

Abstract
In the title compound, C9H9NO3·H2O, the plane of the acetamide group is oriented at 20.52 (8)° with respect to the benzene ring, whereas the plane of the carboxylic acid group is essentially coplanar with the benzene ring [maximum deviation = 0.033 (1) Å]. In the crystal, classical O—H⋯O and N—H⋯O hydrogen bonds and weak C—H⋯O hydrogen bonds link the organic molecules and water molecules of crystallization into a three-dimensional supramolecular architecture.
Keywords: crystal structure, 4-acetamidobenzoic acid, hydrogen bonding, hydrated carboxylic acid
Related literature
For applications of 4-acetamidobenzoic acid in coordination chemistry, see: Yin et al. (2011 ▶); Wang et al. (2009 ▶). For related structures, see: Kashino et al. (1986 ▶); Jedrzejas et al. (1995 ▶).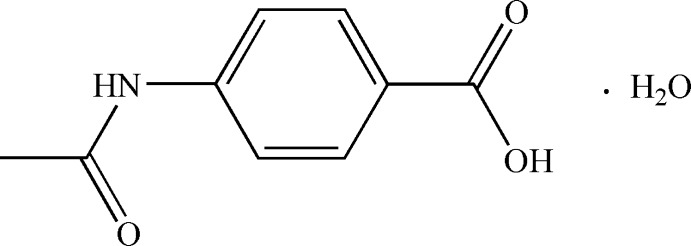
Experimental
Crystal data
C9H9NO3·H2O
M r = 197.19
Monoclinic,

a = 6.6712 (13) Å
b = 28.870 (6) Å
c = 4.992 (1) Å
β = 100.01 (3)°
V = 946.8 (3) Å3
Z = 4
Mo Kα radiation
μ = 0.11 mm−1
T = 293 K
0.31 × 0.28 × 0.26 mm
Data collection
Rigaku MM007-HF CCD (Saturn 724+) diffractometer
7525 measured reflections
1819 independent reflections
1448 reflections with I > 2σ(I)
R int = 0.025
Refinement
R[F 2 > 2σ(F 2)] = 0.040
wR(F 2) = 0.116
S = 1.06
1819 reflections
140 parameters
1 restraint
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.16 e Å−3
Δρmin = −0.21 e Å−3
Data collection: CrystalStructure (Rigaku/MSC, 2006 ▶); cell refinement: CrystalStructure; data reduction: CrystalStructure; program(s) used to solve structure: SHELXTL (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXTL; molecular graphics: DIAMOND (Brandenburg, 1999 ▶); software used to prepare material for publication: SHELXTL.
Supplementary Material
Crystal structure: contains datablock(s) I, new_global_publ_block. DOI: 10.1107/S1600536814021886/xu5824sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536814021886/xu5824Isup2.hkl
Supporting information file. DOI: 10.1107/S1600536814021886/xu5824Isup3.cml
. DOI: 10.1107/S1600536814021886/xu5824fig1.tif
The molecular structure of the title compound, with atom labels and 30% probability displacement ellipsoids.
. DOI: 10.1107/S1600536814021886/xu5824fig2.tif
Hydrogen bonds are shown as brown dashed lines.
. DOI: 10.1107/S1600536814021886/xu5824fig3.tif
A view of the crystal packing.
CCDC reference: 906509
Additional supporting information: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (, ).
| DHA | DH | HA | D A | DHA |
|---|---|---|---|---|
| N1H4O4i | 0.89(2) | 2.17(2) | 3.003(2) | 155.4(17) |
| O1H1O2ii | 0.88(2) | 1.75(2) | 2.6285(18) | 174(2) |
| O4H2WO4iii | 0.92(2) | 1.98(2) | 2.8920(14) | 176(2) |
| O4H1WO3iv | 0.83(3) | 1.93(3) | 2.7549(18) | 173(2) |
| C9H9CO3i | 0.96 | 2.57 | 3.484(2) | 160 |
Symmetry codes: (i)  ; (ii)
; (ii)  ; (iii)
; (iii)  ; (iv)
; (iv)  .
.
Acknowledgments
The work was supported by the Natural Science Foundation of Yunnan Province, China (grant No. 2009CD048).
supplementary crystallographic information
S1. Comment
In the past few years, 4-acetamidobenzoic acid has attracted increasing attention as a multifunctional ligand (Kashino et al., 1986; Jedrzejas et al., 1995). It shows potential diversified coordination and exhibits various coordination modes, such as monodentate, chelating, and multidentate bridging, etc in those reported complexes (Yin et al., 2011; Wang et al., 2009). Herein we report the synthesis and structure of the title compound.
