

Abstract
Bacteria–phage coevolution, the reciprocal evolution between bacterial hosts and the phages that infect them, is an important driver of ecological and evolutionary processes in microbial communities. There is growing evidence from both laboratory and natural populations that coevolution can maintain phenotypic and genetic diversity, increase the rate of bacterial and phage evolution and divergence, affect community structure, and shape the evolution of ecologically relevant bacterial traits. Although the study of bacteria–phage coevolution is still in its infancy, with open questions regarding the specificity of the interaction, the gene networks of coevolving partners, and the relative importance of the coevolving interaction in complex communities and environments, there have recently been major advancements in the field. In this review, we sum up our current understanding of bacteria–phage coevolution both in the laboratory and in nature, discuss recent findings on both the coevolutionary process itself and the impact of coevolution on bacterial phenotype, diversity and interactions with other species (particularly their eukaryotic hosts), and outline future directions for the field.
Keywords: bacteriophage, resistance, host–parasite, antagonistic, species interaction, infection
Introduction
Bacteria–phage interactions are central to the ecology and evolution of microbial communities. Phages are known to alter competition among bacterial strains/species (e.g. Bohannan & Lenski, 2000a, b; Joo et al., 2006; Koskella et al., 2012), maintain bacterial diversity (e.g. Buckling & Rainey, 2002a, b; Rodriguez-Valera et al., 2009), and mediate horizontal gene transfer among bacteria (e.g. Kidambi et al., 1994; Canchaya et al., 2003). Phages have evolved a diversity of life histories and transmission strategies to exploit prokaryotic host cells for their own reproduction (Fig. 1); phages with a temperate lifestyle and filamentous phages are capable of forming long-term associations with bacterial cells, through lysogeny and pseudolysogeny, while phages with an exclusively lytic lifestyle are obligate killers of their host, requiring lysis to transmit to the next host cell (reviewed in Clokie et al., 2011; Abedon, 2012). Phages are abundant in natural ecosystems, often outnumbering coexisting bacteria (Thomas et al., 2011; Williamson et al., 2013; Engelhardt et al., 2014), and can impose significant mortality on their bacterial hosts (Faruque et al., 2005; Allen et al., 2010; Shapiro et al., 2010; Clokie et al., 2011). Bacteria can readily evolve resistance to phage attack by de novo mutation and have a diverse arsenal of other mechanisms with which to defend themselves against phage infections (Fig. 2). Given the abundance of phages and potential impact of phage-mediated selection on bacterial populations, there is growing interest among microbial ecologists to understand the coevolutionary processes underlying interactions between bacteria and lytic phages. The mechanisms of phage infectivity and bacterial resistance to phages have been reviewed extensively elsewhere (Nechaev & Severinov, 2008; Hyman & Abedon, 2010; Labrie et al., 2010; Westra et al., 2012; Molineux & Panja, 2013; Samson et al., 2013; Young, 2013), as has the potential impact of temperate phages on the fitness and phenotype of their bacterial hosts, for example through horizontal transfer of functional genes (Brüssow et al., 2004; Stern & Sorek, 2011; Varani et al., 2013). Therefore, we focus this review on the feedback between microbial ecology and bacteria–phage coevolution.
Fig 1.
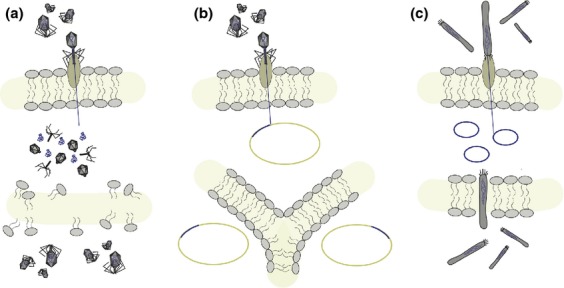
Diversity of phage reproductive strategies. (a) The ‘lytic’ phages replicate within their host cell and must burst the cell open to transmit to the next generation. These phages are therefore obligate killers of their hosts and are necessarily detrimental to host populations. (b) The ‘temperate’ phages (also referred to as ‘lysogenic’ phages or, once within the host genome, ‘prophages’) integrate into the host genome and reproduce along with the host cell. The integration of phage into the host genome can play a significant role in shaping bacterial phenotype and fitness (reviewed in Brüssow et al., 2004). (c) A relatively less common type of phage, the ‘filamentous’ phage, is able to reproduce without lysing the host cell and is continually secreted into the environment. These phages can also significantly alter bacterial phenotype, for example by encoding for toxins (Waldor & Mekalanos, 2012). Finally, the ‘cryptic prophages’ (not shown) are once temperate phages that have lost the ability to reproduce independently of their host (i.e. they can no longer enter the lytic cycle and transmit horizontally).
Fig 2.
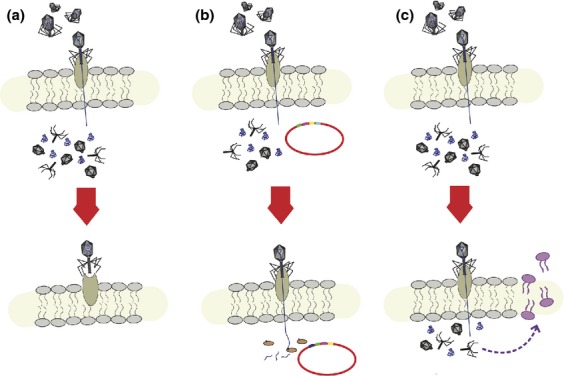
Illustration of bacterial resistance mechanisms against phages in the lytic cycle. There exist very few host-parasite systems for which the underlying mechanisms of infection and resistance are as well understood as bacteria–phage interactions, and yet new research continues to demonstrate the wide variety, complexity and sophistication of these coevolved systems (e.g. Høyland-Kroghsbo et al., 2013; Seed et al., 2013). Bacteria have evolved a number of defense mechanisms against invading phages, including those that (a) prevent phage adsorption, (b) degrade phage DNA inside the cell and/or block replication, and (c) initiate cell death upon infection. First, (a) bacteria can lose or alter the target receptor of phages, can produce an extracellular matrix of polysaccharides that blocks phage attachment, or can produce competitive inhibitors that bind to the phage attachment site. Phages can counter-adapt by altering their tail fiber attachment sites, by producing enzymes to degrade the matrix, or by changing the receptors to which they attach. (b) After successful phage attachment, bacteria can still prevent infection using a restriction-modification system, whereby recognized phage DNA is degraded by restriction enzymes, or through the CRISPR-Cas system, which has been identified in over 40% of bacteria and 90% of archaea (reviewed in Westra et al., 2012). (c) Finally, many bacterial species have mechanisms that lead to cell death, for example by degrading the cell wall, upon successful phage infection, thereby protecting neighboring cells from further infection. However, phages have been known to evolve mechanisms to evade even this level of defense by mimicking the host's defensive system (Blower et al., 2012).
Coevolution is defined as the process of reciprocal adaptation and counter-adaptation between ecologically interacting species (Janzen, 1980). Bacteria–phage systems have led the way as model systems for understanding mechanisms of infection and genome evolution, but have lagged dramatically behind other host-parasite systems in the integration of ecology and coevolution into a working understanding of the interaction. This is unfortunate given the insights gained from other systems to the role that coevolution plays in shaping genome evolution (Hosokawa et al., 2006; Kerstes et al., 2012), driving divergence among populations (Benkman, 1999), maintaining diversity within populations (Koskella & Lively, 2009; Morran et al., 2011), and even ecosystem-level processes (Stouffer & Bascompte, 2011). The evidence from bacteria–phage systems thus far suggests that populations are highly dynamic over time and rapidly (co)evolving. For example, a study following changes in the human gut virome over two and a half years demonstrates both high persistence of phage lineages through time and rapid substitution rates, particularly among those phages known to be lytic (Minot et al., 2013). Analysis of microbiota from 124 human hosts used the integration of phage DNA into the clustered regularly interspaced short palindromic repeats (CRISPR) regions of the bacterial genome to infer past selection pressure from phages and was able to demonstrate both a clear role for phages in shaping the microbiome and a potential reservoir of shared phages among individuals (Stern et al., 2012). Similar dynamical changes have been observed for bacterial and phage communities in the ocean, where associations seem to fluctuate over the course of days and yet are relatively stable over longer time scales (Needham et al., 2013). Indeed, it is now widely accepted that phages act to maintain microbial diversity at many levels and across ecosystems: within genomes (Banfield & Young, 2009; Avrani et al., 2011); within populations (Brockhurst et al., 2004; Benmayor et al., 2008); among populations (Buckling & Rainey, 2002a Morgan et al., 2005); and within communities (Bohannan & Lenski, 2000b; Brockhurst et al., 2006; Duerkop et al., 2012).
In this review, we summarize the empirical evidence for coevolution between bacteria and phages arising both from laboratory experimentation and from studies of natural communities. We report that (1) sustained coevolution is a common albeit not a universal outcome of copropagation of bacteria and phage and can be detected in natural bacteria–phage communities; (2) the dynamics and outcomes of bacteria–phage coevolution are often contingent upon environmental conditions in predictable ways; and (3) the process of coevolution can impact bacteria and phage diversity at all levels (from genomes to communities) and significantly affects bacterial phenotype. We focus primarily on interactions between bacteria and lytic phages, rather than temperate phages or interactions among archaea and their viruses (we refer readers to a previous review of archaea–virus interactions; Snyder & Young, 2011), because the former have proven much more amenable to laboratory testing than the latter and have therefore been the focus of the majority of experimental coevolution studies (for a general overview of this approach, see Brockhurst & Koskella, 2013). In addition, we use the terms ‘host’ and ‘parasite’ throughout the review to refer to an interaction from which one player benefits (the parasite) to the other's detriment (the host), for example via reduced fecundity or death. As obligate killers of their host cells, whereby they must burst the cell open to release infectious virons into the environment and infect future hosts, lytic phages can usefully be considered parasites (or parasitoids) for the sake of applying predictions from theory and comparing the systems to other antagonistic interactions.
Coevolution in the laboratory
Potentially endless cycles of defense and counter-defense
Early laboratory studies of bacteria–phage interactions, largely using Escherichia coli B as a host to one of various tailed-phages [Caudovirales, including the myoviruses T2 (Paynter & Bungay, 1969) & T4 (Horne, 1970), the podovirus T7 (Chao et al., 1977), and the siphoviruses, including T1 (Schrag & Mittler, 1996), T5 (Lenski & Levin, 1985), and λ-vir (Spanakis & Horne, 1987)], suggested that bacteria–phage coevolution was limited to between 0.5 and 1.5 cycles of reciprocal evolutionary change (where a cycle is the evolution of bacterial resistance followed by the evolution of phage infectivity, often termed a host-range mutant). In all cases, coevolution appeared to cease after the emergence of a resistant bacterial genotype that the phage could not evolve to overcome (reviewed in Dennehy, 2012). Resistance in these studies generally derived from de novo mutations causing modifications to the structure of bacterial surface molecules targeted by phages, which prevented phage attachment (Lenski & Levin, 1985). Similar results of constrained coevolution have been documented for cyanobacteria and cyanophages (Cowlishaw & Mrsa, 1975; Cannon et al., 1976; Barnet et al., 1981), Vibrio cholerae and vibriophages (Wei et al., 2010, 2011), and various other marine bacteria (Waterbury & Valois, 1993; Middelboe et al., 2001). Lenski and Levin concluded that bacteria–phage coevolution was fundamentally constrained by an asymmetry of evolutionary potential between bacteria and phages (Lenski & Levin, 1985). They argued that this asymmetry arises because many mutational routes to receptor modification or loss can achieve resistance, while phage infectivity requires the evolution of specific binding to the modified version of the existing receptor or to an entirely different receptor, which is likely to be subject to greater mutational constraint.
