

Abstract
The title compound, C5H6N4O, is approximately planar, with an angle of 11.04 (15)° between the planes of the pyrimidine ring and the non-H atoms of the carboximidamide unit. The molecule adopts an E configuration about the C=N double bond. In the crystal, adjacent molecules are linked by pairs of N—H⋯O hydrogen bonds, forming inversion dimers with an R 2 2(10) ring motif. The dimers are further linked via N—H⋯N and O—H⋯N hydrogen bonds into a sheet structure parallel to the ac plane. The crystal structure also features N—H⋯O and weak C—H⋯O hydrogen bonds and offset π–π stacking interactions between adjacent pyrimidine rings [centroid–centroid distance = 3.622 (1) Å].
Keywords: crystal structure, pyrimidine-2-carboximidamide, non-covalent interactions, hydrogen bonding, π–π stacking interactions, biological activity
Related literature
For details of non-covalent interactions, see: Desiraju (2007 ▶). For the role of intermolecular hydrogen bonds in the design of organic crystals, see: Aakeroy & Seddon (1993 ▶). For background to substituted N′-hydroxybenzamidines as intermediates in the synthesis of 1,2,4-oxadiazole derivatives, see: Kundu et al. (2012 ▶). For the biological activity of substituted N′-hydroxybenzamidines and 1,2,4-oxadiazole derivatives, see: Sakamoto et al. (2007 ▶); Tyrkov & Sukhenko (2004 ▶).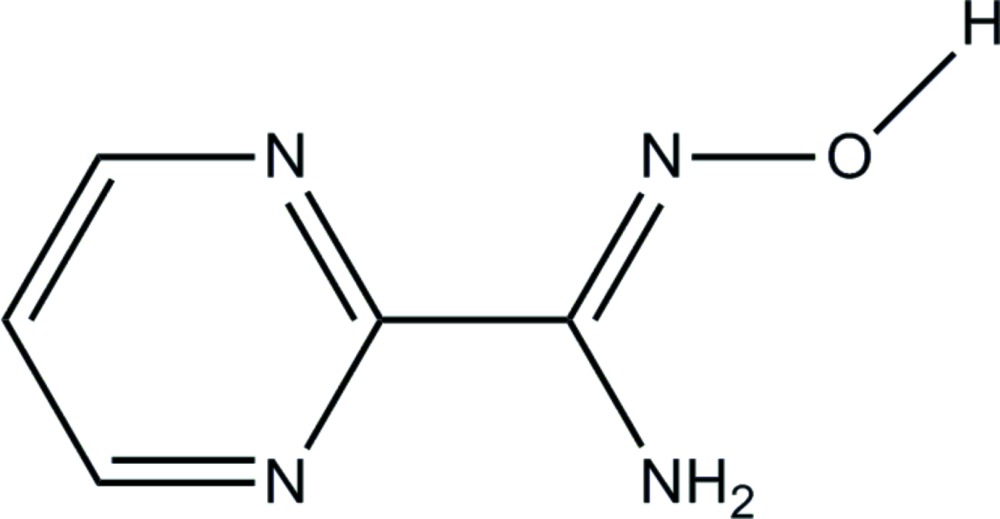
Experimental
Crystal data
C5H6N4O
M r = 138.14
Monoclinic,

a = 7.4066 (7) Å
b = 8.0165 (8) Å
c = 10.2200 (9) Å
β = 101.888 (6)°
V = 593.8 (1) Å3
Z = 4
Mo Kα radiation
μ = 0.12 mm−1
T = 100 K
0.62 × 0.17 × 0.08 mm
Data collection
Bruker SMART APEXII CCD area-detector diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2009 ▶) T min = 0.931, T max = 0.990
4073 measured reflections
1030 independent reflections
831 reflections with I > 2σ(I)
R int = 0.047
Refinement
R[F 2 > 2σ(F 2)] = 0.054
wR(F 2) = 0.181
S = 1.13
1030 reflections
103 parameters
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.29 e Å−3
Δρmin = −0.31 e Å−3
Data collection: APEX2 (Bruker, 2009 ▶); cell refinement: SAINT (Bruker, 2009 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXTL; molecular graphics: SHELXTL; software used to prepare material for publication: SHELXTL and PLATON (Spek, 2009 ▶).
