

The gauche conformation of backbone torsion angles (ϕ, θ) for β3,-Ac6c-OH is observed in the N-protected derivatives of 1-aminocyclohexaneacetic acid.
Keywords: crystal structure, disubstituted-β-amino acids, π–π interaction, hydrogen bonds, conformation
Abstract
N-Protected derivatives of 1-aminocyclohexaneacetic acid (β3,3-Ac6c), namely Valeroyl-β3,3-Ac6c-OH [2-(1-pentanamidocyclohexyl)acetic acid, C13H23NO3], (I), Fmoc-β3,3-Ac6c-OH [2-(1-{[(9H-fluoren-9-yloxy)carbonyl]amino}cyclohexyl)acetic acid, C23H25NO4], (II), and Pyr-β3,3-Ac6c-OH {2-[1-(pyrazine-2-amido)cyclohexyl]acetic acid, C13H17N3O3}, (III), were synthesized and their conformational properties were determined by X-ray diffraction analysis. The backbone torsion angles (ϕ, θ) for β3,3-Ac6c-OH are restricted to gauche conformations in all the derivatives, with a chair conformation of the cyclohexane ring. In the crystal structure of (I), the packing of molecules shows both carboxylic acid R 2 2(8) O—H⋯O and centrosymmetric R 2 2(14) N—H⋯O hydrogen-bonding interactions, giving rise to chains along the c-axis direction. In (II), centrosymmetric carboxylic acid R 2 2(8) O—H⋯O dimers are extended through N—H⋯O hydrogen bonds and together with inter-ring π–π interactions between Fmoc groups [ring centroid distance = 3.786 (2) Å], generate a layered structure lying parallel to (010). In the case of compound (III), carboxylic acid O—H⋯Npyrazine hydrogen bonds give rise to zigzag ribbon structures extending along the c-axis direction.
Chemical context
β-Amino acids are homologues of α-amino acids, which are constituents of several bioactive natural and synthetic products. β-Amino acids have been used as building blocks in peptidomimetic drug design (Cheng et al. 2001 ▶). The introduction of β-amino acids into pharmacologically active peptide sequences has shown improved biological activity and metabolic stability (Yamazaki et al., 1991 ▶; Huang et al., 1993 ▶). The backbone conformation of a β-amino acid is defined by the torsional angles ϕ, θ and ψ (Banerjee & Balaram, 1997 ▶), as shown in Fig. 1 ▶. The monosubstitution at the α- and β-carbon atoms plays an important role in the folding of oligomers of β-amino acids (Seebach et al., 2009 ▶). 
Figure 1.

Definition of backbone torsion angles for β-amino acids.
In order to investigate the effect of protecting groups and disubstitution on the conformation of β-amino acids, N-protected derivatives of 1-aminocyclohexaneacetic acid (β3,3Ac6c), i.e. Valeroyl-β3,3-Ac6c-OH (I), Fmoc-β3,3-Ac6c-OH (II) and Pyr-β3,3-Ac6c-OH (III) were synthesized. The crystal structures of the three compounds were determined and are reported herein, together with their comparative conformational features.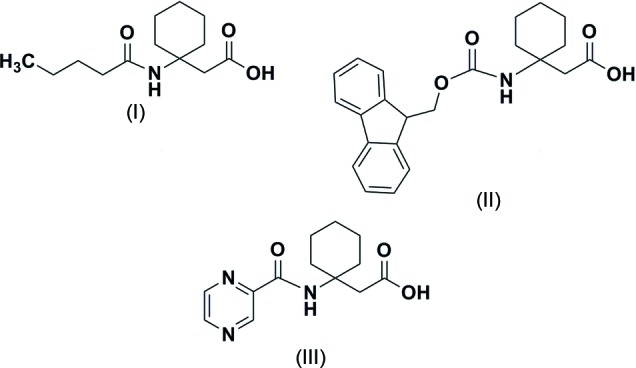
Structural commentary
The molecular conformations of Valeroyl-β3,3-Ac6c-OH (I), Fmoc-β3,3-Ac6c-OH (II) and Pyr-β3,3-Ac6c-OH (III) are shown in Fig. 2 ▶. The backbone torsion angles (ϕ, θ) (C0′—N1—C1B —C1A and N1—C1B—C1A—C1′) adopt a gauche conformation in all three compounds [ϕ = 61.9 (3)°, θ = 57.2 (3)° for (I); ϕ = 56.7 (3)°, θ = 66.1 (3)° for (II) and ϕ = 65.5 (2)°, θ = 55.0 (2)° for (III). The torsional angle ψ restricts the extended (trans) conformation for (I) [166.9 (2)°] and (III) [157.9 (2)°]. In the case of (II), it is restricted to a gauche conformation [i.e. ψ = −63.6 (3)°]. In a 3,3-disubstituted β-amino acid residue, β3,3-Ac6c-OH, the cyclohexane ring imposes a restriction on the torsion angles ϕ and θ. The protecting groups at the N-terminus of (I) adopts a trans geometry [ω0 (C4—C0′—N1—C1B) = 177.4 (2) for (I), ω0 (O—C0′—N1—C1B) = −175.64 (19) for (II) and ω0 (C6—C0—N1— C1A) = −170.04 (17)° for (III)]. In the case of the N-protected tert-butyloxycarbonyl (Boc) group, the protecting group adopts a cis geometry with ω0 = 14.50° (Vasudev et al., 2008 ▶). The cyclohexane ring adopts a chair conformation with axial amino and equatorial CH2CO groups in all the derivatives. In Pyr-β3,3-Ac6c-OH (III), an intramolecular N—H⋯N interaction is observed between NH of the β3,3-Ac6c-OH residue and N3 of the pyrazine ring as shown in Fig. 3 ▶ c. There are no intramolecular hydrogen bonding interactions observed in the crystal structures of derivatives (I) and (II).