Despite the logic and likely generality of this mutational asymmetry, evidence from a range of other bacteria–phage interactions suggests that prolonged bouts of recurrent coevolutionary cycles can and indeed do occur in laboratory culture. The most intensively studied bacteria–phage interaction, in terms of the pattern and process of its coevolution, is that between Pseudomonas fluorescens SBW25 and the T7-like podovirus, Φ2 (Buckling & Rainey, 2002b; Brockhurst et al., 2007a). When propagated by batch culture in rich media, these species undergo persistent coevolution during long-term experiments (c. 450 bacterial generations; Buckling & Rainey, 2002a, b; Hall et al., 2011a). The coevolutionary interaction of P. fluorescens and Φ2 is comprised of two distinct phases (Hall et al., 2011b). During the first 200–250 bacterial generations, coevolution proceeds through a series of recurrent, reciprocal selective sweeps, whereby directional selection drives the fixation of new bacterial resistance mutations followed by new phage mutations restoring infectivity (Hall et al., 2011b). Consequently, bacteria and phages evolve to become, respectively, more broadly resistant (i.e. can resist a greater number of phage genotypes) and more broadly infectious (i.e. can infect a greater number of bacterial genotypes) over time (Buckling & Rainey, 2002a, b). This mode of coevolution is often termed an ‘arms race’ due to the escalation of defense and counter-defense traits by both species (Dawkins & Krebs, 1979; Woolhouse et al., 2002; Gandon et al., 2008). Qualitatively similar arms-race coevolution has also been observed in chemostat cultures of E. coli O157:H7 with the T4-like myovirus, PP01 (Mizoguchi et al., 2003), of Cellulophaga baltica and phi-S (Middelboe et al., 2009), and of the cyanobacterium Synechococcus with the myovirus, RIM8 (Marston et al., 2012).
After c. 250 bacterial generations, the rate of arms-race coevolution between P. fluorescens and Φ2 decelerates, such that both bacterial resistance range and phage infectivity range reach their asymptotes. This occurs due to progressively weaker responses to directional selection over time, due primarily to costs of generalism in both the bacteria and the phages (Hall et al., 2011b). In bacteria, resistance mutations altering cell-surface lipopolysaccharide molecules (Scanlan et al., 2013) are associated with high pleiotropic costs due to impaired function of these molecules, which reduces fitness (Brockhurst et al., 2004; Buckling et al., 2006). For the phages, mutations conferring broader infectivity range often occur in genes encoding host-attachment proteins (Paterson et al., 2010; Scanlan et al., 2011). The precise nature of the associated costs of generalism is poorly characterized but generalist phages appear to suffer impaired growth rates (Poullain et al., 2008). Thus, arms-race coevolution eventually gives way to sustained oscillations of bacterial and phage genotypes with different resistance and infectivity specificities, respectively (Hall et al., 2011b). These oscillations are driven by negative frequency-dependent selection. By this process, phages evolve to infect common bacterial genotypes, giving an advantage to rare bacterial resistance alleles, which rise in frequency, and so on, indefinitely. This mode of coevolution is termed fluctuating selection dynamics (Gandon et al., 2008).
Intriguingly, there is evidence of mutational asymmetry between P. fluorescens and phage Φ2 of the kind hypothesized by Lenski and Levin to impose a fundamental constraint upon bacteria–phage coevolution (Lenski & Levin, 1985). Specifically, whereas bacteria can readily evolve broad resistance ranges via single spontaneous mutations, phage evolution of broad infectivity ranges during coevolution seems to require the stepwise acquisition of multiple mutations and is relatively more constrained (Hall et al., 2011a, b). The implication, therefore, is that despite the mutational asymmetry, extensive bacteria–phage coevolution is possible, even in the absence of environmental heterogeneity or biotic complexity, although further data from alternate systems are required before any broad generalizations can be made. Why then was coevolution so constrained in early studies using E. coli B? One possibility is that E. coli B has a long history of laboratory cultivation (Daegelen et al., 2009), whereas the bacterial strains capable of extensive coevolution were much more recently isolated from the environment [P. fluorescens SBW25 from a sugar beet leaf (Bailey et al., 1995), E. coli O157:H7 from a diarrheal disease outbreak (Mizoguchi et al., 2003), and Synechoccus spp. from the ocean (Marston et al., 2012)]. Indeed, E. coli B has multiple known defects in its lipopolysaccharide (Yoon et al., 2012), a key attachment site for many tailed phages (e.g. T4, T7, and T2; Lenski & Levin, 1985), which may limit its scope for coevolution with certain phages. Similarly, the tailed-phages employed in early studies were also highly laboratory-adapted following many growth cycles on E. coli B prior to the advent of cryogenic storage (Demerec & Fano, 1945). This inadvertent selection for specialization on a particular host receptor may have constrained the subsequent evolutionary potential of the phages.
A fascinating exception comes from recent experiments between E. coli B and λ-vir where an extensive arms race is only observed following the evolution of a ‘key innovation’ by the phage to bind to a new host receptor against which it has no history of prior adaptation (OmpF instead of LamB; Meyer et al., 2012). This, along with the evidence from the other interactions described above, suggests that extensive arms races may be a common feature of newly constituted interactions between tailed-phages and bacteria (or their previously untargeted receptors). Other demographic factors may also counteract the mutational asymmetry constraint. For instance, phages typically have shorter generation times and larger population sizes than their bacterial hosts, both of which are thought to enhance evolutionary potential in host-parasite systems (Gandon & Michalakis, 2002). Moreover, other forms of resistance, such as CRISPR-mediated resistance, require bacteria to specifically match the targeted phage (Barrangou et al., 2007), which reverses the mutational asymmetry in favor of phages. Recent evidence suggests that phages can indeed readily evolve to escape recognition by CRISPR resistance (Levin et al., 2013; Sun et al., 2013). There is an urgent need for more empirical studies across a wide taxonomic range of bacteria–phage interactions to determine whether the patterns observed in studies so far are generalizable.
Coevolution as a driver of diversity
Bacteria–phage coevolution plays an important role in shaping genotypic, phenotypic and community-level diversity (Weinbauer & Rassoulzadegan, 2004; Rodriguez-Valera et al., 2009; Avrani et al., 2011). Coevolution can promote high levels of within population diversity in terms of both bacterial resistance and phage infectivity phenotypes (Poullain et al., 2008) and the underlying genotypes (Paterson et al., 2010; Scanlan et al., 2011). Coevolving lytic phages can increase diversity within bacterial populations by selecting for multiple modes of resistance (Brockhurst et al., 2004, 2005; Benmayor et al., 2008; Forde et al., 2008), and both lytic (Bohannan & Lenski, 2000a, b) and temperate phages (Schwinghamer & Brockwell, 1978; Joo et al., 2006) have been shown to alter apparent competition among bacterial strains. Moreover, the stochasticity of coevolutionary trajectories among populations can drive correlated phenotypic and genetic divergence (Paterson et al., 2010). This has been observed both as variation in the dominant bacterial colony morphologies among populations (which presumably became linked to successful resistance mutations) (Buckling & Rainey, 2002a, b; Brockhurst et al., 2004; Vogwill et al., 2011) and divergence of bacterial resistance and phage infection specificities between populations giving rise to local adaptation (Buckling & Rainey, 2002a, b; Morgan et al., 2005).
Recent work from an experimental Streptococcus thermophiles–phage system tracked metagenomic changes in the bacterial and phage populations after 1 week of coculturing (Sun et al., 2013). The researchers examined acquisition of phage-related spacers in each of the two functional CRISPR loci within the bacterial genome and found that after 1 week, all host cells in the population had acquired at least one spacer that matched the phage genome, with a remarkably high level of CRISPR spacer diversity among individual bacteria within the population. The phage genome was also sequenced and three SNPs were documented, one of which was found in a proto-spacer region. Each of these mutations reached a frequency of over 88% in the phage population, suggesting that phage diversity was relatively minimal compared with that of the bacterial host population. Furthermore, a follow-on experiment shows that the common spacer in the bacterial population fluctuates over a 15-day period and also suggests strong selection pressure determining which regions of the phage genome become incorporated as spacers (Paez-Espino et al., 2013). A similar asymmetry in bacterial and phage diversity was documented during a 6-month chemostat experiment using cyanobacteria and cyanophages (Marston et al., 2012).
Within a community ecology context, phages hold potential to mediate competition among bacterial species, as has been discussed in great detail in light of the ‘kill the winner hypothesis’, in which population growth by otherwise dominant bacterial species is hampered by phage infection (Thingstad, 2000; Winter et al., 2010). Results of several laboratory characterizing the ecological effects of phages on their hosts are consistent with a role for phages in maintaining bacterial diversity (Harcombe & Bull, 2005; Brockhurst et al., 2006), and the introduction of narrow host range phages to replicate experimental, two species communities of marine bacteria significantly altered the biomass of the nonhost species (Middelboe et al., 2003). However, how coevolution between bacteria and phages within a complex community setting might influence the interaction network and microbial diversity remain open questions (Beckett & Williams, 2013).
Ecological contingency of bacteria–phage coevolution
Resistance of bacteria to lytic phages has been found to carry substantial fitness costs (Lenski, 1988; Bohannan et al., 2002), including an increased cost of deleterious mutations (Buckling et al., 2006), decreased ability to metabolize carbon (Middelboe et al., 2009), altered competitive ability (Brockhurst et al., 2005; Lennon et al., 2007; Quance & Travisano, 2009), and increased susceptibility to other phages (Avrani et al., 2011; Marston et al., 2012). Ecological conditions can mediate the costs and benefits of resistance and infectivity for bacteria and phages, respectively (Messenger et al., 1999; Jessup & Bohannan, 2008). Thus, the effects of several key ecological variables on bacteria–phage coevolution have been studied to identify conditions that promote or constrain coevolutionary dynamics. Increasing dispersal, or mixing, within populations can increase contact rates between bacteria and phages, and can be achieved by as simple a manipulation as periodically shaking the culture vessel (e.g. experiments comparing static and shaken cultures of P. fluorescens and Φ2; Brockhurst et al., 2003). Population mixing enhances phage transmission, thereby increasing the benefit of resistance to bacteria. This effect strengthens selection for the evolution of resistance in bacteria, which correspondingly strengthens the selection upon phages to restore infectivity, and, overall, accelerates arms-race coevolution (Brockhurst et al., 2003). Dispersal at larger spatial scales, that is, between populations or patches within a metapopulation, can also accelerate coevolution; however, this effect is critically dependent upon the rate of dispersal. Low to intermediate rates of dispersal (< 1% per generation approximately) of bacteria and phages, or bacteria or phages alone, can accelerate coevolution by increasing the supply of beneficial genetic variation, which enhances the response of the dispersing species to reciprocal selection (Lively 2009; Brockhurst et al., 2007b; Morgan et al., 2007; Vogwill et al., 2008). In contrast, high rates of dispersal (> 10% per generation approximately) act to homogenize genetic variation and synchronize coevolutionary dynamics across populations (Vogwill et al., 2009a; Vogwill et al., 2011), thereby diminishing the benefits of dispersal and leading to rates of arms-race coevolution similar to those observed in isolated populations (Morgan et al., 2007; Vogwill et al., 2008).