Supplementary Material
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S1600536814020285/sj5423sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536814020285/sj5423Isup2.hkl
Supporting information file. DOI: 10.1107/S1600536814020285/sj5423Isup3.cml
. DOI: 10.1107/S1600536814020285/sj5423fig1.tif
The molecular structure of the title compound with atom labels. Displacement ellipsoids are shown at the 50% probability level.
b . DOI: 10.1107/S1600536814020285/sj5423fig2.tif
The crystal packing of the title compound viewed along the b axis die=rection. H atoms not involved in intermolecular interactions (dashed lines) have been omitted for clarity.
CCDC reference: 1018015
Additional supporting information: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N4—H2N4⋯O1i | 0.89 (3) | 2.27 (3) | 2.996 (3) | 139 (3) |
| N4—H1N4⋯N3ii | 0.92 (3) | 2.30 (3) | 3.106 (3) | 146 (3) |
| O1—H1O1⋯N2iii | 0.95 (4) | 1.85 (4) | 2.783 (3) | 167 (3) |
| C3—H3A⋯O1iv | 0.95 | 2.51 | 3.305 (4) | 141 |
Symmetry codes: (i)  ; (ii)
; (ii)  ; (iii)
; (iii)  ; (iv)
; (iv)  .
.
Acknowledgments
NJJ thanks the UGC–SAP for the award of an RFSMS. PTM thanks the UGC for a one-time BSR grant.
supplementary crystallographic information
S1. Comment
Supramolecular architectures assembled via various delicate noncovalent interactions such as hydrogen bonds, π—π stacking and electrostatic interactions, have attracted intense interest in recent years because of their fascinating structural diversity and potential applications for functional materials (Desiraju, 2007). In particular, the application of intermolecular hydrogen bonding is a well known and efficient tool in the field of organic crystal design owing to their strength and directional properties (Aakeroy & Seddon, 1993). Substituted N'-hydroxybenzamidines are important intermediates obtained during the synthesis of pharmaceuticaly important 1,2,4-oxadiazole derivatives (Kundu et al., 2012). 1,2,4-oxadiazole derivatives are well known for their biological activities such as for anti-HIV (Sakamoto et al., 2007) and anti-microbial applications (Tyrkov & Sukhenko, 2004). Herein, we report the crystal structure determination of the title compound, (I).
The asymmetric unit of the title compound is shown in Fig. 1. The essentially planar pyrimidine ring [N1/N2/C1–C4, maximum deviation of 0.009 (2) Å at atom C4] forms a dihedral angle of 11.04 (15)° with the hydroxyacetimidamide (N4/C5/N3/O1). The compound adopts an E configuration across the C5═N3 double bond, as the OH group and benzene ring are on opposite sides of the double bond while the hydrogen atom of the hydroxy group is directed away from the NH2 group. The bond lengths and angles are within normal ranges.
In the crystal packing, molecules are linked by a pair of N4—H2N4···O1i hydrogen bonds (symmetry code in Table 1) into an inversion dimer, forming an R22(10) ring motif. These molecules are self-assembled via N4—H1N4···N3ii hydrogen bonds (graph-set notation C(4); symmetry code as in Table 1), which interconnect the dimers resulting in a sheet parallel to the ac plane as shown in Fig 2. Furthermore, the crystal structure is stabilized by O1–H1O1···N2iii and weak C3—H3A···O1iv hydrogen bonds (symmetry code as in Table 1) and π–π stacking interactions between the pyrimidine (N1/N2/C1-C4) rings [centroid-centroid distance = 3.622 (1) Å; (symmetry code: 1-x, - y, -z)].