Figure 2.

ORTEP view of the molecular conformation with the atom-labelling scheme. for Valeroyl-β3,3-Ac6c-OH (I), (b) Fmoc-β3,3-Ac6c-OH (II) and (c) Pyr-β3,3-Ac6c-OH (III). The displacement ellipsoids are drawn at the 40% probability level. H atoms are shown as small spheres of arbitrary radii.
Figure 3.

(a) Packing of Valeroyl-β3,3-Ac6c-OH (I) down the b-axis showing the alternative hydrophilic and hydrophobic layers (b) space-filling model.
Supramolecular features
In the crystals of compounds (I) and (II), intermolecular hydrogen-bonding interactions generate primary centrosymmetric dimeric but different substructures (Figs. 4 ▶ and 5 ▶). In (I), N1—H⋯O1ii bond pairs (Table 1 ▶) give a cyclic  (14) motif which is extended into a ribbon structure along the c-axis direction through a second but non-centrosymmetric cyclic carboxylic acid (8) O2—H⋯Oi hydrogen-bond motif (Fig. 4 ▶
a). In (II), the intermolecular dimeric association is through the centrosymmetric (8) carboxylic acid hydrogen-bonding motif. Structure extension is through N1—H⋯O1′ (carboxyl) hydrogen bonds (Table 2 ▶), generating a two-dimensional layered structure lying parallel to (010) (Fig. 4 ▶
c). Also present in the structure are π–π interactions between the Fmoc groups with an intercentroid distance of 3.786 (2) Å. Fig. 4 ▶
c shows the aromatic rings of Fmoc groups stacked in a face-to-face and edge-to-face manner, together with inter-plane distances that are within the range for stabilizing π–π interactions (Burley & Petsko, 1985 ▶; Sengupta et al., 2005 ▶) and have been reported to induce self-assembly in peptides (Wang & Chau, 2011 ▶). In the case of (I) and (II), the molecular packing in the crystals leads to the formation of alternating hydrophobic and hydrophilic layers. In the crystals of (III), in which no dimer substructure formation is present, the molecules are linked by an intermolecular carboxylic acid O2—H⋯N2i hydrogen bond (Table 3 ▶) with a pyrazine N-atom acceptor, leading to the formation of a zigzag ribbon structure extending along the c-axis direction.
(14) motif which is extended into a ribbon structure along the c-axis direction through a second but non-centrosymmetric cyclic carboxylic acid (8) O2—H⋯Oi hydrogen-bond motif (Fig. 4 ▶
a). In (II), the intermolecular dimeric association is through the centrosymmetric (8) carboxylic acid hydrogen-bonding motif. Structure extension is through N1—H⋯O1′ (carboxyl) hydrogen bonds (Table 2 ▶), generating a two-dimensional layered structure lying parallel to (010) (Fig. 4 ▶
c). Also present in the structure are π–π interactions between the Fmoc groups with an intercentroid distance of 3.786 (2) Å. Fig. 4 ▶
c shows the aromatic rings of Fmoc groups stacked in a face-to-face and edge-to-face manner, together with inter-plane distances that are within the range for stabilizing π–π interactions (Burley & Petsko, 1985 ▶; Sengupta et al., 2005 ▶) and have been reported to induce self-assembly in peptides (Wang & Chau, 2011 ▶). In the case of (I) and (II), the molecular packing in the crystals leads to the formation of alternating hydrophobic and hydrophilic layers. In the crystals of (III), in which no dimer substructure formation is present, the molecules are linked by an intermolecular carboxylic acid O2—H⋯N2i hydrogen bond (Table 3 ▶) with a pyrazine N-atom acceptor, leading to the formation of a zigzag ribbon structure extending along the c-axis direction.
Figure 4.

(a) Packing of Fmoc-β3,3-Ac6c-OH (II) down the a-axis. (b) Space-filling model showing the alternative hydrophilic and hydrophobic layers (packing down the c-axis). (c) The environment of the Fmoc group showing the aromatic interaction. The centroid–centroid distances are shown.