Increasing the rate of supply of resources (i.e. the concentration of carbon substrates) to bacteria reduces the cost of resistance mutations and accelerates the rate at which such mutations arise and invade populations (Bohannan & Lenski, 1997; Harrison et al., 2013). Bacterial population densities also increase with resource supply, which can lead to elevated bacteria–phage contact rates, potentially enhancing the benefits of resistance. Combined, these population genetic and ecological effects of increased resource supply both act to intensify reciprocal selection and enhance the bacterial response to phage-mediated selection, accelerating coevolutionary dynamics (Lopez-Pascua & Buckling, 2008). As a result of differences in mutational supply, the strength of selection and the relative costs of resistance mutations, different levels of resource supply select for qualitatively different bacterial resistance mutations, and correspondingly, phage infectivity mutations (Forde et al., 2008; Lopez-Pascua et al., 2012). This causes greater divergence among populations of contrasting resource supply levels than is observed among populations with equivalent resource supply levels, causing stronger patterns of local adaptation (Lopez-Pascua et al., 2012). Therefore, resource supply affects not only the rate of bacteria–phage coevolution, but also its trajectory.
Several studies using the P. fluorescens-Φ2 and E. coli-T7 interactions have considered the effects of spatial ecological heterogeneity on bacteria–phage coevolutionary dynamics (Forde et al., 2004, 2007; Vogwill et al., 2009a, b; Lopez-Pascua et al., 2010). In particular, these experiments have manipulated dispersal of bacteria and phages between populations propagated under different ecological conditions that vary in the intensity of coevolution [e.g. between populations with high vs. low resource supply (Forde et al., 2004; Lopez-Pascua et al., 2010) or high vs. low rates of population mixing (Vogwill et al., 2009a)]. In general, these studies reveal that, given dispersal, the rate of coevolution at the ‘landscape level’ (i.e. across all connected populations) is set by the fastest coevolving population, which acts as a coevolutionary pacemaker (Vogwill et al., 2009b). The effects of temporal heterogeneity on bacteria–phage coevolution have been less extensively studied. Recent experiments with P. fluorescens-Φ2 manipulated the frequency of fluctuations in resource supply by propagating populations in environments that alternated between high and low resource supply at different rates. Rapidly fluctuating environments (e.g. alternating every c. 7.5–15 bacterial generations) constrained arms-race coevolution relative to constant environments, whereas slowly fluctuating environments (e.g. alternating every c. 30 bacterial generations) did not. This occurred because selective sweeps of bacterial resistance mutations, which were only ever observed under high resource supply, required c. 25 bacterial generations to occur and were therefore impeded by rapid fluctuations in resource supply (Harrison et al., 2013).
While most experimental coevolution has employed pairs of species, bacteria–phage interactions often occur embedded within a diverse microbial community. Relatively few experimental coevolution studies have attempted to scale up community complexity beyond pairwise interactions. However, recent experiments suggest that addition of multiple exploiters of bacteria, such as other phages or protist predators, may limit the ability of bacteria to evolve defense against a focal phage species. For example, the evolution of phage resistance in the plant-pathogenic bacterium, P. syringae, was found to differ depending on the heterogeneity of the phage population. Those bacterial populations that were coevolving with multiple phage genotypes were able to evolve resistance as readily as populations coevolving with single phage genotypes, but the former paid a greater cost for such resistance (Koskella et al., 2012). Similarly, P. aeruginosa strains evolved in the presence of two phages were found to have decreased growth rate and motility relative to those evolved with a single phage but increased production of siderophores (Hosseinidoust et al., 2013). The addition of a protist predator, Tetrahymena thermophila, to experimental populations of P. fluorescens and Φ2 impeded coevolution between the bacteria and phage, but favored the maintenance of coexisting resistance phenotypes specialized against one or other of these natural enemies (Friman & Buckling, 2012).
Coevolution in the wild
Given the overwhelming data from experimental laboratory studies for the ecological contingency of bacteria–phage coevolution, the key question becomes whether what we know about these interactions in the laboratory can be directly translated to make predictions about what happens in natural microbial communities. There are a number of reasons to think that this might not be straightforward; the additional abiotic and biotic selection pressures, variation in resources, competition among species, and vastly differing population sizes and migration rates are all likely to alter the trajectory of bacteria–phage coevolution relative to what is observed in simple microcosm experiments. Although the underlying process should remain the same, it becomes a daunting task to predict which specific features of the natural world might be most influential in altering the trajectory of bacteria–phage coevolution. The wealth of literature describing bacterial and phage diversity in nature (reviewed in Weinbauer, 2004; Breitbart & Rohwer, 2005; Clokie et al., 2011) is suggestive of on-going coevolutionary dynamics, but the role of coevolution relative to other factors such as dispersal or selection by the abiotic environment in shaping this diversity has rarely been examined.
Seminatural environment microcosm studies
One elegant approach is to bridge the gap between purely experimental and purely observational (and thus correlational) studies by running experiments under seminatural conditions. Gomez and Buckling have applied this approach by measuring coevolution between bacteria and phages in soil microcosms that are either sterile prior to introduction of the target bacteria and phage clones or that contain a natural community of microorganisms (Gómez & Buckling, 2011). In this way, they have successfully demonstrated that coevolution between marked strains of P. fluorescens and phage Φ2 in soil is more in line with fluctuating selection than arms-race dynamics, as neither the bacteria nor phage become increasingly resistant/infective over time. This is in stark contrast to previous laboratory studies using the same system but run in nutrient-rich media broth (Brockhurst et al., 2003, 2007a, b). One explanation for the observed difference is that resistance is more costly in the soil environment than in the broth environment and therefore that resistance to previous phage types is lost in favor of specific resistance to contemporary phage. This increased cost of resistance would both constrain the continual arms-race selection toward increased resistance/infectivity and lead to fluctuating dynamics as new resistances are gained and old resistances are lost. Indeed, Gómez and Buckling (2011) go on to show that resistant bacterial strains evolved in broth were no less fit than their sensitive ancestor, while those which had evolved resistance in soil had a 36% reduction in fitness. Intriguingly, they found no substantial difference in coevolutionary outcome between those bacteria and phages coevolving in the presence vs. absence of the natural microbial community. This result suggests that phage-mediated selection may be strong enough to override selection arising from interspecific competition. Furthermore, the work demonstrates that experiments with added complexity have a great deal to offer for elucidating both which components of natural systems are important in predicting the outcome of coevolution and those that might be less important than predicted. More recently, this same seminatural system has been used to demonstrate that coevolving phages do not select for increased bacterial mutation rate (Gómez & Buckling, 2013), as had been previously observed from studies in vitro (Pal et al., 2007). Similarly, seminatural mesocosm studies of phytoplankton and their associated viral populations have demonstrated long-term genotypic stability over time in a natural fjord (Martínez et al., 2007) and arms-race dynamics during bloom development (Vardi et al., 2012).
Evidence for bacteria–phage coevolution in nature
Given the multiple competing selection pressures and the fact that bacteria–phage encounter rates in nature are likely to be dramatically different than those observed in liquid microcosms, it is unclear exactly how important of an evolutionary force phages are in shaping natural microbial populations. However, there is building evidence from across a number of bacteria–phage systems that coevolution is occurring and having significant ecological impact in nature. The first critical step in demonstrating a role for phages in driving bacterial evolution has been quantifying both their prevalence and host range in nature. One way to indirectly test for the impact of lytic phages in shaping microbial diversity is to remove them from natural populations/communities and determine whether there is a corresponding change in genotypic or community composition. For example, the depletion of viral particles from seawater led to a marked change in the relative abundances of marine bacterioplankton (Bouvier & Del Giorgio, 2007), and the manipulation of the presence of viral and protist predators in seminatural communities of bacterioplankton found both increased bacterial richness in the presence of phages and a complex interaction between phages and protists on bacterial mortality that ranged from synergistic to antagonistic (Weinbauer et al., 2007).
Thanks to the power of metagenomics, the true extent of phage prevalence is being uncovered, and it is now clear that phages are ubiquitous across natural systems and can account for the turnover of c. 20% of the living biomass in the sea (Suttle, 2007; Clokie et al., 2011). For example, a newly discovered phage (HMO-2011) capable of infecting the most abundant lineage of marine bacteria, the SAR116 clade, can account for up to 25% of all viral genome reads in the ocean (Kang et al., 2013). Sequence-based estimates of phage abundance are likely to be significantly higher than estimates based on quantification of infective phage particles [for example, abundance estimates of cyanophages infecting cyanobacteria in the open ocean were found to be two orders of magnitude lower than that of their hosts (Sullivan et al., 2003)], but do not allow for a complete understanding of the infection network within natural populations and communities. In particular, sequence-based approaches do not allow for estimates of infection prevalence, as it is typically not possible to determine the host range of a given phage. For example, evidence from Salmonella bacteria and their associated phages on a dairy farm suggests both great diversity of phages, and a high density of multiple specialist phage types, each capable of infecting common bacterial strains (Moreno Switt et al., 2013). A statistical analysis of large phage host range datasets has recently been introduced and applied to bacteria–phage interaction networks from soil (Poisot et al., 2013), the ocean (Flores et al., 2013) and a meta-analysis of 38 laboratory-tested networks (Flores et al., 2011). Overall, these data support the idea that phages in nature span the continuum from specialist to generalist, resulting in what is known as a ‘nested’ structure (reviewed in Weitz et al., 2013; Martiny et al., 2014). The next key steps in the study of bacteria–phage coevolution can be broken down into those data supporting: (1) dynamical change in bacterial and phage communities over time; (2) phage adaptation to bacterial populations; (3) bacterial response to phage-mediated selection; and (4) finally, reciprocal change of phage and bacterial communities over time. Each of which we review below.
There are a number of sophisticated studies that support fluctuations in bacterial populations and corresponding phage populations over time (Waterbury & Valois, 1993; Mathias et al., 1995; Cochran & Paul, 1998; Marston & Sallee, 2003; Faruque et al., 2005). Phage abundance in a bioreactor was observed to change frequently and was correlated with changes in the corresponding bacterial taxonomic group (Shapiro et al., 2010). Cyanobacteria and their associated cyanophages were also found to be highly dynamic. By tracking mutational change in a phage tail sheath gene over both 5-year and 1-day sampling periods, Kimura and coauthors were able to demonstrate both short- and long-term oscillations of the five major cyanophage genotypes, suggestive of frequency-dependent dynamics (Kimura et al., 2013). In concert with previous findings from the same system showing highly diverse CRISPR sequences in the host population (Kuno et al., 2012), this work reveals strong associations and rapid evolution of the bacterial and phage populations. Similarly, dynamic change was reported for a population of Sphingomonas sp. and its lytic phage in a freshwater lake in Northern Germany over the course of 3 months (Jost & Wiese, 2013). The population dynamics observed will of course vary both according to the timeframe and the resolution of analysis. For example, phage and bacterial communities from four aquaculture or solar saltern environments were monitored over time at both a course and fine-grain taxonomic scale. At the scale of the species, both microbial and viral communities remained stable, with the top microbial and viral taxa persisting over periods ranging from a single day to over a year (Rodriguez-Brito et al., 2010). At the strain level, however, the authors observed continuous variation in the abundance of both viral and bacterial genotypes.