S2. Experimental
A hot methanol solution (20 ml) of N'-hydroxypyrimidine- 2-carboximidamide (69 mg, Aldrich) was warmed over a magnetic stirrer hotplate for a few minutes. The resulting solution was allowed to cool slowly at room temperature. Single crystals of the title compound (I) appeared from the mother liquor after a few days.
S3. Refinement
O- and N-bound H atoms were located in a difference Fourier map and refined freely [O–H = 0.94 (4) Å and N–H = 0.89 (3) Å and 0.92 (3) Å]. The remaining hydrogen atoms were positioned geometrically [C–H= 0.95 Å] and were refined using a riding model, with Uiso(H)=1.2 Ueq(C).
Figures
Fig. 1.

The molecular structure of the title compound with atom labels. Displacement ellipsoids are shown at the 50% probability level.
Fig. 2.
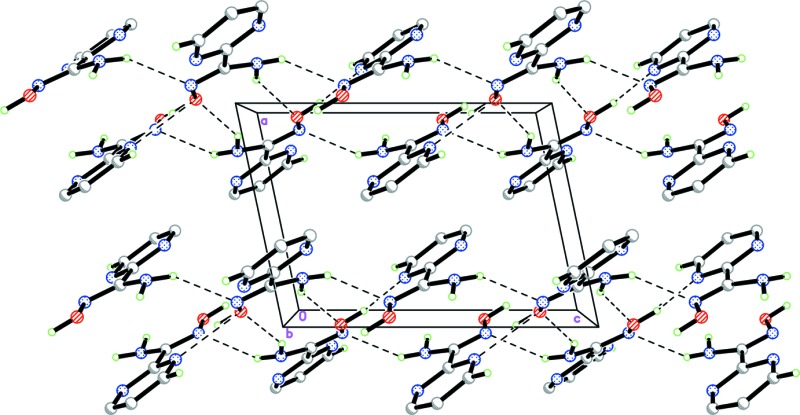
The crystal packing of the title compound viewed along the b axis die=rection. H atoms not involved in intermolecular interactions (dashed lines) have been omitted for clarity.
Crystal data
| C5H6N4O | F(000) = 288 |
| Mr = 138.14 | Dx = 1.545 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 1905 reflections |
| a = 7.4066 (7) Å | θ = 2.8–29.9° |
| b = 8.0165 (8) Å | µ = 0.12 mm−1 |
| c = 10.2200 (9) Å | T = 100 K |
| β = 101.888 (6)° | Plate, colourless |
| V = 593.8 (1) Å3 | 0.62 × 0.17 × 0.08 mm |
| Z = 4 |
Data collection
| Bruker SMART APEXII CCD area-detector diffractometer | 1030 independent reflections |
| Radiation source: fine-focus sealed tube | 831 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.047 |
| φ and ω scans | θmax = 25.0°, θmin = 2.8° |
| Absorption correction: multi-scan (SADABS; Bruker, 2009) | h = −8→8 |
| Tmin = 0.931, Tmax = 0.990 | k = −9→8 |
| 4073 measured reflections | l = −12→12 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.054 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.181 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.13 | w = 1/[σ2(Fo2) + (0.093P)2 + 0.7207P] where P = (Fo2 + 2Fc2)/3 |
| 1030 reflections | (Δ/σ)max < 0.001 |
| 103 parameters | Δρmax = 0.29 e Å−3 |
| 0 restraints | Δρmin = −0.31 e Å−3 |
Special details
| Experimental. The crystal was placed in the cold stream of an Oxford Cryosystems Cobra open-flow nitrogen cryostat operating at 100.0 (1) K. |
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | 0.9616 (3) | 0.4128 (2) | 0.15856 (18) | 0.0171 (6) | |
| N1 | 0.6515 (3) | 0.0148 (3) | −0.1252 (2) | 0.0159 (6) | |
