. J Surg Oncol. 2005;89(3):193–201. [DOI] [PubMed] [Google Scholar]
- 5. Harding JL, Yeh MW, Robinson BG, Delbridge LW, Sidhu SB. Potential pitfalls in the diagnosis of phaeochromocytoma. Med J Aust. 2005;182(12):637–640. [PubMed] [Google Scholar]
- 6. Kimura N, Takayanagi R, Takizawa N, et al. Pathological grading for predicting metastasis in phaeochromocytoma and paraganglioma. Endocr Relat Cancer. 2014;21(3):405–414. [DOI] [PubMed] [Google Scholar]
- 7. Lee TK, Murthy SR, Cawley NX, et al. An N-terminal truncated carboxypeptidase E splice isoform induces tumor growth and is a biomarker for predicting future metastasis in human cancers. J Clin Invest. 2011;121(3):880–892. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8. Zinnamosca L, Petramala L, Cotesta D, et al. Neurofibromatosis type 1 (NF1) and pheochromocytoma: Prevalence, clinical and cardiovascular aspects. Arch Dermatol Res. 2011;303(5):317–325. [DOI] [PubMed] [Google Scholar]
- 9. Pacak K, Fojo T, Goldstein DS, et al. Radiofrequency ablation: A novel approach for treatment of metastatic pheochromocytoma. J Natl Cancer Inst. 2001;93(8):648–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ghayee HK, Bhagwandin VJ, Stastny V, et al. Progenitor cell line (hPheo1) derived from a human pheochromocytoma tumor. PLoS One. 2013;8(6):e65624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fritz A, Walch A, Piotrowska K, et al. Recessive transmission of a multiple endocrine neoplasia syndrome in the rat. Cancer Res. 2002;62(11):3048–3051. [PubMed] [Google Scholar]
- 12. Molatore S, Liyanarachchi S, Irmler M, et al. Pheochromocytoma in rats with multiple endocrine neoplasia (MENX) shares gene expression patterns with human pheochromocytoma. Proc Natl Acad Sci USA. 2010;107(43):18493–18498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jacks T, Shih TS, Schmitt EM, Bronson RT, Bernards A, Weinberg RA. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat Genet. 1994;7(3):353–361. [DOI] [PubMed] [Google Scholar]
- 14. Powers JF, Evinger MJ, Tsokas P, Bedri S, Alroy J, Shahsavari M, Tischler AS. Pheochromocytoma cell lines from heterozygous neurofibromatosis knockout mice. Cell Tissue Res. 2000;302(3):309–320. [DOI] [PubMed] [Google Scholar]
- 15. Giubellino A, Woldemichael GM, Sourbier C, et al. Characterization of two mouse models of metastatic pheochromocytoma using bioluminescence imaging. Cancer Lett. 2012;316(1):46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martiniova L, Lai EW, Elkahloun AG, et al. Characterization of an animal model of aggressive metastatic pheochromocytoma linked to a specific gene signature. Clin Exp Metastasis. 2009;26(3):239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ohta S, Lai EW, Morris JC, et al. Metastasis-associated gene expression profile of liver and subcutaneous lesions derived from mouse pheochromocytoma cells. Mol Carcinog. 2008;47(4):245–251. [DOI] [PubMed] [Google Scholar]
- 18. Korpershoek E, Pacak K, Martiniova L. Murine models and cell lines for the investigation of pheochromocytoma: Applications for future therapies? Endocr Pathol. 2012;23(1):43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ziegler CG, Ullrich M, Schally AV, et al. Anti-tumor effects of peptide analogs targeting neuropeptide hormone receptors on mouse pheochromocytoma cells. Mol Cell Endocrinol. 2013;371(1–2):189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Powers JF, Schelling K, Brachold JM, et al. High-level expression of receptor tyrosine kinase Ret and responsiveness to Ret-activating ligands in pheochromocytoma cell lines from neurofibromatosis knockout mice. Mol Cell Neurosci. 2002;20(3):382–389. [DOI] [PubMed] [Google Scholar]