There is empirical data to support that phages in nature are locally adapted to their bacterial hosts (Fig. 3; Vos et al., 2009; Gómez & Buckling, 2011; Koskella et al., 2011). This is in contrast to laboratory studies where bacterial local adaptation (i.e. where the phages do particularly poorly on their coevolving hosts relative to others) is often found (Buckling & Rainey, 2002a, b; Morgan et al., 2005; Vogwill et al., 2010). In soil from a flood plain near Oxford, phages collected from only 1 cm apart were found to be more infective to bacterial hosts (mainly Stenotrophomonas) from those same samples than they were to neighboring bacterial isolates (Vos et al., 2009). This study confirms that bacteria–phage interactions can be very local indeed and suggest that phages are adapting rapidly to bacterial strains in close proximity. In contrast, phage populations collected from the leaves of horse chestnut trees were found to be just as infective to bacterial hosts from other leaves within the same tree canopy as those from their own leaf (Koskella et al., 2011). In this case, phage local adaptation was only apparent when comparing phage infectivity on bacterial hosts from neighboring trees, and this was true both for the whole bacterial community and also for those isolates that were identified as P. syringae. These patterns observed in terrestrial systems may not translate to aquatic and marine communities, where evidence from co-existing populations of cyanobacteria and phages has demonstrated that, despite a high abundance of phage, most circulating host types are in fact resistant to local phages (Waterbury & Valois, 1993).
Fig 3.
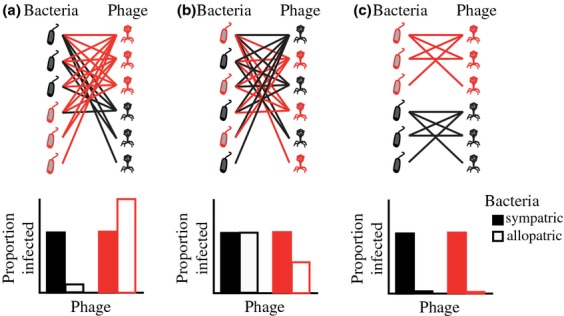
An illustration of phage local adaptation across two populations. In the first case, (a) the bacteria–phage network across the two populations is nested, and the population containing phages with the broadest host range (in red) also contains the most resistant bacteria. This structure, which is loosely based on directional, arms-race selection, does not result in an overall pattern of local adaptation. In the second case, (b) the network remains nested, but phage host range and bacterial resistance are not correlated with population. Again, this structure does not result in an overall pattern of local adaptation. In the final case, (c) phages from one population are only infective to bacteria from the same population, although the network structure within each population remains nested. Unlike the others, this structure would lead to an overall pattern of phage local adaptation. Overall, we suggest that if phage-mediated selection typically leads to an increase in generalized resistance to phage attack, phages from one population should be most infective to hosts from populations under relaxed phage-mediated selection and least infective to those from populations under relatively strong phage-mediated selection, regardless of sympatry. Phage local adaptation therefore suggests both that hosts are unable to evolve resistance rapidly enough to escape their local phages and that phage infectivity/bacterial resistance is relatively specific.
Insight into the scale of bacteria–phage coevolutionary dynamics can also be gleaned from studies that attempt to match the CRISPR spacers in bacterial genomes to phage DNA found from the same location. For example, in this way, researchers have examined datasets from the human microbiome project and found segregation of CRISPR sequence according to the body-site from which the sample was taken (Rho et al., 2012). Furthermore, by comparing the CRISPR spacer sequences across body sites and among individuals, they were able to demonstrate that, while resampling of the same site from the same individual recovered many of the same spacers, different individuals had almost no spacers in common. A similar approach was taken to investigate adaptation of Candidatus Accumulibacter phosphatis across geographically separated bioreactors (Kunin et al., 2008). In this case, despite evidence for high dispersal among the sites and relatively little divergence among bacterial genotypes overall, the authors were able to demonstrate rapid divergence of CRISPR sequences which correlated with local phage-mediated selection. Furthermore, the two regions of the bacterial genome suggested to be evolving under phage-mediated selection, the CRISPR elements and the EPS gene clusters, were also the regions found to be most different among closely related strains of S. thermophilus isolated from yogurt manufactured in France and in the United Kingdom (Bolotin et al., 2004). Together, these genomic and phenotypic studies suggest that phages are capable of rapidly adapting to their bacterial hosts and that natural bacterial host populations will respond to this selection by evolving increased resistance.
Direct evidence for a response to phage-mediated selection in nature has proven somewhat more difficult to obtain. A recent study from microbial communities living within the leaves of horse chestnut trees utilized a ‘time-shift’ approach (Gandon et al., 2008; Gaba & Ebert, 2009) to determine whether bacteria were more resistant to phages from relatively earlier in the season compared with those from later in the season (Koskella, 2013). Indeed, bacterial isolates were found to be most resistant to phages from the prior month and least resistant to phages from 1 month in the future, suggesting a rapid response to phage-mediated selection that could be explained by mutational change or immigration of new and resistant strains and species. Similarly, phages were found to be most infective to bacteria from earlier in the season and least infective to those from the future, to which they had not yet adapted.
Coevolving gene networks
The measure of bacteria–phage coevolution in nature has been hampered by our lack of understanding of the interaction networks of the host and parasite. As mentioned above, this understanding is increasing with sophisticated statistical models and meta-analyses of large datasets (Weitz et al., 2013). Specifically, knowing whether phages commonly have narrow host ranges, infecting only a single or multiple genotypes of a single host species, or broad host ranges, infecting multiple species or even species spanning multiple genera, is a prerequisite for elucidating the impact that phages will have on their host population and community and vice versa (Fig. 3; Sullivan et al., 2003). To illustrate this point, imagine that a single phage is capable of infecting two different bacterial species in its local environment. These two bacteria may evolve resistance via different mechanisms, or by incorporating different phage-derived spacers, but they may also evolve resistance via a shared mechanism or spacer region. Under the former scenario, the phage may also diversify, creating the potential for two new pairwise coevolving bacteria–phage lineages. However, under the latter scenario, the phage may well adapt to overcome the shared mechanism and the process of coevolution is pairwise at the gene level, but not the species level. Thus, the need for resolution of infection networks at the gene level, rather than species level, is pressing.
There are reasons to think that phage specificity (i.e. narrow host range), as opposed to generalism (broad host range), is the rule rather than the exception in nature (reviewed in Hyman & Abedon, 2010; Koskella & Meaden, 2013; Martiny et al., 2014). Although our understanding of what constitutes a narrow or broad host range is necessarily hampered by the reference panel against which specificity is measured, the infection networks that have been analyzed to date illustrate a continuum between narrow and broad host range, with most phages infecting some but not all of the panel of bacteria they are confronted with, suggesting some constraint upon host range even among the most broadly infectious phages. Constraints on host range could arise due to fitness trade-offs (Duffy et al., 2006), differences between hosts in terms of overall quality (Heineman et al., 2008), or intracellular defenses against successfully adsorbed phages (Sieber & Gudelj, 2014). Regardless of the exact degree of specificity of the interaction, it is clear that many phages infect more than one bacterial strain or species and that many bacteria can act as host to a number of different phages in the environment (Flores et al., 2011). Thus, bacteria–phage coevolution is unlikely to be purely pairwise and we might instead think of bacterial and phage communities as coevolving gene networks, whereby particular phage genes are under selection by corresponding bacterial genes, regardless of which host genome they might be found. The breakdown of simple pairwise coevolution in bacteria–phage systems is further exacerbated by the process of horizontal transfer of genes among bacterial genomes (Smillie et al., 2011), much of which is phage-mediated (Canchaya et al., 2003), and the potential for recombination among phage genomes during coinfection (Benbow et al., 1974; Riede, 1986; Shcherbakov et al., 1992; Worobey & Holmes, 1999; Lin et al., 2012). Evidence from cyanobacterial populations even supports the movement of mutations conferring resistance to phage among bacterial genomes via HGT of a hypervariable genomic island (Avrani et al., 2011). Importantly, recombination among phage genomes has been shown to alter host range (Riede, 1986; Lin et al., 2012), and early theory set out to describe phage evolution as a process occurring at the modular level, where interchangeable genetic elements were recombined to create the optimal phenotype at any given time (Botstein, 1980). The complexity of untangling interactions among genes from interactions among organisms has been highlighted in a recent review of the microbiome (Boon et al., 2014), and many of the same complexities exist when exploring bacteria–phage networks in nature.
Future directions
Throughout the review, we have attempted to highlight the progress that has been made in our understanding of coevolution between bacteria and phages, as well as to emphasize that our understanding is still rapidly developing. Even our comprehension of bacterial resistance mechanisms and phage infectivity is continually improving. The relatively recent discovery of CRISPRs as a defense against phage (Barrangou et al., 2007; Andersson & Banfield, 2008) has already been expanded to include anti-CRISPR counter-measures by phages (Bondy-Denomy et al., 2012) and to show that phages can also carry a CRISPR-cas system to target a chromosomal island of the bacterial host (Seed et al., 2013). Furthermore, the importance of bacterial suicide upon phage infection has recently been demonstrated in E. coli and was found to be a low cost strategy for reducing the population-wide impact of phage (Refardt et al., 2013) that is favored in spatially structured environments (Berngruber et al., 2013). However, the coevolutionary implications of these new mechanisms have not yet been explored, and this avenue is ripe for empirical testing using an experimental coevolution approach and for examination of natural patterns in the field.
Another key advance of the field will be incorporation of both theoretical and empirical examination of coevolution between bacteria and temperate, as well as filamentous phages. There are a number of reasons to expect the coevolutionary process to differ for these interactions relative to those with lytic phage. Primarily, many of these phages confer a strong fitness benefit to their hosts and thus will act more as mutualists than parasites. This can shift dynamics from parasite-mediated negative frequency-dependent selection (where hosts are constantly evolving to defend themselves against the common parasite) to positive frequency-dependent selection, where for example, carrying the lysogenic phage confers resistance to the same phage in the lytic form and therefore the benefit of being a lysogen increases with the frequency of other lysogens. Similarly, filamentous phages can increase the fitness of their hosts through toxin production and increased pathogenicity, as has been found for V. cholerae, the causative agent of cholera (Waldor & Mekalanos, 1996). Both filamentous and temperate phage systems have proven amenable to in vitro experimentation, but, to our knowledge, have not been used to test for coevolution. One-sided experimental evolution of the filamentous phage f1 demonstrated increased virulence (in terms of larger impact on population density) when horizontal transmission among hosts was increased relative to vertical transmission within a dividing bacterial lineage (Messenger et al., 1999), but it remains to be determined whether the bacterial population would respond by evolving increased resistance under these same conditions. One-sided experimental evolution of the lysogenic phage λ was also used to select for altered sensitivity and threshold for the switch from lysogenic to lytic phage life cycle (Refardt & Rainey, 2010).