| N2 | 0.8106 (3) | −0.0836 (3) | 0.0883 (2) | 0.0140 (6) | |
| N3 | 0.9064 (3) | 0.2423 (3) | 0.1497 (2) | 0.0150 (6) | |
| N4 | 0.8007 (3) | 0.3212 (3) | −0.0758 (2) | 0.0155 (6) | |
| C1 | 0.5875 (4) | −0.1406 (4) | −0.1538 (3) | 0.0183 (7) | |
| H1A | 0.5107 | −0.1612 | −0.2389 | 0.022* | |
| C2 | 0.6283 (4) | −0.2713 (4) | −0.0652 (3) | 0.0198 (7) | |
| H2A | 0.5801 | −0.3800 | −0.0867 | 0.024* | |
| C3 | 0.7425 (4) | −0.2368 (4) | 0.0560 (3) | 0.0183 (7) | |
| H3A | 0.7743 | −0.3244 | 0.1191 | 0.022* | |
| C4 | 0.7592 (3) | 0.0362 (3) | −0.0043 (2) | 0.0127 (6) | |
| C5 | 0.8291 (3) | 0.2095 (3) | 0.0268 (2) | 0.0126 (7) | |
| H2N4 | 0.869 (4) | 0.413 (4) | −0.057 (3) | 0.012 (7)* | |
| H1N4 | 0.784 (4) | 0.281 (4) | −0.162 (3) | 0.025 (8)* | |
| H1O1 | 1.029 (5) | 0.429 (4) | 0.247 (4) | 0.022 (8)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0221 (11) | 0.0123 (11) | 0.0157 (11) | −0.0038 (8) | 0.0012 (9) | −0.0005 (7) |
| N1 | 0.0142 (12) | 0.0173 (13) | 0.0169 (12) | −0.0003 (10) | 0.0048 (10) | −0.0023 (9) |
| N2 | 0.0130 (12) | 0.0135 (12) | 0.0156 (12) | 0.0008 (9) | 0.0033 (10) | 0.0000 (9) |
| N3 | 0.0164 (12) | 0.0098 (12) | 0.0185 (12) | −0.0026 (10) | 0.0027 (10) | −0.0001 (9) |
| N4 | 0.0196 (13) | 0.0134 (13) | 0.0130 (12) | −0.0015 (11) | 0.0023 (10) | 0.0016 (9) |
| C1 | 0.0139 (14) | 0.0209 (15) | 0.0212 (14) | −0.0018 (12) | 0.0058 (12) | −0.0060 (12) |
| C2 | 0.0166 (15) | 0.0129 (14) | 0.0319 (16) | −0.0030 (11) | 0.0096 (13) | −0.0071 (12) |
| C3 | 0.0177 (15) | 0.0147 (14) | 0.0246 (15) | 0.0015 (12) | 0.0096 (12) | 0.0004 (11) |
| C4 | 0.0092 (13) | 0.0160 (15) | 0.0138 (13) | 0.0006 (11) | 0.0043 (11) | −0.0032 (10) |
| C5 | 0.0099 (13) | 0.0137 (14) | 0.0160 (13) | 0.0011 (11) | 0.0065 (11) | 0.0002 (10) |
Geometric parameters (Å, º)
| O1—N3 | 1.424 (3) | N4—H2N4 | 0.89 (3) |
| O1—H1O1 | 0.94 (4) | N4—H1N4 | 0.92 (3) |
| N1—C4 | 1.336 (3) | C1—C2 | 1.378 (4) |
| N1—C1 | 1.343 (4) | C1—H1A | 0.9500 |
| N2—C3 | 1.343 (4) | C2—C3 | 1.376 (4) |
| N2—C4 | 1.347 (3) | C2—H2A | 0.9500 |
| N3—C5 | 1.295 (3) | C3—H3A | 0.9500 |
| N4—C5 | 1.362 (3) | C4—C5 | 1.494 (4) |
| N3—O1—H1O1 | 106.1 (18) | C3—C2—H2A | 121.6 |
| C4—N1—C1 | 116.0 (2) | C1—C2—H2A | 121.6 |
| C3—N2—C4 | 116.2 (2) | N2—C3—C2 | 122.4 (3) |
| C5—N3—O1 | 108.7 (2) | N2—C3—H3A | 118.8 |
| C5—N4—H2N4 | 112.9 (18) | C2—C3—H3A | 118.8 |
| C5—N4—H1N4 | 118 (2) | N1—C4—N2 | 125.9 (2) |
| H2N4—N4—H1N4 | 117 (3) | N1—C4—C5 | 115.6 (2) |
