

Abstract
Background
Polydactyly is a highly common congenital limb defect. Extra digits may appear as an isolated anomaly or as a part of a syndrome. Mutations in GLI3 have been shown to cause Greig cephalopolysyndactyly, Pallister-Hall syndrome and non-syndromic polydactyly. Genotype-phenotype correlation studies of GLI3 mutations suggest a model by which mutations in the zinc-finger domain (ZFD) of GLI3 likely lead to syndromic polydactyly. Here we describe a rare case of autosomal dominant heterozygous missense mutation in the ZFD of GLI3 leading to a variable polydactyly-syndactyly complex.
Case presentation
A large Jewish Moroccan family presented with apparently autosomal dominant heredity of bilateral thumb polydactyly in hands and feet combined with post-axial polydactyly type B or type A. Syndactyly was evident in most patients’ hands and feet. Apart from head circumference beyond 90th percentile in some of the affected individuals, none had craniofacial dysmorphism. A novel GLI3 c.1802A > G (p.His601Arg) mutation was found in all affected individuals.
Conclusion
We demonstrate that a mutation in the ZFD domain of GLI3 leads to phenotypic variability, including an isolated limb phenotype. Thus, the variability in phenotypes caused by mutations in this master developmental regulator is more profound than has been previously suggested.
Electronic supplementary material
The online version of this article (doi:10.1186/s12881-014-0110-9) contains supplementary material, which is available to authorized users.
Keywords: Polydactyly, Syndactyly, GCPS, GLI3, Mutation, C2H2, Zinc finger
Background
Polydactyly, one of the most common congenital hand/foot malformations encountered in clinical genetics [1,2], can occur as an isolated entity or as part of pleiotropic developmental anomaly syndromes [3]. Post-axial polydactyly is far more common than pre-axial and central polydactyly; occasionally, concomitant syndactyly is seen with some forms of polydactyly [2]. The GLI3 protein is a zinc finger transcription factor expressed early in development [4]. It is required for the specification of dorsal cell types and for suppression of ventral cell types in the forebrain [5]. GLI3 expression in the anterior half of the limb buds appears to play an important role in suppressing digit formation [5]. These observations, together with the diverse phenotypes resulting from molecular defects in GLI3, serve as a classical example of pleiotropy: GLI3 mutations are known to cause different clinical entities: Greig Cephalopolysyndactyly Syndrome (GCPS) (MIM ID #175700), Pallister-Hall Syndrome (MIM ID #146510), Acrocallosal Syndrome (MIM ID #200990), Pre-axial Polydactyly type IV (MIM ID #174700) and Post-axial Polydactyly type A (MIM ID #174200). The great majority of known GLI3 mutations cause loss of function [4]. Some reports describe GLI3 mutations as a cause of isolated polydactyly [6], while others consider all GLI3 mutations as causing polydacyly within the GCPS spectrum [7]. It has been suggested that mutations in different domains of the gene underlie the different evolving phenotypes, and that mutations 5′ to or within the zinc finger domain (ZFD) of GLI3 specifically cause GCPS [6,8]. The classic clinical presentation of GCPS is a triad of polysyndactyly, macrocephaly and hypertelorism [9]. Here we report a new heterozygous missense mutation in the zinc finger domain of GLI3 leading to a variable phenotype presenting in some cases as an isolated polydactyly-syndactyly complex.
Case presentation
A large Jewish Moroccan kindred presented with apparently autosomal dominant heredity of polydactyly (Figure 1A). Affected and unaffected family members underwent thorough clinical and molecular evaluation following Soroka Medical Center IRB approval and informed consent. Phenotypic variability among the 14 affected individuals was evident: most had hands and feet bilateral thumb polydactyly and post-axial polydactyly type B. Syndactyly was found in the feet of all affected individuals and in the hands of most (Figure 1D,E). A single case of thumb polydactyly combined with post-axial polydactyly type A (well developed separated digit) was evident (Figure 1A, IV:1; Figure 1B,C). None of the affected individuals had apparent craniofacial dysmorphism. Occipitofrontal head circumference (OFC) and interpupillary distance (IPD) were measured in 13 (8 affected and 5 unaffected) family members. The measurements were most variable (see Additional file 1). Five patients had an OFC at or above the 90th percentile, while all the healthy individuals showed OFC beneath 90th percentile. As to IPD measurements, 5 of 8 affected as well as 4 of 5 unaffected individuals were above 97th percentile.
Figure 1.
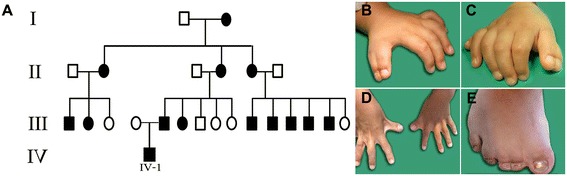
The clinical phenotype. A. Family tree of the kindred studied. B,C. Pre-axial polydactyly combined with post-axial polydactyly (individual IV:1). D,E. Various hands/feet syndactyly phenotypes of affected individuals (each photograph is of a different affected individual).