Finally, further exploration of the similarities and differences between bacteriophages and other viruses will both help inform the utility of in vitro coevolution studies as a basis for building predictions for other virus–cell interactions, and uncover any unique adaptations of phages to their bacterial hosts. For example, examination of the archaeon, Sulfolobus islandicus, and its associated viral parasites isolated from hot springs suggests a clear biogeographic structure, such that viral genomes were found to be specifically associated with each local host population (Held & Whitaker, 2009). This system has led the way in uncovering the parallel role of CRISPR systems in archaea–virus interactions and reinforces evidence from bacteria–phage systems that demonstrate a role for viruses in maintaining host diversity (Held et al., 2010). In addition, an examination of temporal dynamics of archaea–virus interactions in a hypersaline lake suggests ample change over the course of both months and years, indicating similar timescales and mechanisms for these interactions as observed with bacteria–phage systems (Emerson et al., 2013). The other similarities between archaea–virus and bacteria–phage interaction have been reviewed elsewhere (Snyder & Young, 2011). Finally, it remains to be seen whether our increasing understanding of bacteria–phage coevolution will prove useful in studies of eukaryote-virus interactions, but at the least, each body of work could help shape the questions addressed in and techniques utilized by the other (Brockhurst et al., 2007a; Sharp & Simmonds, 2011).
Acknowledgments
The authors wish to acknowledge the helpful comments of four anonymous reviewers. This work was supported by a NERC Independent research fellowship to B.K. (NE/K00879X/1), and grants to M.A.B. from NERC (NE/H005080/1) and the ERC (COEVOCON 311490).
References
- Abedon ST. Phages. In: ST Abedon, P Hyman., editors. Bacteriophages in Health and Disease. Vol. 24. MA: CABI, Cambridge; 2012. pp. 1–5. [Google Scholar]
- Allen B, Willner D, Oechel WC. Lipson D. Top-down control of microbial activity and biomass in an Arctic soil ecosystem. Environ Microbiol. 2010;12:642–648. doi: 10.1111/j.1462-2920.2009.02104.x. [DOI] [PubMed] [Google Scholar]
- Andersson AF. Banfield JF. Virus population dynamics and acquired virus resistance in natural microbial communities. Science. 2008;320:1047–1050. doi: 10.1126/science.1157358. [DOI] [PubMed] [Google Scholar]
- Avrani S, Wurtzel O, Sharon I, Sorek R. Lindell D. Genomic island variability facilitates Prochlorococcus-virus coexistence. Nature. 2011;474:604–608. doi: 10.1038/nature10172. [DOI] [PubMed] [Google Scholar]
- Bailey MJ, Lilley AK, Thompson IP, Rainey PB. Ellis RJ. Site directed chromosomal marking of a fluorescent pseudomonad isolated from the phytosphere of sugar beet; Stability and potential for marker gene transfer. Mol Ecol. 1995;4:755–763. doi: 10.1111/j.1365-294x.1995.tb00276.x. [DOI] [PubMed] [Google Scholar]
- Banfield JF. Young M. Variety—the splice of life—in microbial communities. Science. 2009;326:1198–1199. doi: 10.1126/science.1181501. [DOI] [PubMed] [Google Scholar]
- Barnet YM, Daft M. Stewart W. Cyanobacteria-cyanophage interactions in continuous culture. J Appl Microbiol. 1981;51:541–552. doi: 10.1111/j.1365-2672.1984.tb04701.x. [DOI] [PubMed] [Google Scholar]
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA. Horvath P. CRISPR provides acquired resistance against viruses in Prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- Beckett SJ. Williams HT. Coevolutionary diversification creates nested-modular structure in phage–bacteria interaction networks. Interface Focus. 2013;3:20130033. doi: 10.1098/rsfs.2013.0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benbow RM, Zuccarelli AJ, Davis GC. Sinsheimer RL. Genetic recombination in bacteriophage φX174. J Virol. 1974;13:898–907. doi: 10.1128/jvi.13.4.898-907.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkman CW. The selection mosaic and diversifying coevolution between crossbills and lodgepole pine. Am Nat. 1999;153:S75–S91. doi: 10.1086/303213. [DOI] [PubMed] [Google Scholar]
- Benmayor R, Buckling A, Bonsall MB, Brockhurst MA. Hodgson DJ. The interactive effects of parasites disturbance, and productivity on experimental adaptive radiations. Evolution. 2008;62:467–477. doi: 10.1111/j.1558-5646.2007.00268.x. [DOI] [PubMed] [Google Scholar]
- Berngruber TW, Lion S. Gandon S. Evolution of suicide as a defence strategy against pathogens in a spatially structured environment. Ecol Lett. 2013;16:446–453. doi: 10.1111/ele.12064. [DOI] [PubMed] [Google Scholar]
- Blower TR, Evans TJ, Przybilski R, Fineran PC. Salmond GP. Viral evasion of a bacterial suicide system by RNA–based molecular mimicry enables infectious altruism. PLoS Genet. 2012;8:e1003023. doi: 10.1371/journal.pgen.1003023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohannan BJM. Lenski RE. Effect of resource enrichment on a chemostat community of bacteria and bacteriophage. Ecology. 1997;78:2303–2315. [Google Scholar]
- Bohannan BJM. Lenski RE. The relative importance of competition and predation varies with productivity in a model community. Am Nat. 2000a;156:329–340. doi: 10.1086/303393. [DOI] [PubMed] [Google Scholar]
- Bohannan BJM. Lenski RE. Linking genetic change to community evolution: insights from studies of bacteria and bacteriophage. Ecol Lett. 2000b;3:362–377. [Google Scholar]
- Bohannan BJM, Kerr B, Jessup CM, Hughes JB. Sandvik G. Trade-offs and coexistence in microbial microcosms. Antonie Van Leeuwenhoek. 2002;81:107–115. doi: 10.1023/a:1020585711378. [DOI] [PubMed] [Google Scholar]
- Bolotin A, Quinquis B, Renault P, Sorokin A, Ehrlich SD, Kulakauskas S, Lapidus A, Goltsman E, Mazur M. Pusch GD. Complete sequence and comparative genome analysis of the dairy bacterium Streptococcus thermophilus. Nat Biotechnol. 2004;22:1554–1558. doi: 10.1038/nbt1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondy-Denomy J, Pawluk A, Maxwell KL. Davidson AR. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature. 2012;493:429–432. doi: 10.1038/nature11723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon E, Meehan CJ, Whidden C, Wong DHJ, Langille MGI. Beiko RG. Interactions in the microbiome: communities of organisms and communities of genes. FEMS Microbiol Rev. 2014;38:90–118. doi: 10.1111/1574-6976.12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botstein D. A therory of modular evolution for bacteriophages. Ann NY Acad Sci. 1980;354:484–491. doi: 10.1111/j.1749-6632.1980.tb27987.x. [DOI] [PubMed] [Google Scholar]
- Bouvier T. Del Giorgio P. Key role of selective viral-induced mortality in determining marine bacterial community composition. Environ Microbiol. 2007;9:287–297. doi: 10.1111/j.1462-2920.2006.01137.x. [DOI] [PubMed] [Google Scholar]
- Breitbart M. Rohwer F. Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 2005;13:278–284. doi: 10.1016/j.tim.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Brockhurst MA. Koskella B. Experimental coevolution of species interactions. Trends Ecol Evol. 2013;28:367–375. doi: 10.1016/j.tree.2013.02.009. [DOI] [PubMed] [Google Scholar]
- Brockhurst MA, Morgan AD, Rainey PB. Buckling A. Population mixing accelerates coevolution. Ecol Lett. 2003;6:975–979. [Google Scholar]
- Brockhurst MA, Rainey PB. Buckling A. The effect of spatial heterogeneity and parasites on the evolution of host diversity. Proc Biol Sci. 2004;271:107–111. doi: 10.1098/rspb.2003.2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockhurst MA, Buckling A. Rainey PB. The effect of a bacteriophage on diversification of the opportunistic bacterial pathogen, Pseudomonas aeruginosa. Proc Biol Sci. 2005;272:1385–1391. doi: 10.1098/rspb.2005.3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockhurst M, Fenton A, Roulston B. Rainey P. The impact of phages on interspecific competition in experimental populations of bacteria. BMC Ecol. 2006;6:19. doi: 10.1186/1472-6785-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockhurst MA, Morgan AD, Fenton A. Buckling A. Experimental coevolution with bacteria and phage The Pseudomonas fluorescens – Phi 2 model system. Infect Genet Evol. 2007a;7:547–552. doi: 10.1016/j.meegid.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Brockhurst MA, Buckling A, Poullain V. Hochberg ME. The impact of migration from parasite-free patches on antagonistic host-parasite coevolution. Evolution. 2007b;61:1238–1243. doi: 10.1111/j.1558-5646.2007.00087.x. [DOI] [PubMed] [Google Scholar]
- Brüssow H, Canchaya C. Hardt W-D. Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol Mol Biol Rev. 2004;68:560–602. doi: 10.1128/MMBR.68.3.560-602.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckling A. Rainey PB. The role of parasites in sympatric and allopatric host diversification. Nature. 2002a;420:496–499. doi: 10.1038/nature01164. [DOI] [PubMed] [Google Scholar]
- Buckling A. Rainey PB. Antagonistic coevolution between a bacterium and a bacteriophage. Proc Biol Sci. 2002b;269:931–936. doi: 10.1098/rspb.2001.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckling A, Wei Y, Massey RC, Brockhurst MA. Hochberg ME. Antagonistic coevolution with parasites increases the cost of host deleterious mutations. Proc Biol Sci. 2006;273:45–49. doi: 10.1098/rspb.2005.3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canchaya C, Fournous G, Chibani-Chennoufi S, Dillmann M-L. Brüssow H. Phage as agents of lateral gene transfer. Curr Opin Microbiol. 2003;6:417–424. doi: 10.1016/s1369-5274(03)00086-9. [DOI] [PubMed] [Google Scholar]
- Cannon RE, Shane MS. Whitaker JM. (Interaction of Plectonema boryanum Cyanophyceae) and the LPP –Cyanophages in continuous culture. J Phycol. 1976;12:418–421. [Google Scholar]
- Chao L, Levin BR. Stewart FM. Complex community in a simple habitat – Experimental study with bacteria and phage. Ecology. 1977;58:369–378. [Google Scholar]