Figure 5.

(a) Packing of Pyr-β3,3-Ac6c-OH (III) down the a-axis showing the ribbon structure. (b) Zigzag arrangement of the ribbons along the c-axis.
Table 1. Hydrogen-bond geometry (, ) for (I) .
| DHA | DH | HA | D A | DHA |
|---|---|---|---|---|
| O2H2OO0i | 0.87(4) | 1.74(4) | 2.599(3) | 166(4) |
| N1H1NO1ii | 0.82(3) | 2.16(3) | 2.981(3) | 172(2) |
Symmetry codes: (i)  ; (ii)
; (ii)  .
.
Table 2. Hydrogen-bond geometry (, ) for (II) .
| DHA | DH | HA | D A | DHA |
|---|---|---|---|---|
| N1H1NO1i | 0.86(2) | 2.35(2) | 3.182(3) | 161(2) |
| O2H2OO1ii | 0.84(3) | 1.83(3) | 2.673(3) | 177(1) |
Symmetry codes: (i)  ; (ii) .
; (ii) .
Table 3. Hydrogen-bond geometry (, ) for (III) .
| DHA | DH | HA | D A | DHA |
|---|---|---|---|---|
| O2H21N2i | 0.93(4) | 1.86(4) | 2.791(3) | 177(4) |
| N1H1NN3 | 0.79(2) | 2.34(2) | 2.729(2) | 111.3(19) |
Symmetry code: (i)  .
.
Synthesis and crystallization
Preparation of Valeroyl-β3,3Ac6c-OH (I): β3,3Ac6c-OH (5 mmol, 785 mg) was dissolved in 5 ml of a 2M NaOH solution and a solution of 5 mmol of valeric anhydride (931 mg) dissolved in 1,4-dioxane was added, after which the mixture was stirred for 4 h at room temperature. On completion of the reaction, the 1,4-dioxane was evaporated and the product was extracted with diethyl ether (3 × 5 ml). The aqueous layer was acidified with 2M HCl and extracted with ethyl acetate (3 × 10ml) and the combined organic layer was washed with brine solution. The organic layer was passed over anhydrous Na2SO4 and evaporated to give Valeroyl-β3Ac6c-OH (yield: 1.1 g, 85.2%). Single crystals were grown by slow evaporation from a solution in methanol/water.
Preparation of Fmoc-β3,3Ac6c-OH (II): β3,3Ac6c-OH (10 mmol, 1.57 g) was dissolved in 1M Na2CO3 solution and Fmoc-OSu (10 mmol, 3.37 g) dissolved in CH3CN was added. The reaction mixture was stirred at room temperature for 6 h. After completion of the reaction, the CH3CN was evaporated and the residue was extracted with diethyl ether (3 × 10 ml). The aqueous layer was acidified with 2M HCl and extracted with ethyl acetate (3 × 15 ml). The combined organic layer was washed with brine solution. The ethyl acetate layer was passed over anhydrous Na2SO4 and evaporated. The residue was purified by crystallization in ethyl acetate/n-hexane, affording Fmoc-β3,3Ac6c-OH (yield: 3.0 g, 79%). Single crystals were obtained by slow evaporation from an ethyl acetate/n-hexane solution.
Preparation of Pyr-β3,3Ac6c-OH (III): Pyrazine carboxylic acid (3 mmol, 372 mg) was dissolved in dry CH2Cl2 and then 200 µl of N-methylmorpholine was added, followed by β3,3Ac6c-OMe. HCl (3 mmol, 622.5 mg) and EDCI. HCl (3 mmol,576 mg) at 273 K. The reaction mixture was stirred at room temperature for 12 h. After completion of the reaction, water was added and the reaction mixture was extracted with CH2Cl2 (3 × 5ml). The combined organic layer was washed with 2M HCl (2 × 5ml), Na2CO3 (2 × 5ml) and brine solution (2 × 5ml). The organic layer was passed over anhydrous Na2SO4 and evaporated to give Pyr-β3,3Ac6c-OMe (Yield: 600 mg, 72.2%). Pyr-β3,3Ac6c-OMe (2 mmol, 554 mg) was dissolved in 2 ml of methanol and 1 ml of 2M NaOH, and the reaction mixture was stirred at room temperature for 4 h. Methanol was evaporated and the residue was extracted with diethyl ether (2 × 5ml). The aqueous layer was acidified with 2M HCl and extracted with ethyl acetate (3 × 5ml). The combined organic layer was washed with brine solution (1 × 5ml). The ethyl acetate layer was passed over anhydrous Na2SO4 and evaporated to give Pyr-β3,3Ac6c-OH (yield: 370 mg, 70.3%). Single crystals were grown from an ethanol/water solution.