The structure of the title compound is shown in Fig. 1, Fig. 2 and hydrogen bond geometry is given in Table 1. In the title compound, the acetamide moiety is oriented with respect to the benzene ring at 20.52 (8)° whereas the carboxyl group is essentially co-planar with the benzene ring [the maximum deviation = 0.033 (1) Å]. In the crystal, classic O—H···O, N—H···O hydrogen bonds and weak C—H···O hydrogen bond link organic molecules and crystalline water molecules into the three dimensional supramolecular architecture.
S2. Experimental
4-Aminobenzoic acid (5 mmol, 686 mg) was added to 20 ml acetic anhydride. The mixture was then refluxed under argon for 8 h. The excess of acetic anhydride was then evaporated under reduced pressure. Water was then added to the resulting solid. The colorless block crystals were obtained after 48 h. Yield: 90% (based on 4-aminobenzoic acid).
S3. Refinement
H atoms attached to N and O atoms were located in a difference Fourier map and refined with positional parameters, Uiso(H) = 1.2Ueq(N) and Uiso(H) = 1.5Ueq(O). H atoms attached to carbons were geometrically fixed and allowed to ride on the corresponding non-H atom with C—H = 0.96 Å, Uiso(H) = 1.5Ueq(C) for methyl H atoms and 1.2Ueq(C) for other.
Figures
Fig. 1.

The molecular structure of the title compound, with atom labels and 30% probability displacement ellipsoids.
Fig. 2.

Hydrogen bonds are shown as brown dashed lines.
Fig. 3.

A view of the crystal packing.
Crystal data
| C9H9NO3·H2O | F(000) = 416 |
| Mr = 197.19 | Dx = 1.383 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 8729 reflections |
| a = 6.6712 (13) Å | θ = 3.1–26.0° |
| b = 28.870 (6) Å | µ = 0.11 mm−1 |
| c = 4.992 (1) Å | T = 293 K |
| β = 100.01 (3)° | Block, colorless |
| V = 946.8 (3) Å3 | 0.31 × 0.28 × 0.26 mm |
| Z = 4 |
Data collection
| Rigaku MM007-HF CCD (Saturn 724+) diffractometer | 1448 reflections with I > 2σ(I) |
| Radiation source: rotating anode | Rint = 0.025 |
| Confocal monochromator | θmax = 26.0°, θmin = 3.1° |
| ω scans at fixed χ = 45° | h = −8→8 |
| 7525 measured reflections | k = −35→35 |
| 1819 independent reflections | l = −6→5 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.040 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.116 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.06 | w = 1/[σ2(Fo2) + (0.0607P)2 + 0.1929P] where P = (Fo2 + 2Fc2)/3 |
| 1819 reflections | (Δ/σ)max < 0.001 |
| 140 parameters | Δρmax = 0.16 e Å−3 |
| 1 restraint | Δρmin = −0.21 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| C1 | 0.6802 (3) | 0.43025 (5) | 0.5354 (3) | 0.0370 (4) | |
| C2 | 0.8849 (3) | 0.42983 (5) | 0.6534 (3) | 0.0432 (4) | |
| H2 | 0.9751 | 0.4497 | 0.5886 | 0.052* | |
| C3 | 0.9563 (3) | 0.40000 (5) | 0.8671 (3) | 0.0420 (4) | |
| H3 | 1.0932 | 0.4000 | 0.9459 | 0.050* | |
| C4 | 0.8203 (2) | 0.37010 (5) | 0.9622 (3) | 0.0346 (4) | |
| C5 | 0.6157 (3) | 0.37085 (6) | 0.8459 (4) | 0.0430 (4) | |
| H5 | 0.5249 | 0.3511 | 0.9107 | 0.052* | |