- Clokie MRJ, Millard AD, Letarov AV. Heaphy S. Phages in nature. Bacteriophage. 2011;1:31–45. doi: 10.4161/bact.1.1.14942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran PK. Paul JH. Seasonal abundance of lysogenic bacteria in a subtropical estuary. Appl Environ Microbiol. 1998;64:2308–2312. doi: 10.1128/aem.64.6.2308-2312.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowlishaw J. Mrsa M. Co-evolution of a virus-alga system. Appl Microbiol. 1975;29:234–239. doi: 10.1128/am.29.2.234-239.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daegelen P, Studier FW, Lenski RE, Cure S. Kim JF. Tracing ancestors and relatives of Escherichia coli B, and the derivation of B strains REL606 and BL21(DE3) J Mol Biol. 2009;394:634–643. doi: 10.1016/j.jmb.2009.09.022. [DOI] [PubMed] [Google Scholar]
- Dawkins R. Krebs JR. Arms races between and within species. Proc R Soc Lond B Biol Sci. 1979;205:489–511. doi: 10.1098/rspb.1979.0081. [DOI] [PubMed] [Google Scholar]
- Demerec M. Fano U. Bacteriophage-resistant mutants in Escherichia coli. Genetics. 1945;30:119–136. doi: 10.1093/genetics/30.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennehy JJ. What can phages tell us about host-pathogen coevolution? Int J Evol Biol. 2012;2012:396165. doi: 10.1155/2012/396165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerkop BA, Clements CV, Rollins D, Rodrigues JLM. Hooper LV. A composite bacteriophage alters colonization by an intestinal commensal bacterium. P Natl Acad Sci USA. 2012;109:17621–17626. doi: 10.1073/pnas.1206136109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy S, Turner PE. Burch CL. Pleiotropic costs of niche expansion in the RNA bacteriophage Φ6. Genetics. 2006;172:751–757. doi: 10.1534/genetics.105.051136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson JB, Andrade K, Thomas BC, Norman A, Allen EE, Heidelberg KB. Banfield JF. Virus-host and CRISPR dynamics in archaea-dominated hypersaline Lake Tyrrell, Victoria, Australia. Archaea. 2013;2013:12. doi: 10.1155/2013/370871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt T, Kallmeyer J, Cypionka H. Engelen B. High virus-to-cell ratios indicate ongoing production of viruses in deep subsurface sediments. ISME J. 2014 doi: 10.1038/ismej.2013.245. (online early). doi: 10.1038/ismej.2013.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faruque SM, Islam MJ, Ahmad QS, Faruque ASG, Sack DA, Nair GB. Mekalanos JJ. Self-limiting nature of seasonal cholera epidemics: role of host-mediated amplification of phage. P Natl Acad Sci USA. 2005;102:6119–6124. doi: 10.1073/pnas.0502069102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores CO, Meyer JR, Valverde S, Farr L. Weitz JS. Statistical structure of host–phage interactions. P Natl Acad Sci USA. 2011;108:E288–E297. doi: 10.1073/pnas.1101595108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores CO, Valverde S. Weitz JS. Multi-scale structure and geographic drivers of cross-infection within marine bacteria and phages. ISME J. 2013;7:520–532. doi: 10.1038/ismej.2012.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forde SE, Thompson JN. Bohannan BJM. Adaptation varies through space and time in a coevolving host-parasitoid interaction. Nature. 2004;431:841–844. doi: 10.1038/nature02906. [DOI] [PubMed] [Google Scholar]
- Forde SE, Thompson JN. Bohannan BJM. Gene flow reverses an adaptive cline in a coevolving host-parasitoid interaction. Am Nat. 2007;169:794–801. doi: 10.1086/516848. [DOI] [PubMed] [Google Scholar]
- Forde SE, Thompson JN, Holt RD. Bohannan BJM. Coevolution drives temporal changes in fitness and diversity across environments in a bacteria-bacteriophage interaction. Evolution. 2008;62:1830–1839. doi: 10.1111/j.1558-5646.2008.00411.x. [DOI] [PubMed] [Google Scholar]
- Friman VP. Buckling A. Effects of predation on real-time host-parasite coevolutionary dynamics. Ecol Lett. 2012;16:39–46. doi: 10.1111/ele.12010. [DOI] [PubMed] [Google Scholar]
- Gaba S. Ebert D. Time-shift experiments as a tool to study antagonistic coevolution. Trends Ecol Evol. 2009;24:226–232. doi: 10.1016/j.tree.2008.11.005. [DOI] [PubMed] [Google Scholar]
- Gandon S. Michalakis Y. Local adaptation, evolutionary potential and host-parasite coevolution: interactions between migration, mutation, population size and generation time. J Evol Biol. 2002;15:451–462. [Google Scholar]
- Gandon S, Buckling A, Decaestecker E. Day T. Host-parasite coevolution and patterns of adaptation across time and space. J Evol Biol. 2008;21:1861–1866. doi: 10.1111/j.1420-9101.2008.01598.x. [DOI] [PubMed] [Google Scholar]
- Gómez P. Buckling A. Bacteria-phage antagonistic coevolution in soil. Science. 2011;332:106–109. doi: 10.1126/science.1198767. [DOI] [PubMed] [Google Scholar]
- Gómez P. Buckling A. Coevolution with phages does not influence the evolution of bacterial mutation rates in soil. ISME J. 2013;7:2242. doi: 10.1038/ismej.2013.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AR, Scanlan PD. Buckling A. Bacteria-phage coevolution and the emergence of generalist pathogens. Am Nat. 2011a;177:44–53. doi: 10.1086/657441. [DOI] [PubMed] [Google Scholar]
- Hall AR, Scanlan PD, Morgan AD. Buckling A. Host-parasite coevolutionary arms races give way to fluctuating selection. Ecol Lett. 2011b;14:635–642. doi: 10.1111/j.1461-0248.2011.01624.x. [DOI] [PubMed] [Google Scholar]
- Harcombe WR. Bull JJ. Impact of phages on two-species bacterial communities. Appl Environ Microbiol. 2005;71:5254–5259. doi: 10.1128/AEM.71.9.5254-5259.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison E, Laine A-L, Hietala M. Brockhurst MA. Rapidly fluctuating environments constrain coevolutionary arms races by impeding selective sweeps. Proc Biol Sci. 2013;280:20130937. doi: 10.1098/rspb.2013.0937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heineman RH, Springman R. Bull JJ. Optimal foraging by bacteriophages through host avoidance. Am Nat. 2008;171:E149–E157. doi: 10.1086/528962. [DOI] [PubMed] [Google Scholar]
- Held N. Whitaker RJ. Viral biogeography revealed by signatures in Sulfolobus islandicus genomes. Environ Microbiol. 2009;11:457–466. doi: 10.1111/j.1462-2920.2008.01784.x. [DOI] [PubMed] [Google Scholar]
- Held NL, Herrera A, Cadillo-Quiroz H. Whitaker RJ. CRISPR associated diversity within a population of Sulfolobus islandicus. PLoS One. 2010;5:e12988. doi: 10.1371/journal.pone.0012988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horne MT. Coevolution of Escherichia coli and bacteriophages in chemostat culture. Science. 1970;168:992. doi: 10.1126/science.168.3934.992-a. [DOI] [PubMed] [Google Scholar]
- Hosokawa T, Kikuchi Y, Nikoh N, Shimada M. Fukatsu T. Strict host-symbiont cospeciation and reductive genome evolution in insect gut bacteria. PLoS Biol. 2006;4:e337. doi: 10.1371/journal.pbio.0040337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseinidoust Z, Tufenkji N. van de Ven TGM. Predation in homogeneous and heterogeneous phage environments affects virulence determinants of Pseudomonas aeruginosa. Appl Environ Microbiol. 2013;79:2862–2871. doi: 10.1128/AEM.03817-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Høyland-Kroghsbo NM, Mærkedahl RB. Svenningsen SL. A quorum-sensing-induced bacteriophage defense mechanism. MBio. 2013;4:e00362–12. doi: 10.1128/mBio.00362-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman P. Abedon ST. Bacteriophage host range and bacterial resistance. Adv Appl Microbiol. 2010;70:217–248. doi: 10.1016/S0065-2164(10)70007-1. [DOI] [PubMed] [Google Scholar]
- Janzen DH. When is it coevolution? Evolution. 1980;34:611–612. doi: 10.1111/j.1558-5646.1980.tb04849.x. [DOI] [PubMed] [Google Scholar]
- Jessup CM. Bohannan BJ. The shape of an ecological trade-off varies with environment. Ecol Lett. 2008;11:947–959. doi: 10.1111/j.1461-0248.2008.01205.x. [DOI] [PubMed] [Google Scholar]
- Joo J, Gunny M, Cases M, Hudson P, Albert R. Harvill E. Bacteriophage-mediated competition in Bordetella bacteria. Proc R Soc Lond B Biol Sci. 2006;273:1843–1848. doi: 10.1098/rspb.2006.3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost G. Wiese J. Temporal variations in the concentrations of bacteria and their lytic phages: an example of an indigenous phage host system in Lake Plusharpsee, Germany. Fundam Appl Limnol. 2013;182:183–190. [Google Scholar]
- Kang I, Oh HM, Kang D. Cho JC. Genome of a SAR116 bacteriophage shows the prevalence of this phage type in the oceans. P Natl Acad Sci USA. 2013;110:12343–12348. doi: 10.1073/pnas.1219930110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerstes N, Berenos C, Schmid-Hempel P. Wegner KM. Antagonistic experimental coevolution with a parasite increases host recombination frequency. BMC Evol Biol. 2012;12:18. doi: 10.1186/1471-2148-12-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidambi SP, Ripp S. Miller RV. Evidence for phage-mediated gene transfer among Pseudomonas aeruginosa strains on the phylloplane. Appl Environ Microbiol. 1994;60:496–500. doi: 10.1128/aem.60.2.496-500.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S, Sako Y. Yoshida T. Rapid Microcystis cyanophage gene diversification revealed by long- and short-term genetic analyses of the tail sheath gene in a natural pond. Appl Environ Microbiol. 2013;79:2789–2795. doi: 10.1128/AEM.03751-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koskella B. Phage-mediated selection on microbiota of a long-lived host. Curr Biol. 2013;23:1256–1260. doi: 10.1016/j.cub.2013.05.038. [DOI] [PubMed] [Google Scholar]
- Koskella B. Lively CM. Evidence for negative frequency-dependent selection during experimental coevolution of a freshwater snail and a sterilizing trematode. Evolution. 2009;63:2213–2221. doi: 10.1111/j.1558-5646.2009.00711.x. [DOI] [PubMed] [Google Scholar]
- Koskella B. Meaden S. Understanding bacteriophage specificity in natural microbial communities. Viruses. 2013;5:806–823. doi: 10.3390/v5030806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koskella B, Thompson JN, Preston GM. Buckling A. Local biotic environment shapes the spatial scale of bacteriophage adaptation to bacteria. Am Nat. 2011;177:440–451. doi: 10.1086/658991. [DOI] [PubMed] [Google Scholar]