Methods and results
Blood samples were obtained from 13 affected and 6 unaffected family members and genomic DNA was extracted by routine techniques. Linkage to genes known to be associated with non-syndromic polydactyly was tested using 2 polymorphic markers flanking each candidate gene. Association of the phenotype with HOXD13, ZPA regulatory sequence (ZRS) and FBLN1 was ruled out (data not shown). Polymorphic markers D7S1526 and D7S691 flanking GLI3 identified a heterozygous haplotype shared by all affected family members (data not shown). Sanger sequencing of all GLI3 exons and their flanking exon-intron boundaries and comparison (NCBI BLAST) to the published GLI3 sequence (GenBank reference number NM_000168.5), identified a single heterozygous missense mutation in exon 12: c.1802A > G, p.His601Arg (Figure 2A,B). This novel mutation was found to segregate within the kindred as expected, demonstrating full penetrance of the phenotype.
Figure 2.
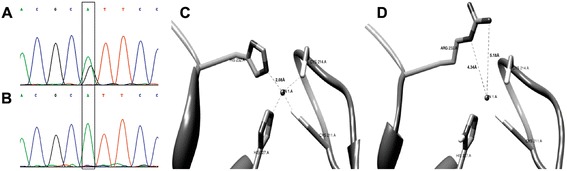
The GLI3 mutation and structural modeling of the predicted mutant protein: A,B. Chromatograms illustrating the GLI3 c.1802A > G heterozygous mutation (A = Affected individual; B = Unaffected individual). C,D. Structural modeling of the predicted wild type (C) and mutant p.His601Arg (D) proteins. Four C2H2 comprising residues are shown. Fragmented lines indicate the coordinative bonds between C2H2 residues and the zinc (marked as small spherical shape in the center). Note that in the wild type protein (C) the bond distance between the histidine in the upper left side and the metal ion is 2.08 Å, while the shortest bond possible between the zinc and the arginine residue in the predicted p.His601Arg mutant protein is 4.34 Å (D).
The GLI3 ZFD consists of 5 C2H2 type zinc fingers. The histidine to arginine substitution caused by the mutation is of the second histidine within one of the C2H2 zinc fingers comprising this domain. In zinc finger domains, a zinc ion forms four coordinate bonds generating a stable fold [10]. Since the GLI3 601His is one of the four residues directly interacting with the Zn2+ ion in a C2H2 zinc finger motif, the p.His601Arg substitution is likely to have functional consequences. Using crystallographic structure of the consensus motif of C2H2 type zinc finger (PDB:3IUF), we used UCSF Chimera software [11] to construct a simplified model illustrating the change in interactions with the Zinc cation. As shown in Figure 2, the mutation is predicted to cause destabilization of the Zn2+ ion, as the stable coordinative bond of histidine 232 (comparable to GLI3 histidine 601) with the metal ion (Figure 2C) is replaced in the mutated protein by a much longer unrealized bond with an arginine residue (Figure 2D). We speculate that the p.His601Arg mutation might alter the ability of GLI3 to bind its downstream targets within the SHH pathway, such as the posterior Hoxd genes [12].
Conclusions
In the kindred we describe, the OFC and IPD findings possibly stem from intrafamilial variability. The craniofacial manifestations of GCPS are highly variable and not all patients with GCPS have obvious macrocephaly [9] as observed in the family described here. In fact, the mild end of the GCPS spectrum is a continuum with isolated polydactyly [7]. Thus, in some cases there is unclear clinical delineation of GCPS versus non-syndromic polydactyly. The effects of truncating mutations (leading to loss of functional regions) on digit number and identity were largely discussed in the context of GLI3′s role as mediator of the hedgehog pathway, whereas connection between defect in the DNA binding domain (DBD) and the phenotype of our pedigree is more elusive. Besides the posterior Hoxd genes, other factors known to be downstream of GLI3 are FGF8, FGF4, GREM1, HAND2 and JAG1 [12]. It is plausible that changes in expression patterns of these GLI3 targets (as a result of an obstructed DBD) can contribute to the polydactyly phenotype. Both the precise mechanism through which the specific mutation leads to the disease phenotype, and the molecular mechanisms underlying the familial phenotypic variability are yet to be elucidated.
In summary, we describe a large kindred with a novel heterozygous GLI3 ZFD domain missense mutation leading to polydactyly-syndactyly complex. The phenotype described is within the wide range of phenotypic spectrum of GCPS established by Biesecker [13]. With recent emerging evidence of GLI3 mutations causing non-syndromic limb defects [14,15], our data highlight the fact that a GLI3 mutation within the ZFD domain can cause a mild form of GCPS with no prominent facial dysmorphism. This report strengthens the need to screen for GLI3 mutations in patients with polydactyly-syndactyly phenotype, even when syndromic features are not evident.
Consent
Written informed consent was obtained from the patients for publication of this Case report and any accompanying images.