| C6 | 0.5461 (3) | 0.40082 (6) | 0.6342 (3) | 0.0439 (4) | |
| H6 | 0.4086 | 0.4012 | 0.5578 | 0.053* | |
| C7 | 0.6019 (3) | 0.46170 (5) | 0.3049 (3) | 0.0394 (4) | |
| C8 | 1.0650 (2) | 0.32340 (5) | 1.2903 (3) | 0.0344 (3) | |
| C9 | 1.0717 (3) | 0.29285 (6) | 1.5361 (3) | 0.0423 (4) | |
| H9A | 1.1507 | 0.2657 | 1.5161 | 0.063* | |
| H9B | 0.9358 | 0.2839 | 1.5528 | 0.063* | |
| H9C | 1.1329 | 0.3095 | 1.6961 | 0.063* | |
| H1 | 0.684 (3) | 0.5062 (7) | 0.080 (4) | 0.063* | |
| H4 | 0.775 (3) | 0.3275 (7) | 1.251 (4) | 0.051* | |
| H1W | 0.452 (4) | 0.2928 (8) | 0.293 (5) | 0.063* | |
| H2W | 0.545 (4) | 0.2597 (7) | 0.471 (5) | 0.063* | |
| N1 | 0.8780 (2) | 0.33893 (5) | 1.1819 (3) | 0.0386 (3) | |
| O1 | 0.7308 (2) | 0.48727 (4) | 0.2144 (3) | 0.0554 (4) | |
| O2 | 0.4148 (2) | 0.46159 (4) | 0.2106 (3) | 0.0546 (4) | |
| O3 | 1.21944 (18) | 0.33300 (4) | 1.1992 (2) | 0.0475 (3) | |
| O4 | 0.5523 (2) | 0.27530 (5) | 0.3128 (3) | 0.0486 (3) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| C1 | 0.0446 (9) | 0.0327 (7) | 0.0322 (8) | 0.0049 (6) | 0.0030 (7) | 0.0027 (6) |
| C2 | 0.0428 (9) | 0.0409 (8) | 0.0444 (9) | −0.0030 (7) | 0.0037 (7) | 0.0100 (7) |
| C3 | 0.0355 (8) | 0.0426 (8) | 0.0452 (9) | −0.0011 (6) | −0.0006 (7) | 0.0091 (7) |
| C4 | 0.0379 (8) | 0.0341 (7) | 0.0311 (7) | 0.0040 (6) | 0.0041 (6) | 0.0028 (6) |
| C5 | 0.0369 (9) | 0.0463 (9) | 0.0445 (9) | −0.0028 (7) | 0.0031 (7) | 0.0120 (7) |
| C6 | 0.0384 (9) | 0.0465 (9) | 0.0428 (9) | 0.0018 (7) | −0.0037 (7) | 0.0064 (7) |
| C7 | 0.0490 (10) | 0.0339 (7) | 0.0338 (8) | 0.0049 (7) | 0.0033 (7) | 0.0020 (6) |
| C8 | 0.0387 (8) | 0.0331 (7) | 0.0297 (7) | 0.0021 (6) | 0.0012 (6) | −0.0010 (5) |
| C9 | 0.0467 (10) | 0.0463 (9) | 0.0326 (8) | 0.0083 (7) | 0.0035 (7) | 0.0071 (6) |
| N1 | 0.0358 (7) | 0.0428 (7) | 0.0369 (7) | 0.0029 (6) | 0.0056 (6) | 0.0113 (5) |
| O1 | 0.0595 (9) | 0.0540 (7) | 0.0495 (7) | −0.0018 (6) | 0.0003 (7) | 0.0215 (6) |
| O2 | 0.0504 (8) | 0.0573 (7) | 0.0515 (7) | 0.0061 (6) | −0.0043 (6) | 0.0177 (6) |
| O3 | 0.0349 (6) | 0.0563 (7) | 0.0500 (7) | 0.0022 (5) | 0.0041 (5) | 0.0156 (5) |
| O4 | 0.0409 (7) | 0.0569 (8) | 0.0496 (7) | 0.0028 (5) | 0.0122 (6) | 0.0053 (6) |
Geometric parameters (Å, º)
| C1—C6 | 1.386 (2) | C7—O2 | 1.255 (2) |
| C1—C2 | 1.390 (2) | C7—O1 | 1.274 (2) |
| C1—C7 | 1.487 (2) | C8—O3 | 1.228 (2) |
| C2—C3 | 1.389 (2) | C8—N1 | 1.347 (2) |
| C2—H2 | 0.9300 | C8—C9 | 1.505 (2) |
| C3—C4 | 1.394 (2) | C9—H9A | 0.9600 |
| C3—H3 | 0.9300 | C9—H9B | 0.9600 |
| C4—C5 | 1.388 (2) | C9—H9C | 0.9600 |
| C4—N1 | 1.4193 (19) | N1—H4 | 0.89 (2) |
| C5—C6 | 1.382 (2) | O1—H1 | 0.881 (16) |
| C5—H5 | 0.9300 | O4—H1W | 0.83 (3) |
| C6—H6 | 0.9300 | O4—H2W | 0.92 (2) |
| C6—C1—C2 | 119.44 (14) | O2—C7—O1 | 123.84 (14) |
| C6—C1—C7 | 119.22 (16) | O2—C7—C1 | 118.76 (15) |
| C2—C1—C7 | 121.34 (15) | O1—C7—C1 | 117.40 (16) |
| C3—C2—C1 | 120.74 (15) | O3—C8—N1 | 123.64 (14) |
| C3—C2—H2 | 119.6 | O3—C8—C9 | 121.75 (15) |