- Koskella B, Lin DM, Buckling A. Thompson JN. The costs of evolving resistance in heterogeneous parasite environments. Proc R Soc Lond B Biol Sci. 2012;279:1896–1903. doi: 10.1098/rspb.2011.2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunin V, He S, Warnecke F, Peterson SB, Martin HG, Haynes M, Ivanova N, Blackall LL, Breitbart M. Rohwer F. A bacterial metapopulation adapts locally to phage predation despite global dispersal. Genome Res. 2008;18:293–297. doi: 10.1101/gr.6835308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuno S, Yoshida T, Kaneko T. Sako Y. Intricate interactions between the bloom-forming cyanobacterium Microcystis aeruginosa and foreign genetic elements, revealed by diversified clustered regularly interspaced short palindromic repeat (CRISPR) signatures. Appl Environ Microbiol. 2012;78:5353–5360. doi: 10.1128/AEM.00626-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrie SJ, Samson JE. Moineau S. Bacteriophage resistance mechanisms. Nat Rev Microbiol. 2010;8:317–327. doi: 10.1038/nrmicro2315. [DOI] [PubMed] [Google Scholar]
- Lennon JT, Khatana SAM, Marston MF. Martiny JB. Is there a cost of virus resistance in marine cyanobacteria? ISME J. 2007;1:300–312. doi: 10.1038/ismej.2007.37. [DOI] [PubMed] [Google Scholar]
- Lenski RE. Experimental studies of pleiotropy and epistasis in Escherichia coli. II. Compensation for maladaptive effects associated with resistance to virus T4. Evolution. 1988;42:433–440. doi: 10.1111/j.1558-5646.1988.tb04150.x. [DOI] [PubMed] [Google Scholar]
- Lenski RE. Levin BR. Constraints on the coevolution of bacteria and virulent phage – a model, some experiments, and predictions for natural communities. Am Nat. 1985;125:585–602. [Google Scholar]
- Levin BR, Moineau S, Bushman M. Barrangou R. The population and evolutionary dynamics of phage and bacteria with CRISPR-mediated immunity. PLoS Genet. 2013;9:e1003312. doi: 10.1371/journal.pgen.1003312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T-Y, Lo Y-H, Tseng P-W, Chang S-F, Lin Y-T. Chen T-S. A T3 and T7 recombinant phage acquires efficient adsorption and a broader host range. PLoS One. 2012;7:e30954. doi: 10.1371/journal.pone.0030954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lively CM. Migration, virulence, and the geographic mosaic of adaptation by parasites. Am Nat. 1999;153:S34–S47. doi: 10.1086/303210. [DOI] [PubMed] [Google Scholar]
- Lopez-Pascua LDC. Buckling A. Increasing productivity accelerates host-parasite coevolution. J Evol Biol. 2008;21:853–860. doi: 10.1111/j.1420-9101.2008.01501.x. [DOI] [PubMed] [Google Scholar]
- Lopez-Pascua LDC, Brockhurst MA. Buckling A. Antagonistic coevolution across productivity gradients: an experimental test of the effects of dispersal. J Evol Biol. 2010;23:207–211. doi: 10.1111/j.1420-9101.2009.01877.x. [DOI] [PubMed] [Google Scholar]
- Lopez-Pascua L, Gandon S. Buckling A. Abiotic heterogeneity drives parasite local adaptation in coevolving bacteria and phages. J Evol Biol. 2012;25:187–195. doi: 10.1111/j.1420-9101.2011.02416.x. [DOI] [PubMed] [Google Scholar]
- Marston MF. Sallee JL. Genetic diversity and temporal variation in the Cyanophage community infecting marine Synechococcus species in Rhode Island's coastal waters. Appl Environ Microbiol. 2003;69:4639–4647. doi: 10.1128/AEM.69.8.4639-4647.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marston MF, Pierciey FJ, Shepard A, Gearin G, Qi J, Yandava C, Schuster SC, Henn MR. Martiny JBH. Rapid diversification of coevolving marine Synechococcus and a virus. P Natl Acad Sci USA. 2012;109:4544–4549. doi: 10.1073/pnas.1120310109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez JM, Schroeder DC, Larsen A, Bratbak G. Wilson WH. Molecular dynamics of Emiliania huxleyi and cooccurring viruses during two separate mesocosm studies. Appl Environ Microbiol. 2007;73:554–562. doi: 10.1128/AEM.00864-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martiny JB, Riemann L, Marston MF. Middelboe M. Antagonistic coevolution of marine planktonic viruses and their hosts. Ann Rev Mar Sci. 2014;6:393–414. doi: 10.1146/annurev-marine-010213-135108. [DOI] [PubMed] [Google Scholar]
- Mathias CB, Kirschner A. Velimirov B. Seasonal variations of virus abundance and viral control of the bacterial production in a backwater system of the Danube river. Appl Environ Microbiol. 1995;61:3734–3740. doi: 10.1128/aem.61.10.3734-3740.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messenger SL, Molineux IJ. Bull J. Virulence evolution in a virus obeys a trade off. Proc Biol Sci. 1999;266:397–404. doi: 10.1098/rspb.1999.0651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer JR, Dobias DT, Weitz JS, Barrick JE, Quick RT. Lenski RE. Repeatability and contingency in the evolution of a key innovation in phage Lambda. Science. 2012;335:428–432. doi: 10.1126/science.1214449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middelboe M, Hagström A, Blackburn N, Sinn B, Fischer U, Borch N, Pinhassi J, Simu K. Lorenz M. Effects of bacteriophages on the population dynamics of four strains of pelagic marine bacteria. Microb Ecol. 2001;42:395–406. doi: 10.1007/s00248-001-0012-1. [DOI] [PubMed] [Google Scholar]
- Middelboe M, Riemann L, Steward GF, Hansen V. Nybroe O. Virus-induced transfer of organic carbon between marine bacteria in a model community. Aquat Microb Ecol. 2003;33:1–10. [Google Scholar]
- Middelboe M, Holmfeldt K, Riemann L, Nybroe O. Haaber J. Bacteriophages drive strain diversification in a marine Flavobacterium: implications for phage resistance and physiological properties. Environ Microbiol. 2009;11:1971–1982. doi: 10.1111/j.1462-2920.2009.01920.x. [DOI] [PubMed] [Google Scholar]
- Minot S, Bryson A, Chehoud C, Wu GD, Lewis JD. Bushman FD. Rapid evolution of the human gut virome. P Natl Acad Sci USA. 2013;110:12450–12455. doi: 10.1073/pnas.1300833110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizoguchi K, Morita M, Fischer CR, Yoichi M, Tanji Y. Unno H. Coevolution of bacteriophage PP01 and Escherichia coli O157:H7 in continuous culture. Appl Environ Microbiol. 2003;69:170–176. doi: 10.1128/AEM.69.1.170-176.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molineux IJ. Panja D. Popping the cork: mechanisms of phage genome ejection. Nat Rev Microbiol. 2013;11:194–204. doi: 10.1038/nrmicro2988. [DOI] [PubMed] [Google Scholar]
- Moreno Switt AI, den Bakker HC, Vongkamjan K, Hoelzer K, Warnick LD, Cummings KJ. Wiedmann M. Salmonella bacteriophage diversity reflects host diversity on dairy farms. Food Microbiol. 2013;36:275–285. doi: 10.1016/j.fm.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AD, Gandon S. Buckling A. The effect of migration on local adaptation in a coevolving host-parasite system. Nature. 2005;437:253–256. doi: 10.1038/nature03913. [DOI] [PubMed] [Google Scholar]
- Morgan AD, Brockhurst MA, Lopez-Pascua LDC, Pal C. Buckling A. Differential impact of simultaneous migration on coevolving hosts and parasites. BMC Evol Biol. 2007;7:1. doi: 10.1186/1471-2148-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morran LT, Schmidt OG, Gelarden IA, Parrish RC. Lively CM. Running with the Red Queen: host-parasite coevolution selects for biparental sex. Science. 2011;333:216–218. doi: 10.1126/science.1206360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechaev S. Severinov K. The elusive object of desire—Interactions of bacteriophages and their hosts. Curr Opin Microbiol. 2008;11:186–193. doi: 10.1016/j.mib.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needham DM, Chow C-ET, Cram JA, Sachdeva R, Parada A. Fuhrman JA. Short-term observations of marine bacterial and viral communities: patterns, connections and resilience. ISME J. 2013;7:1274–1285. doi: 10.1038/ismej.2013.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez-Espino D, Morovic W, Sun CL, Thomas BC, K-i Ueda, Stahl B, Barrangou R. Banfield JF. Strong bias in the bacterial CRISPR elements that confer immunity to phage. Nat Commun. 2013;4:1430. doi: 10.1038/ncomms2440. [DOI] [PubMed] [Google Scholar]
- Pal C, Maciá MD, Oliver A, Schachar I. Buckling A. Coevolution with viruses drives the evolution of bacterial mutation rates. Nature. 2007;450:1079–1081. doi: 10.1038/nature06350. [DOI] [PubMed] [Google Scholar]
- Paterson S, Vogwill T, Buckling A, et al. Antagonistic coevolution accelerates molecular evolution. Nature. 2010;464:275–278. doi: 10.1038/nature08798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paynter MJB. Bungay HR. Dynamics of coliphage infections. In: Perlman D, editor; Fermentation Advances. New York, NY: Academic Press; 1969. pp. 323–336. [Google Scholar]
- Poisot T, Lounnas M. Hochberg M. The structure of natural microbial enemy-victim networks. Ecol Process. 2013;2:1–9. [Google Scholar]
- Poullain V, Gandon S, Brockhurst MA, Buckling A. Hochberg ME. The evolution of specificity in evolving and coevolving antagonistic interactions between a bacteria and its phage. Evolution. 2008;62:1–11. doi: 10.1111/j.1558-5646.2007.00260.x. [DOI] [PubMed] [Google Scholar]
- Quance MA. Travisano M. Effects of temperature on the fitness cost of resistance to bacteriophage T4 in Escherichia coli. Evolution. 2009;63:1406–1416. doi: 10.1111/j.1558-5646.2009.00654.x. [DOI] [PubMed] [Google Scholar]
- Refardt D. Rainey PB. Tuning a genetic switch: experimental evolution and natural variation of prophage induction. Evolution. 2010;64:1086–1097. doi: 10.1111/j.1558-5646.2009.00882.x. [DOI] [PubMed] [Google Scholar]
- Refardt D, Bergmiller T. Kümmerli R. Altruism can evolve when relatedness is low: evidence from bacteria committing suicide upon phage infection. Proc R Soc Lond B. 2013;280:1759. doi: 10.1098/rspb.2012.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rho M, Wu Y-W, Tang H, Doak TG. Ye Y. Diverse CRISPRs evolving in human microbiomes. PLoS Genet. 2012;8:e1002441. doi: 10.1371/journal.pgen.1002441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riede I. T-even type phages can change their host range by recombination with gene34 (tail fibre) or gene23 (head) Mol Gen Genet. 1986;205:160–163. doi: 10.1007/BF02428046. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Brito B, Li L, Wegley L, et al. Viral and microbial community dynamics in four aquatic environments. ISME J. 2010;4:739–751. doi: 10.1038/ismej.2010.1. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Valera F, Martin-Cuadrado A-B, Rodriguez-Brito B, Pašić L, Thingstad TF, Rohwer F. Mira A. Explaining microbial population genomics through phage predation. Nat Rev Microbiol. 2009;7:828–836. doi: 10.1038/nrmicro2235. [DOI] [PubMed] [Google Scholar]