Acknowledgements
This research was supported by the Israel Science Foundation (grant No. 1689/12). We deeply thank the Morris Kahn family foundation for the generous support of this study, and the patients and their families for kind participation. We would also like to acknowledge Dotan Amar for his assistance in drawing some of the figures. Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR001081).
Additional file
Clinical data. Gender, affected (+/−), age, OFC, IPD and malformations data of family members.
Footnotes
Michael Volodarsky and Yshaia Langer contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MV carried out the molecular genetic studies and drafted the manuscript. YL was involved in acquisition and analysis of clinical data and helped drafting the manuscript. OSB conceived and coordinated the study and took part in writing the manuscript. All authors read and approved the final manuscript.
Contributor Information
Michael Volodarsky, Email: volodarm@bgu.ac.il.
Yshaia Langer, Email: langeria@gmail.com.
Ohad S Birk, Email: obirk@bgu.ac.il.
References
- 1.Shubha RP, Sankar VH. Polydactyly and genes. Indian J Pediatr. 2010;77(3):277–281. doi: 10.1007/s12098-010-0033-1. [DOI] [PubMed] [Google Scholar]
- 2.Chong AK. Common congenital hand conditions. Singapore Med J. 2010;51(12):965–971. [PubMed] [Google Scholar]
- 3.Biesecker LG. Polydactyly: how many disorders and how many genes? Am J Med Genet. 2002;112(3):279–283. doi: 10.1002/ajmg.10779. [DOI] [PubMed] [Google Scholar]
- 4.Biesecker LG. What you can learn from one gene: GLI3. J Med Genet. 2006;43:465–469. doi: 10.1136/jmg.2004.029181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Motoyoma J. Essential roles of Gli3 and sonic hedgehog in pattern formation and developmental anomalies caused by their dysfunction. Congenit Anom (Kyoto) 2006;46:123–128. doi: 10.1111/j.1741-4520.2006.00114.x. [DOI] [PubMed] [Google Scholar]
- 6.Johnston JJ, Sapp JC, Turner JT, Amor D, Aftimos S, Aleck KA, Bocian M, Bodurtha JN, Cox GF, Curry CJ, Day R, Donnai D, Field M, Fujiwara I, Gabbett M, Gal M, Graham JM, Hedera P, Hennekam RC, Hersh JH, Hopkin RJ, Kayserili H, Kidd AM, Kimonis V, Lin AE, Lynch SA, Maisenbacher M, Mansour S, McGaughran J, Mehta L, et al. Molecular analysis expands the spectrum of phenotypes associated with GLI3 mutations. Hum Mutat. 2010;31:1142–1154. doi: 10.1002/humu.21328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balk K, Biesecker LG. The clinical atlas of Greig cephalopolysyndactyly syndrome. Am J Med Genet A. 2008;1;146A(5):548–557. doi: 10.1002/ajmg.a.32167. [DOI] [PubMed] [Google Scholar]
- 8.De’murger F, Ichkou A, Mougou-Zerelli S, Le Merrer M, Goudefroye G, Delezoide AL, Que’lin C, Manouvrier S, Baujat G, Fradin M, Pasquier L, Megarbane’ A, Faivre L, Baumann C, Nampoothiri S, Roume J, Isidor B, Lacombe D, Delrue MA, Mercier S, Philip N, Schaefer E, Holder M, Krause A, Laffargue F, Sinico M, Amram D, Andre’ G, Liquier A, Rossi M: New insights into genotype-phenotype correlation for GLI3 mutations.Eur J Hum Genet 2014. 10.1038/ejhg.2014.62.
- 9.Biesecker LG. The Greig cephalopolysyndactyly syndrome. Orphanet J Rare Dis. 2008;24(3):10. doi: 10.1186/1750-1172-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takafumi N, Makiko S, Kiyoshi A. Correlated mutation analysis of C2H2 zinc finger domains. Genome Inform. 2003;14:565–566. [Google Scholar]
- 11.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 12.Sheth R, Bastida MF, Ros M. Hoxd and Gli3 interactions modulate digit number in the amniote limb. Dev Biol. 2007;310(2):430–441. doi: 10.1016/j.ydbio.2007.07.023. [DOI] [PubMed] [Google Scholar]
- 13.Biesecker LG. GeneReviews™ (Internet) 2001. Greig Cephalopolysyndactyly Syndrome. [Google Scholar]
- 14.Wang Z, Wang J, Li Y, Geng J, Fu Q, Xu, Shen Y. Novel frame-shift mutations of GLI3 gene in non-syndromic postaxial polydactyly patients. Clin Chim Acta. 2014;10(433):195–199. doi: 10.1016/j.cca.2014.03.012. [DOI] [PubMed] [Google Scholar]
- 15.Sethi SK, Goyal D, Khalil S, Yadav DK. Two Indian families with Greig cephalosyndactyly with non-syndromic phenotype. Eur J Pediatr. 2013;172(8):1131–1135. doi: 10.1007/s00431-013-1938-2. [DOI] [PubMed] [Google Scholar]