| C1—C2—H2 | 119.6 | N1—C8—C9 | 114.60 (14) |
| C2—C3—C4 | 119.27 (16) | C8—C9—H9A | 109.5 |
| C2—C3—H3 | 120.4 | C8—C9—H9B | 109.5 |
| C4—C3—H3 | 120.4 | H9A—C9—H9B | 109.5 |
| C5—C4—C3 | 119.93 (14) | C8—C9—H9C | 109.5 |
| C5—C4—N1 | 116.64 (14) | H9A—C9—H9C | 109.5 |
| C3—C4—N1 | 123.41 (15) | H9B—C9—H9C | 109.5 |
| C6—C5—C4 | 120.34 (15) | C8—N1—C4 | 128.98 (14) |
| C6—C5—H5 | 119.8 | C8—N1—H4 | 116.6 (13) |
| C4—C5—H5 | 119.8 | C4—N1—H4 | 114.4 (13) |
| C5—C6—C1 | 120.27 (16) | C7—O1—H1 | 117.5 (15) |
| C5—C6—H6 | 119.9 | H1W—O4—H2W | 104 (2) |
| C1—C6—H6 | 119.9 | ||
| C6—C1—C2—C3 | 0.6 (2) | C7—C1—C6—C5 | 179.16 (15) |
| C7—C1—C2—C3 | −179.47 (15) | C6—C1—C7—O2 | 1.9 (2) |
| C1—C2—C3—C4 | 0.3 (3) | C2—C1—C7—O2 | −178.08 (15) |
| C2—C3—C4—C5 | −0.9 (2) | C6—C1—C7—O1 | −177.98 (15) |
| C2—C3—C4—N1 | −178.96 (15) | C2—C1—C7—O1 | 2.1 (2) |
| C3—C4—C5—C6 | 0.6 (3) | O3—C8—N1—C4 | −3.9 (3) |
| N1—C4—C5—C6 | 178.79 (15) | C9—C8—N1—C4 | 176.06 (14) |
| C4—C5—C6—C1 | 0.3 (3) | C5—C4—N1—C8 | 163.45 (15) |
| C2—C1—C6—C5 | −0.9 (2) | C3—C4—N1—C8 | −18.4 (2) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H4···O4i | 0.89 (2) | 2.17 (2) | 3.003 (2) | 155.4 (17) |
| O1—H1···O2ii | 0.88 (2) | 1.75 (2) | 2.6285 (18) | 174 (2) |
| O4—H2W···O4iii | 0.92 (2) | 1.98 (2) | 2.8920 (14) | 176 (2) |
| O4—H1W···O3iv | 0.83 (3) | 1.93 (3) | 2.7549 (18) | 173 (2) |
| C9—H9C···O3i | 0.96 | 2.57 | 3.484 (2) | 160 |
Symmetry codes: (i) x, y, z+1; (ii) −x+1, −y+1, −z; (iii) x, −y+1/2, z+1/2; (iv) x−1, y, z−1.
Footnotes
Supporting information for this paper is available from the IUCr electronic archives (Reference: XU5824).
References
- Brandenburg, K. (1999). DIAMOND. Crystal Impact GbR, Bonn, Germany.
- Jedrzejas, M. J., Luo, M., Singh, S., Brouillette, W. J. & Air, G. M. (1995). Acta Cryst. C51, 1910–1912. [DOI] [PubMed]
- Kashino, S., Matsushita, T., Iwamoto, T., Yamaguchi, K. & Haisa, M. (1986). Acta Cryst. C42, 457–462.
- Rigaku/MSC. (2006). CrystalStructure. Rigaku/MSC, The Woodlands, Texas, USA.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Wang, Z. H., Fan, J. & Zhang, W. G. (2009). Z. Anorg. Allg. Chem. 635, 2333–2339.
- Yin, X., Fan, J., Wang, Z.-H. & Zhang, W.-G. (2011). Z. Anorg. Allg. Chem. 637, 773–777.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, new_global_publ_block. DOI: 10.1107/S1600536814021886/xu5824sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536814021886/xu5824Isup2.hkl
Supporting information file. DOI: 10.1107/S1600536814021886/xu5824Isup3.cml
. DOI: 10.1107/S1600536814021886/xu5824fig1.tif
The molecular structure of the title compound, with atom labels and 30% probability displacement ellipsoids.
. DOI: 10.1107/S1600536814021886/xu5824fig2.tif
Hydrogen bonds are shown as brown dashed lines.
. DOI: 10.1107/S1600536814021886/xu5824fig3.tif
A view of the crystal packing.
CCDC reference: 906509
Additional supporting information: crystallographic information; 3D view; checkCIF report