Refinement details
Crystal data, data collection and structure refinement details are summarized in Table 4 ▶. For derivative (I), H atoms for N1 and O2 were located in a difference Fourier map and both their coordinates and U iso values were refined. The remaining H atoms were positioned geometrically and were treated as riding on their parent C atoms, with C—H distances of 0.96–0.98 Å and with U iso(H) = 1.2U eq(C) or 1.5U eq(methyl C). For derivatives (II) and (III), all hydrogen atoms were located from a difference Fourier map and both their coordinates and U iso values were refined. In (II), the carboxyl O—H distance was constrained to 0.84 Å. Although not of consequence with the achiral molecule of (III), which crystallized in the non-centrosymmetric space group Pca21, the structure was inverted in the final cycles of refinement as the Flack parameter was 0.8 (14). The inverted structure gave a value of 0.2 (14) for 1585 Friedel pairs.
Table 4. Experimental details.
| (I) | (II) | (III) | |
|---|---|---|---|
| Crystal data | |||
| Chemical formula | C13H23NO3 | C23H25NO4 | C13H17N3O3 |
| M r | 241.32 | 379.44 | 263.30 |
| Crystal system, space group | Monoclinic, P21/c | Triclinic, P

|
Orthorhombic, P c a21 |
| Temperature (K) | 291 | 291 | 291 |
| a, b, c () | 9.5894(5), 12.5007(7), 12.3709(8) | 6.0834(4), 12.7642(9), 12.8399(9) | 8.7135(1), 10.5321(1), 14.3907(2) |
| , , () | 90, 109.984(7), 90 | 94.018(6), 92.295(6), 100.489(6) | 90, 90, 90 |
| V (3) | 1393.66(14) | 976.53(12) | 1320.66(3) |
| Z | 4 | 2 | 4 |
| Radiation type | Mo K | Mo K | Mo K |
| (mm1) | 0.08 | 0.09 | 0.10 |
| Crystal size (mm) | 0.30 0.08 0.08 | 0.30 0.05 0.03 | 0.25 0.25 0.25 |
| Data collection | |||
| Diffractometer | Oxford Diffraction Xcalibur, Sapphire3 CCD | Oxford Diffraction Xcalibur, Sapphire3 CCD | Oxford Difraction Xcalibur, Sapphire3 CCD |
| Absorption correction | Multi-scan (CrysAlis PRO; Oxford Diffraction, 2010 ▶) | Multi-scan (CrysAlis PRO; Oxford Diffraction, 2010 ▶) | Multi-scan (CrysAlis PRO; Oxford Diffraction, 2010 ▶) |
| T min, T max | 0.797, 1.000 | 0.947, 1.000 | 0.931, 1.000 |
| No. of measured, independent and observed [I > 2(I)] reflections | 14087, 2737, 1717 | 7781, 4166, 2037 | 68869, 2878, 2670 |
| R int | 0.047 | 0.047 | 0.034 |
| (sin /)max (1) | 0.617 | 0.639 | 0.639 |
| Refinement | |||
| R[F 2 > 2(F 2)], wR(F 2), S | 0.068, 0.213, 1.03 | 0.054, 0.086, 0.97 | 0.042, 0.106, 1.04 |
| No. of reflections | 2737 | 4166 | 2878 |
| No. of parameters | 162 | 353 | 240 |
| No. of restraints | 0 | 1 | 1 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement | All H-atom parameters refined | All H-atom parameters refined |
| max, min (e 3) | 0.36, 0.30 | 0.15, 0.20 | 0.27, 0.26 |
| Absolute structure | (Flack, 1983 ▶): 1585 Friedel pairs | ||
| Absolute structure parameter | 0.2(14) | ||
Supplementary Material
Crystal structure: contains datablock(s) I, II, III, New_Global_Publ_Block. DOI: 10.1107/S1600536814020777/zs2313sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536814020777/zs2313Isup2.hkl
Structure factors: contains datablock(s) II. DOI: 10.1107/S1600536814020777/zs2313IIsup3.hkl
Structure factors: contains datablock(s) III. DOI: 10.1107/S1600536814020777/zs2313IIIsup4.hkl
Supporting information file. DOI: 10.1107/S1600536814020777/zs2313Isup5.cml
Supporting information file. DOI: 10.1107/S1600536814020777/zs2313IIsup6.cml
Supporting information file. DOI: 10.1107/S1600536814020777/zs2313IIIsup7.cml
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
RR acknowledges the Council of Scientific and Industrial Research (CSIR), India, for financial assistance under MLP5009and BSC-120. RK wishes to acknowledge the Department of Science and Technology, India, for sanctioning the single-crystal X-ray diffractometer as a National Facility under project No: SR/S2 /CMP/47.