- 21. Cascón A, Robledo M. MAX and MYC: A heritable breakup. Cancer Res. 2012;72(13):3119–3124. [DOI] [PubMed] [Google Scholar]
- 22. Ziegler CG, Brown JW, Schally AV, et al. Expression of neuropeptide hormone receptors in human adrenal tumors and cell lines: Antiproliferative effects of peptide analogues. Proc Natl Acad Sci USA. 2009;106(37):15879–15884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Denorme M, Yon L, Roux C, Gonzalez BJ, Baudin E, Anouar Y, Dubessy C. Both sunitinib and sorafenib are effective treatments for pheochromocytoma in a xenograft model. Cancer Lett. 2014. [DOI] [PubMed] [Google Scholar]
- 24. Martiniova L, Kotys MS, Thomasson D, et al. Noninvasive monitoring of a murine model of metastatic pheochromocytoma: A comparison of contrast-enhanced microCT and nonenhanced MRI. J Magn Reson Imaging. 2009;29(3):685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Martiniova L, Ohta S, Guion P, et al. Anatomical and functional imaging of tumors in animal models: Focus on pheochromocytoma. Ann NY Acad Sci. 2006;1073:392–404. [DOI] [PubMed] [Google Scholar]
- 26. Peitzsch M, Pelzel D, Glöckner S, et al. Simultaneous liquid chromatography tandem mass spectrometric determination of urinary free metanephrines and catecholamines, with comparisons of free and deconjugated metabolites. Clin Chim Acta. 2013;418:50–58. [DOI] [PubMed] [Google Scholar]
- 27. Ho YP, Schnabel V, Swiersy A, Stirnnagel K, Lindemann D. A small-molecule-controlled system for efficient pseudotyping of prototype foamy virus vectors. Mol Ther. 2012;20(6):1167–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stirnnagel K, Luftenegger D, Stange A, et al. Analysis of prototype foamy virus particle-host cell interaction with autofluorescent retroviral particles. Retrovirology. 2010;7:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Morgenroth A, Cartellieri M, Schmitz M, et al. Targeting of tumor cells expressing the prostate stem cell antigen (PSCA) using genetically engineered T-cells. Prostate. 2007;67(10):1121–1131. [DOI] [PubMed] [Google Scholar]
- 30. Tomayko MM, Reynolds CP. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol. 1989;24(3):148–154. [DOI] [PubMed] [Google Scholar]
- 31. Ohta S, Lai EW, Taniguchi S, Tischler AS, Alesci S, Pacak K. Animal models of pheochromocytoma including NIH initial experience. Ann NY Acad Sci. 2006;1073:300–305. [DOI] [PubMed] [Google Scholar]
- 32. Henriquez NV, van Overveld PG, Que I, Buijs JT, et al. Advances in optical imaging and novel model systems for cancer metastasis research. Clin Exp Metastasis. 2007;24(8):699–705. [DOI] [PubMed] [Google Scholar]
- 33. Deliolanis NC, Kasmieh R, Wurdinger T, Tannous BA, Shah K, Ntziachristos V. Performance of the red-shifted fluorescent proteins in deep-tissue molecular imaging applications. J Biomed Opt. 2008;13(4):044008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Licha K. Contrast Agents for Optical Imaging. Top Curr Chem. 2002;222:1–29. [Google Scholar]
- 35. Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat Methods. 2005;2(12):905–909. [DOI] [PubMed] [Google Scholar]
- 36. Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22(12):1567–1572. [DOI] [PubMed] [Google Scholar]
- 37. Budzynski W, Radzikowski C. Cytotoxic cells in immunodeficient athymic mice. Immunopharmacol Immunotoxicol. 1994;16(3):319–346. [DOI] [PubMed] [Google Scholar]
- 38. Jose PA, Eisner GM, Felder RA. Regulation of blood pressure by dopamine receptors. Nephron Physiol. 2003;95(2):19–27. [DOI] [PubMed] [Google Scholar]