| N1—C1—C2 | 122.8 (2) | N2—C4—C5 | 118.5 (2) |
| N1—C1—H1A | 118.6 | N3—C5—N4 | 125.5 (2) |
| C2—C1—H1A | 118.6 | N3—C5—C4 | 117.4 (2) |
| C3—C2—C1 | 116.7 (3) | N4—C5—C4 | 117.1 (2) |
| C4—N1—C1—C2 | 0.2 (4) | C3—N2—C4—C5 | 179.3 (2) |
| N1—C1—C2—C3 | −1.1 (4) | O1—N3—C5—N4 | −1.7 (3) |
| C4—N2—C3—C2 | 0.8 (4) | O1—N3—C5—C4 | −179.51 (19) |
| C1—C2—C3—N2 | 0.5 (4) | N1—C4—C5—N3 | 168.3 (2) |
| C1—N1—C4—N2 | 1.3 (4) | N2—C4—C5—N3 | −12.7 (3) |
| C1—N1—C4—C5 | −179.8 (2) | N1—C4—C5—N4 | −9.7 (3) |
| C3—N2—C4—N1 | −1.8 (4) | N2—C4—C5—N4 | 169.3 (2) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N4—H2N4···O1i | 0.89 (3) | 2.27 (3) | 2.996 (3) | 139 (3) |
| N4—H1N4···N3ii | 0.92 (3) | 2.30 (3) | 3.106 (3) | 146 (3) |
| O1—H1O1···N2iii | 0.95 (4) | 1.85 (4) | 2.783 (3) | 167 (3) |
| C3—H3A···O1iv | 0.95 | 2.51 | 3.305 (4) | 141 |
Symmetry codes: (i) −x+2, −y+1, −z; (ii) x, −y+1/2, z−1/2; (iii) −x+2, y+1/2, −z+1/2; (iv) x, y−1, z.
Footnotes
Supporting information for this paper is available from the IUCr electronic archives (Reference: SJ5423).
References
- Aakeroy, C. B. & Seddon, K. R. (1993). Chem. Soc. Rev. 22, 397–407.
- Bruker (2009). SADABS, APEX2 and SAINT Bruker AXS Inc., Madison, Wisconsin, USA.
- Desiraju, G. R. (2007). Angew. Chem. Int. Ed. 46, 8342–8356. [DOI] [PubMed]
- Kundu, M., Singh, B., Ghosh, T., Maiti, B. C. & Maity, T. K. (2012). Indian J. Chem. Sect. B, 51, 493–497.
- Sakamoto, T., Cullen, M. D., Hartman, T. L., Watson, K. M., Buckheit, R. W., Pannecouque, C., DeClercq, E. & Cushman, M. (2007). J. Med. Chem. 50, 3314–3319. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Tyrkov, A. G. & Sukhenko, L. T. (2004). Pharm. Chem. J. 38, 30–38.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S1600536814020285/sj5423sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536814020285/sj5423Isup2.hkl
Supporting information file. DOI: 10.1107/S1600536814020285/sj5423Isup3.cml
. DOI: 10.1107/S1600536814020285/sj5423fig1.tif
The molecular structure of the title compound with atom labels. Displacement ellipsoids are shown at the 50% probability level.
b . DOI: 10.1107/S1600536814020285/sj5423fig2.tif
The crystal packing of the title compound viewed along the b axis die=rection. H atoms not involved in intermolecular interactions (dashed lines) have been omitted for clarity.
CCDC reference: 1018015
Additional supporting information: crystallographic information; 3D view; checkCIF report