- Samson JE, Magadán AH, Sabri M. Moineau S. Revenge of the phages: defeating bacterial defences. Nat Rev Microbiol. 2013;11:675–687. doi: 10.1038/nrmicro3096. [DOI] [PubMed] [Google Scholar]
- Scanlan PD, Hall AR, Lopez-Pascua LDC. Buckling A. Genetic basis of infectivity evolution in a bacteriophage. Mol Ecol. 2011;20:981–989. doi: 10.1111/j.1365-294X.2010.04903.x. [DOI] [PubMed] [Google Scholar]
- Scanlan PD, Hall AR, Burlinson P, Preston G. Buckling A. No effect of host–parasite co-evolution on host range expansion. J Evol Biol. 2013;26:205–209. doi: 10.1111/jeb.12021. [DOI] [PubMed] [Google Scholar]
- Schrag SJ. Mittler JE. Host-parasite coexistence: the role of spatial refuges in stabilizing bacteria-phage interactions. Am Nat. 1996;148:348–377. [Google Scholar]
- Schwinghamer E. Brockwell J. Competitive advantage of bacteriocin and phage-producing strains of Rhizobium trifolii in mixed culture. Soil Biol Biochem. 1978;10:383–387. [Google Scholar]
- Seed KD, Lazinski DW, Calderwood SB. Camilli A. A bacteriophage encodes its own CRISPR/Cas adaptive response to evade host innate immunity. Nature. 2013;494:489–491. doi: 10.1038/nature11927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro OH, Kushmaro A. Brenner A. Bacteriophage predation regulates microbial abundance and diversity in a full-scale bioreactor treating industrial wastewater. ISME J. 2010;4:327–336. doi: 10.1038/ismej.2009.118. [DOI] [PubMed] [Google Scholar]
- Sharp PM. Simmonds P. Evaluating the evidence for virus/host co-evolution. Curr Opin Virol. 2011;1:436–441. doi: 10.1016/j.coviro.2011.10.018. [DOI] [PubMed] [Google Scholar]
- Shcherbakov VP, Plugina L. Nesheva M. Genetic recombination in bacteriophage T4: single-burst analysis of cosegregants and evidence in favor of a splice/patch coupling model. Genetics. 1992;131:769–781. doi: 10.1093/genetics/131.4.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber M. Gudelj I. Do-or-die life cycles and diverse post-infection resistance mechanisms limit the evolution of parasite host ranges. Ecol Lett. 2014;17:491–498. doi: 10.1111/ele.12249. [DOI] [PubMed] [Google Scholar]
- Smillie CS, Smith MB, Friedman J, Cordero OX, David LA. Alm EJ. Ecology drives a global network of gene exchange connecting the human microbiome. Nature. 2011;480:241–244. doi: 10.1038/nature10571. [DOI] [PubMed] [Google Scholar]
- Snyder JC. Young MJ. Advances in understanding archaea-virus interactions in controlled and natural environments. Curr Opin Microbiol. 2011;14:497–503. doi: 10.1016/j.mib.2011.07.007. [DOI] [PubMed] [Google Scholar]
- Spanakis E. Horne MT. Coadaptation of Escherichia coli and coliphage Lambda-vir in continuous culture. J Gen Microbiol. 1987;133:353–360. doi: 10.1099/00221287-133-2-353. [DOI] [PubMed] [Google Scholar]
- Stern A. Sorek R. The phage-host arms race: shaping the evolution of microbes. BioEssays. 2011;33:43–51. doi: 10.1002/bies.201000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern A, Mick E, Tirosh I, Sagy O. Sorek R. CRISPR targeting reveals a reservoir of common phages associated with the human gut microbiome. Genome Res. 2012;22:1985–1994. doi: 10.1101/gr.138297.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stouffer DB. Bascompte J. Compartmentalization increases food-web persistence. P Natl Acad Sci USA. 2011;108:3648–3652. doi: 10.1073/pnas.1014353108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MB, Waterbury JB. Chisholm SW. Cyanophages infecting the oceanic cyanobacterium Prochlorococcus. Nature. 2003;424:1047–1051. doi: 10.1038/nature01929. [DOI] [PubMed] [Google Scholar]
- Sun CL, Barrangou R, Thomas BC, Horvath P, Fremaux C. Banfield JF. Phage mutations in response to CRISPR diversification in a bacterial population. Environ Microbiol. 2013;15:463–470. doi: 10.1111/j.1462-2920.2012.02879.x. [DOI] [PubMed] [Google Scholar]
- Suttle CA. Marine viruses — major players in the global ecosystem. Nat Rev Microbiol. 2007;5:801–812. doi: 10.1038/nrmicro1750. [DOI] [PubMed] [Google Scholar]
- Thingstad TF. Elements of a theory for the mechanisms controlling abundance, diversity, and biogeochemical role of lytic bacterial viruses in aquatic systems. Limnol Oceanogr. 2000;45:1320–1328. [Google Scholar]
- Thomas R, Berdjeb L, Sime-Ngando T. Jacquet S. Viral abundance, production, decay rates and life strategies (lysogeny versus lysis) in Lake Bourget (France) Environ Microbiol. 2011;13:616–630. doi: 10.1111/j.1462-2920.2010.02364.x. [DOI] [PubMed] [Google Scholar]
- Varani AM, Monteiro-Vitorello CB, Nakaya HI. Van Sluys M-A. The role of prophage in plant-pathogenic bacteria. Annu Rev Phytopathol. 2013;51:429–451. doi: 10.1146/annurev-phyto-081211-173010. [DOI] [PubMed] [Google Scholar]
- Vardi A, Haramaty L, Van Mooy BAS, Fredricks HF, Kimmance SA, Larsen A. Bidle KD. Host–virus dynamics and subcellular controls of cell fate in a natural coccolithophore population. P Natl Acad Sci USA. 2012;109:19327–19332. doi: 10.1073/pnas.1208895109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogwill T, Fenton A. Brockhurst MA. The impact of parasite dispersal on antagonistic host-parasite coevolution. J Evol Biol. 2008;21:1252–1258. doi: 10.1111/j.1420-9101.2008.01574.x. [DOI] [PubMed] [Google Scholar]
- Vogwill T, Fenton A. Brockhurst MA. Dispersal and natural enemies interact to drive spatial synchrony and decrease stability in patchy populations. Ecol Lett. 2009a;12:1194–1200. doi: 10.1111/j.1461-0248.2009.01374.x. [DOI] [PubMed] [Google Scholar]
- Vogwill T, Fenton A, Buckling A, Hochberg ME. Brockhurst MA. Source populations act as coevolutionary pacemakers in experimental selection mosaics containing hotspots and coldspots. Am Nat. 2009b;173:E171–E176. doi: 10.1086/597374. [DOI] [PubMed] [Google Scholar]
- Vogwill T, Fenton A. Brockhurst MA. How does spatial dispersal network affect the evolution of parasite local adaptation? Evolution. 2010;64:1795–1801. doi: 10.1111/j.1558-5646.2009.00937.x. [DOI] [PubMed] [Google Scholar]
- Vogwill T, Fenton A. Brockhurst MA. Coevolving parasites enhance the diversity-decreasing effect of dispersal. Biol Lett. 2011;7:578–580. doi: 10.1098/rsbl.2011.0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos M, Birkett PJ, Birch E, Griffiths RI. Buckling A. Local adaptation of bacteriophages to their bacterial hosts in soil. Science. 2009;325:833. doi: 10.1126/science.1174173. [DOI] [PubMed] [Google Scholar]
- Waldor MK. Mekalanos JJ. Lysogenic conversion by a filamentous phage encoding Cholera toxin. Science. 1996;272:1910–1914. doi: 10.1126/science.272.5270.1910. [DOI] [PubMed] [Google Scholar]
- Waterbury JB. Valois FW. Resistance to co-occurring phages enables marine Synechococcus communities to coexist with cyanophages abundant in seawater. Appl Environ Microbiol. 1993;59:3393–3399. doi: 10.1128/aem.59.10.3393-3399.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Ocampo P. Levin BR. An experimental study of the population and evolutionary dynamics of Vibrio cholerae O1 and the bacteriophage JSF4. Proc R Soc Lond B. 2010;277:3247–3254. doi: 10.1098/rspb.2010.0651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Kirby A. Levin BR. The population and evolutionary dynamics of Vibrio cholerae and its bacteriophage: conditions for maintaining phage-limited communities. Am Nat. 2011;178:715–725. doi: 10.1086/662677. [DOI] [PubMed] [Google Scholar]
- Weinbauer MG. Ecology of prokaryotic viruses. FEMS Microbiol Rev. 2004;28:127–181. doi: 10.1016/j.femsre.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Weinbauer MG. Rassoulzadegan F. Are viruses driving microbial diversification and diversity? Environ Microbiol. 2004;6:1–11. doi: 10.1046/j.1462-2920.2003.00539.x. [DOI] [PubMed] [Google Scholar]
- Weinbauer MG, Hornák K, Jezbera J, Nedoma J, Dolan JR. Šimek K. Synergistic and antagonistic effects of viral lysis and protistan grazing on bacterial biomass, production and diversity. Environ Microbiol. 2007;9:777–788. doi: 10.1111/j.1462-2920.2006.01200.x. [DOI] [PubMed] [Google Scholar]
- Weitz JS, Poisot T, Meyer JR, Flores CO, Valverde S, Sullivan MB. Hochberg ME. Phage–bacteria infection networks. Trends Microbiol. 2013;21:82–91. doi: 10.1016/j.tim.2012.11.003. [DOI] [PubMed] [Google Scholar]
- Westra ER, Swarts DC, Staals RH, Jore MM, Brouns SJ. van der Oost J. The CRISPRs, they are a-changin': how prokaryotes generate adaptive immunity. Annu Rev Genet. 2012;46:311–339. doi: 10.1146/annurev-genet-110711-155447. [DOI] [PubMed] [Google Scholar]
- Williamson KE, Corzo KA, Drissi CL, Buckingham JM, Thompson CP. Helton RR. Estimates of viral abundance in soils are strongly influenced by extraction and enumeration methods. Biol Fertil Soils. 2013;49:857–869. [Google Scholar]
- Winter C, Bouvier T, Weinbauer MG. Thingstad TF. Trade-Offs between competition and defense specialists among unicellular planktonic organisms: the “Killing the Winner” hypothesis revisited. Microbiol Mol Biol Rev. 2010;74:42–57. doi: 10.1128/MMBR.00034-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolhouse MEJ, Webster JP, Domingo E, Charlesworth B. Levin BR. Biological and biomedical implications of the co-evolution of pathogens and their hosts. Nat Genet. 2002;32:569–577. doi: 10.1038/ng1202-569. [DOI] [PubMed] [Google Scholar]
- Worobey M. Holmes EC. Evolutionary aspects of recombination in RNA viruses. J Gen Virol. 1999;80:2535–2543. doi: 10.1099/0022-1317-80-10-2535. [DOI] [PubMed] [Google Scholar]
- Yoon SH, Han MJ, Jeong H, Lee CH, Xia XX, Lee DH, Shim JH, Lee SY, Oh TK. Kim JF. Comparative multi-omics systems analysis of Escherichia coli strains B and K-12. Genome Biol. 2012;13:R37. doi: 10.1186/gb-2012-13-5-r37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young R. Phage lysis: do we have the hole story yet? Curr Opin Microbiol. 2013;16:790–797. doi: 10.1016/j.mib.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]