supplementary crystallographic information
Crystal data
| C13H17N3O3 | F(000) = 560 |
| Mr = 263.30 | Dx = 1.324 Mg m−3 |
| Orthorhombic, Pca21 | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: P 2c -2ac | Cell parameters from 28796 reflections |
| a = 8.7135 (1) Å | θ = 3.7–27.0° |
| b = 10.5321 (1) Å | µ = 0.10 mm−1 |
| c = 14.3907 (2) Å | T = 291 K |
| V = 1320.66 (3) Å3 | Cube, colorless |
| Z = 4 | 0.25 × 0.25 × 0.25 mm |
Data collection
| Oxford Difraction Xcalibur, Sapphire3 CCD diffractometer | 2878 independent reflections |
| Radiation source: fine-focus sealed tube | 2670 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.034 |
| Detector resolution: 16.1049 pixels mm-1 | θmax = 27.0°, θmin = 3.9° |
| ω scans | h = −11→11 |
| Absorption correction: multi-scan (CrysAlis PRO; Oxford Diffraction, 2010) | k = −13→13 |
| Tmin = 0.931, Tmax = 1.000 | l = −18→18 |
| 68869 measured reflections |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.042 | All H-atom parameters refined |
| wR(F2) = 0.106 | w = 1/[σ2(Fo2) + (0.048P)2 + 0.4913P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.04 | (Δ/σ)max = 0.032 |
| 2878 reflections | Δρmax = 0.27 e Å−3 |
| 240 parameters | Δρmin = −0.26 e Å−3 |
| 1 restraint | Absolute structure: (Flack, 1983): 1585 Friedel pairs |
| Primary atom site location: structure-invariant direct methods | Absolute structure parameter: 0.2 (14) |
Special details
| Experimental. CrysAlis PRO, Oxford Diffraction Ltd., Version 1.171.34.40 (release 27–08-2010 CrysAlis171. NET) (compiled Aug 27 2010,11:50:40) Empirical absorption correction using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm. |
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2sigma(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| H1 | 0.902 (2) | 0.412 (2) | 1.0592 (15) | 0.030 (5)* | |
| H1B2 | 1.400 (3) | 0.435 (2) | 1.2329 (16) | 0.041 (6)* | |
| H1B5 | 1.272 (3) | 0.384 (2) | 1.4161 (18) | 0.049 (7)* | |
| H1B6 | 1.183 (3) | 0.442 (2) | 1.3342 (17) | 0.048 (6)* | |
| H1B1 | 1.499 (2) | 0.380 (2) | 1.3191 (16) | 0.033 (5)* | |
| H1D2 | 1.651 (3) | 0.100 (3) | 1.200 (2) | 0.061 (7)* | |
| H1N | 1.161 (2) | 0.172 (2) | 1.2259 (16) | 0.036 (6)* | |
| H1G1 | 1.481 (3) | 0.256 (2) | 1.1391 (19) | 0.045 (6)* | |
| H2 | 0.709 (3) | 0.107 (2) | 0.971 (2) | 0.051 (7)* | |
| H3 | 0.894 (3) | −0.021 (2) | 1.0429 (17) | 0.047 (6)* | |
| H1B4 | 1.269 (3) | 0.107 (2) | 1.3669 (17) | 0.045 (6)* | |
| H1G4 | 1.401 (3) | 0.050 (3) | 1.232 (2) | 0.061 (7)* | |
| H1G2 | 1.616 (3) | 0.323 (3) | 1.173 (2) | 0.066 (8)* | |
| H1B3 | 1.415 (3) | 0.192 (2) | 1.3989 (19) | 0.046 (6)* | |
| H1G3 | 1.502 (3) | 0.005 (3) | 1.3184 (19) | 0.065 (8)* | |
| H1D1 | 1.667 (4) | 0.176 (3) | 1.298 (2) | 0.071 (9)* | |
| H21 | 0.885 (5) | 0.352 (4) | 1.458 (3) | 0.106 (13)* | |
| C1 | 0.9028 (2) | 0.3259 (2) | 1.05370 (16) | 0.0401 (4) | |
| N2 | 0.7893 (2) | 0.27249 (19) | 1.00502 (14) | 0.0442 (4) | |
| C3 | 0.7875 (3) | 0.1459 (2) | 1.00191 (15) | 0.0433 (5) | |
| C4 | 0.8954 (3) | 0.0736 (2) | 1.04730 (16) | 0.0450 (5) | |
| N3 | 1.0074 (2) | 0.12584 (16) | 1.09722 (13) | 0.0398 (4) | |
| C6 | 1.0105 (2) | 0.25193 (18) | 1.10026 (14) | 0.0343 (4) | |
| C0 | 1.1318 (2) | 0.31669 (18) | 1.15876 (15) | 0.0382 (4) | |
| O0' | 1.1604 (2) | 0.42890 (15) | 1.14626 (15) | 0.0658 (6) | |
| N1 | 1.19610 (18) | 0.24153 (15) | 1.22264 (12) | 0.0348 (4) | |
| C1A | 1.2995 (2) | 0.28157 (17) | 1.29864 (13) | 0.0311 (4) | |
| C1B | 1.2100 (2) | 0.36660 (19) | 1.36671 (16) | 0.0381 (4) | |
| C1' | 1.0648 (2) | 0.31013 (19) | 1.40619 (15) | 0.0423 (5) | |
| O1 | 1.0357 (3) | 0.2011 (2) | 1.4129 (3) | 0.1197 (13) | |
| O2 | 0.9700 (2) | 0.39599 (18) | 1.43574 (16) | 0.0677 (6) | |
| C1B1 | 1.4369 (2) | 0.3562 (2) | 1.26078 (16) | 0.0394 (4) | |
| C1G1 | 1.5373 (3) | 0.2777 (2) | 1.19617 (18) | 0.0504 (5) | |
| C1D | 1.5948 (3) | 0.1582 (3) | 1.2448 (2) | 0.0575 (6) | |
| C1B2 | 1.3586 (2) | 0.15966 (19) | 1.34564 (14) | 0.0366 (4) | |
| C1G2 | 1.4634 (3) | 0.0817 (2) | 1.28311 (18) | 0.0483 (5) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| C1 | 0.0386 (10) | 0.0391 (10) | 0.0428 (10) | 0.0036 (9) | −0.0049 (9) | 0.0025 (10) |
| N2 | 0.0374 (8) | 0.0538 (11) | 0.0414 (9) | 0.0036 (8) | −0.0067 (7) | 0.0039 (8) |
| C3 | 0.0402 (11) | 0.0513 (13) | 0.0386 (11) | −0.0063 (9) | −0.0045 (9) | −0.0018 (10) |
| C4 | 0.0510 (12) | 0.0399 (11) | 0.0440 (10) | −0.0049 (10) | −0.0066 (9) | −0.0016 (9) |
| N3 | 0.0445 (9) | 0.0361 (8) | 0.0389 (9) | −0.0002 (7) | −0.0048 (8) | 0.0035 (7) |
| C6 | 0.0340 (9) | 0.0368 (10) | 0.0321 (9) | 0.0012 (7) | 0.0002 (7) | 0.0023 (8) |
| C0 | 0.0388 (10) | 0.0335 (9) | 0.0422 (11) | 0.0024 (8) | −0.0048 (8) | 0.0032 (8) |
| O0' | 0.0769 (12) | 0.0360 (8) | 0.0843 (13) | −0.0095 (8) | −0.0359 (11) | 0.0160 (8) |
| N1 | 0.0365 (8) | 0.0291 (8) | 0.0388 (9) | −0.0040 (6) | −0.0074 (7) | 0.0016 (6) |
| C1A | 0.0299 (8) | 0.0305 (9) | 0.0329 (9) | −0.0015 (7) | 0.0010 (7) | −0.0030 (7) |
| C1B | 0.0352 (10) | 0.0327 (10) | 0.0464 (11) | −0.0002 (8) | 0.0058 (9) | −0.0069 (8) |
| C1' | 0.0418 (11) | 0.0358 (10) | 0.0494 (12) | −0.0009 (9) | 0.0134 (9) | −0.0037 (9) |
| O1 | 0.0886 (16) | 0.0440 (10) | 0.227 (3) | −0.0088 (10) | 0.099 (2) | −0.0111 (15) |
| O2 | 0.0533 (10) | 0.0462 (9) | 0.1035 (15) | 0.0071 (8) | 0.0373 (10) | 0.0115 (9) |
| C1B1 | 0.0322 (9) | 0.0395 (11) | 0.0464 (11) | −0.0066 (8) | 0.0044 (9) | −0.0020 (9) |
| C1G1 | 0.0453 (12) | 0.0557 (13) | 0.0500 (13) | −0.0058 (11) | 0.0180 (11) | −0.0036 (11) |
| C1D | 0.0454 (12) | 0.0660 (16) | 0.0611 (16) | 0.0172 (11) | 0.0121 (12) | −0.0050 (12) |
| C1B2 | 0.0371 (10) | 0.0398 (9) | 0.0329 (10) | 0.0042 (8) | −0.0012 (8) | 0.0026 (8) |
| C1G2 | 0.0551 (13) | 0.0404 (11) | 0.0493 (12) | 0.0171 (10) | 0.0039 (11) | −0.0012 (9) |
Geometric parameters (Å, º)
| C1—N2 | 1.337 (3) | C1B—H1B6 | 0.95 (3) |
| C1—C6 | 1.391 (3) | C1'—O1 | 1.180 (3) |
| C1—H1 | 0.91 (2) | C1'—O2 | 1.297 (3) |
| N2—C3 | 1.334 (3) | O2—H21 | 0.93 (5) |
| C3—C4 | 1.375 (3) | C1B1—C1G1 | 1.521 (3) |
| C3—H2 | 0.91 (3) | C1B1—H1B2 | 0.97 (2) |
| C4—N3 | 1.331 (3) | C1B1—H1B1 | 1.03 (2) |
| C4—H3 | 1.00 (3) | C1G1—C1D | 1.525 (4) |
| N3—C6 | 1.329 (3) | C1G1—H1G1 | 0.98 (3) |
| C6—C0 | 1.514 (3) | C1G1—H1G2 | 0.90 (3) |
| C0—O0' | 1.221 (2) | C1D—C1G2 | 1.505 (4) |
| C0—N1 | 1.336 (3) | C1D—H1D2 | 1.01 (3) |
| N1—C1A | 1.478 (2) | C1D—H1D1 | 1.00 (3) |
| N1—H1N | 0.79 (2) | C1B2—C1G2 | 1.523 (3) |
| C1A—C1B1 | 1.532 (3) | C1B2—H1B4 | 1.00 (2) |
| C1A—C1B | 1.539 (2) | C1B2—H1B3 | 0.97 (3) |
| C1A—C1B2 | 1.540 (3) | C1G2—H1G4 | 0.97 (3) |
| C1B—C1' | 1.509 (3) | C1G2—H1G3 | 1.01 (3) |
| C1B—H1B5 | 0.91 (3) | ||
| N2—C1—C6 | 121.03 (19) | O1—C1'—C1B | 126.5 (2) |
| N2—C1—H1 | 117.5 (14) | O2—C1'—C1B | 112.51 (18) |
| C6—C1—H1 | 121.3 (14) | C1'—O2—H21 | 106 (3) |
| C3—N2—C1 | 116.57 (18) | C1G1—C1B1—C1A | 112.85 (17) |
| N2—C3—C4 | 122.0 (2) | C1G1—C1B1—H1B2 | 113.6 (14) |
| N2—C3—H2 | 118.2 (16) | C1A—C1B1—H1B2 | 109.0 (14) |
| C4—C3—H2 | 119.8 (16) | C1G1—C1B1—H1B1 | 108.9 (12) |
| N3—C4—C3 | 121.9 (2) | C1A—C1B1—H1B1 | 104.3 (12) |
| N3—C4—H3 | 117.3 (15) | H1B2—C1B1—H1B1 | 107.7 (18) |
| C3—C4—H3 | 120.8 (15) | C1B1—C1G1—C1D | 110.9 (2) |
| C6—N3—C4 | 116.44 (19) | C1B1—C1G1—H1G1 | 110.5 (15) |
| N3—C6—C1 | 122.0 (2) | C1D—C1G1—H1G1 | 111.0 (15) |
| N3—C6—C0 | 118.85 (18) | C1B1—C1G1—H1G2 | 112.3 (19) |
| C1—C6—C0 | 119.08 (17) | C1D—C1G1—H1G2 | 111.1 (19) |
| O0'—C0—N1 | 126.08 (19) | H1G1—C1G1—H1G2 | 101 (2) |
| O0'—C0—C6 | 119.79 (18) | C1G2—C1D—C1G1 | 111.1 (2) |
| N1—C0—C6 | 114.12 (17) | C1G2—C1D—H1D2 | 106.1 (16) |
| C0—N1—C1A | 126.55 (16) | C1G1—C1D—H1D2 | 111.2 (16) |
| C0—N1—H1N | 115.3 (17) | C1G2—C1D—H1D1 | 107.4 (17) |
| C1A—N1—H1N | 116.8 (17) | C1G1—C1D—H1D1 | 113.3 (18) |
| N1—C1A—C1B1 | 111.06 (16) | H1D2—C1D—H1D1 | 107 (2) |
| N1—C1A—C1B | 109.15 (15) | C1G2—C1B2—C1A | 113.00 (17) |
| C1B1—C1A—C1B | 108.90 (15) | C1G2—C1B2—H1B4 | 110.6 (13) |
| N1—C1A—C1B2 | 106.91 (15) | C1A—C1B2—H1B4 | 109.3 (13) |
| C1B1—C1A—C1B2 | 108.81 (16) | C1G2—C1B2—H1B3 | 110.4 (15) |
| C1B—C1A—C1B2 | 112.02 (16) | C1A—C1B2—H1B3 | 102.9 (15) |
| C1'—C1B—C1A | 115.79 (16) | H1B4—C1B2—H1B3 | 110 (2) |
| C1'—C1B—H1B5 | 106.5 (16) | C1D—C1G2—C1B2 | 112.6 (2) |
| C1A—C1B—H1B5 | 108.4 (17) | C1D—C1G2—H1G4 | 109.7 (17) |
| C1'—C1B—H1B6 | 108.0 (15) | C1B2—C1G2—H1G4 | 107.0 (16) |
| C1A—C1B—H1B6 | 107.2 (15) | C1D—C1G2—H1G3 | 111.0 (17) |
| H1B5—C1B—H1B6 | 111 (2) | C1B2—C1G2—H1G3 | 109.4 (16) |
| O1—C1'—O2 | 121.0 (2) | H1G4—C1G2—H1G3 | 107 (2) |
| C6—C1—N2—C3 | −1.5 (3) | C0—N1—C1A—C1B2 | −173.2 (2) |
| C1—N2—C3—C4 | 0.8 (3) | N1—C1A—C1B—C1' | 55.0 (2) |
| N2—C3—C4—N3 | 0.3 (4) | C1B1—C1A—C1B—C1' | 176.42 (19) |
| C3—C4—N3—C6 | −0.6 (3) | C1B2—C1A—C1B—C1' | −63.2 (2) |
| C4—N3—C6—C1 | −0.2 (3) | C1A—C1B—C1'—O1 | 23.6 (4) |
| C4—N3—C6—C0 | 177.82 (19) | C1A—C1B—C1'—O2 | −157.9 (2) |
| N2—C1—C6—N3 | 1.3 (3) | N1—C1A—C1B1—C1G1 | −62.6 (2) |
| N2—C1—C6—C0 | −176.68 (19) | C1B—C1A—C1B1—C1G1 | 177.20 (19) |
| N3—C6—C0—O0' | 162.9 (2) | C1B2—C1A—C1B1—C1G1 | 54.8 (2) |
| C1—C6—C0—O0' | −19.1 (3) | C1A—C1B1—C1G1—C1D | −57.0 (3) |
| N3—C6—C0—N1 | −18.5 (3) | C1B1—C1G1—C1D—C1G2 | 55.1 (3) |
| C1—C6—C0—N1 | 159.53 (19) | N1—C1A—C1B2—C1G2 | 67.3 (2) |
| O0'—C0—N1—C1A | 8.4 (4) | C1B1—C1A—C1B2—C1G2 | −52.7 (2) |
| C6—C0—N1—C1A | −170.04 (17) | C1B—C1A—C1B2—C1G2 | −173.13 (18) |
| C0—N1—C1A—C1B1 | −54.6 (2) | C1G1—C1D—C1G2—C1B2 | −53.8 (3) |
| C0—N1—C1A—C1B | 65.5 (2) | C1A—C1B2—C1G2—C1D | 53.7 (3) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O2—H21···N2i | 0.93 (4) | 1.86 (4) | 2.791 (3) | 177 (4) |
| N1—H1N···N3 | 0.79 (2) | 2.34 (2) | 2.729 (2) | 111.3 (19) |
Symmetry code: (i) −x+3/2, y, z+1/2.
Footnotes
IIIM Communication number: IIIM/1553/2013.
References
- Banerjee, A. & Balaram, P. (1997). Curr. Sci. 73, 1067–1077.
- Burley, S. K. & Petsko, G. A. (1985). Science, 229, 23–28. [DOI] [PubMed]
- Cheng, R. P., Gellman, S. H. & DeGrado, W. F. (2001). Chem. Rev. 101, 3219–3232. [DOI] [PubMed]
- Farrugia, L. J. (2012). J. Appl. Cryst. 45, 849–854.
- Flack, H. D. (1983). Acta Cryst. A39, 876–881.
- Huang, Z., Pröbstl, A., Spencer, J. R., Yamazaki, T. & Goodman, M. (1993). Int. J. Pept. Protein Res. 42, 352–365. [DOI] [PubMed]
- Oxford Diffraction (2010). CrysAlis PRO. Oxford Diffraction Ltd, Yarnton, England.
- Seebach, D., Beck, A. K., Capone, S., Deniau, G., Grošelj, U. & Zass, E. (2009). Synthesis, 1, 1–32.
- Sengupta, A., Mahalakshmi, R., Shamala, N. & Balaram, P. (2005). J. Pept. Res. 65, 113–129. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Vasudev, P. G., Rai, R., Shamala, N. & Balaram, P. (2008). Biopolymers, 90, 138–150. [DOI] [PubMed]
- Wang, W. & Chau, Y. (2011). Chem. Commun. 47, 10224. [DOI] [PubMed]
- Yamazaki, T., Pröbsti, A., Schiller, P. W. & Goodman, M. (1991). Int. J. Pept. Protein Res. 37, 364–381. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, II, III, New_Global_Publ_Block. DOI: 10.1107/S1600536814020777/zs2313sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536814020777/zs2313Isup2.hkl
Structure factors: contains datablock(s) II. DOI: 10.1107/S1600536814020777/zs2313IIsup3.hkl
Structure factors: contains datablock(s) III. DOI: 10.1107/S1600536814020777/zs2313IIIsup4.hkl
Supporting information file. DOI: 10.1107/S1600536814020777/zs2313Isup5.cml
Supporting information file. DOI: 10.1107/S1600536814020777/zs2313IIsup6.cml
Supporting information file. DOI: 10.1107/S1600536814020777/zs2313IIIsup7.cml
Additional supporting information: crystallographic information; 3D view; checkCIF report