

The title compound has an E conformation about the azobenzene linkage and the benzene rings are almost coplanar to one another [dihedral angle = 1.36 (7)°]. In the crystal, a combination of O—H⋯O and C—H⋯O hydrogen bonds and C—H⋯π interactions leads to the formation of slabs parallel to (001).
Keywords: crystal structure, azobenzene, benzoic acid, liquid crystal, nematic phase
Abstract
The title compound, C16H14N2O3, has an E conformation about the azobenzene [—N=N– = 1.2481 (16) Å] linkage. The benzene rings are almost coplanar [dihedral angle = 1.36 (7)°]. The O atoms of the carboxylic acid group are disordered over two sets of sites and were refined with an occupancy ratio of 0.5:0.5. The two disordered components of the carboxylic acid group make dihedral angles of 1.5 (14) and 3.8 (12)° with the benzene ring to which they are attached. In the crystal, molecules are linked via pairs of O—H⋯O hydrogen bonds, forming inversion dimers. The dimers are connected via C—H⋯O hydrogen bonds, forming ribbons lying parallel to [120]. These ribbons are linked via C—H⋯π interactions, forming slabs parallel to (001).
Chemical context
It is interesting to note that the title compound shows a nematic phase (Cr 190 N 218 I) . Hence, liquid crystallinity may be induced by the formation of hydrogen-bonded dimers. A number of liquid crystal (LC) systems containing hydrogen bonds that function between identical molecules have been reported (Kang & Samulski, 2000 ▶; Rahman et al., 2012 ▶). Much attention has been paid to hydrogen-bonded supramolecular LCs, including LC dimers based on hydrogen-bonding interactions and several supramolecular LC trimers based on hydrogen-bonding interactions (Lee et al., 2001 ▶; Paleos & Tsiourvas, 2001 ▶; Takahashi et al., 2003 ▶; Bai et al., 2007 ▶). A particular aspect of photonics, in which the molecular geometry can be controlled by light, is being proposed as a future technology for optical storage devices (Ikeda & Tsutsumi, 1995 ▶; Jayalaxmi et al., 2009 ▶). The heart of the phenomenon in such systems is the reversible photo-induced shape transformation of the molecules containing the photochromic azobenzene groups. The title compound contains an azo (—N=N—) linkage, it was easy to synthesize and hence cost-effective for the possibility of photochromism and photoisomerization usage (Lutfor et al., 2013a ▶,b
▶). We report herein on its synthesis and crystal structure.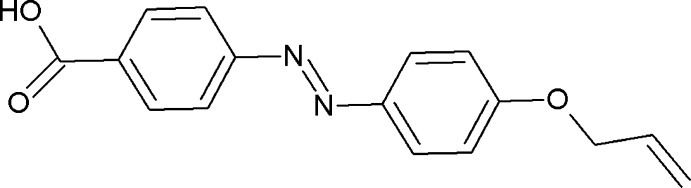
Structural commentary
The molecular structure of the title molecule is illustrated in Fig. 1 ▶. The oxygen atoms forming the carboxylic acid group are each disordered over two positions and were refined with half occupancy. The carboxylic acid group (C16/O2/O3) is almost coplanar with the attached benzene ring (C10–C15), making dihedral angles of 3.44 (9) and 3.65 (8)° for the two disorder components. The title compound has an E conformation about the azobenzene (—N=N—) linkage, the length of the N1—N2 bond is 1.2481 (16) Å and the torsion angle for the azo unit (C7—N1=N2—C10) is 179.99 (10)°, which is comparable with the values of ca ±180° observed in 4,4-azinodibenzoic acid (Yu & Liu, 2009 ▶) and (E)-ethyl-4-{[4-(decanoxloxy)phenyl]diazenly} benzoate (Lai et al., 2002 ▶). The benzene rings (C4–C9) and (C10–C15) are almost coplanar, making a dihedral angle of 1.38 (7)°, compared with 6.79 (9)° in the previously reported compound 4-{(E)-2-[4-(but-3-en-1-yloxy)phenyl]-diazen-1-yl}benzoic acid, (Rahman et al., 2012 ▶).
Figure 1.

The molecular structure of the title compound, showing the atom labelling. Displacement ellipsoids are drawn at the 30% probability level. Only one component of the disordered carboxylic acid group is shown.
Supramolecular features
In the crystal, molecules are linked via pairs of O—H⋯O hydrogen bonds, forming inversion dimers (Table 1 ▶ and Fig. 2 ▶). The dimers are connected via C—H⋯O hydrogen bonds, forming two-molecule-thick ribbons lying parallel to [120]; see Table 1 ▶ and Fig. 3 ▶. Adjacent ribbons are linked via C—H⋯π interactions, forming slabs parallel to (001), as shown in Fig. 3 ▶ (Table 1 ▶).
Table 1. Hydrogen-bond geometry (, ).
Cg1 is the centroid of the C4C9 ring.
| DHA | DH | HA | D A | DHA |
|---|---|---|---|---|
| O3H3O2i | 0.82 | 1.90 | 2.71(3) | 166 |
| C6H6AO2ii | 0.93 | 2.59 | 3.367(15) | 145 |
| C3H3A Cg1iii | 0.97 | 2.66 | 3.504(2) | 145 |
Symmetry codes: (i)  ; (ii)
; (ii)  ; (iii)
; (iii)  .
.
Figure 2.

A partial view along the a-axis of the crystal packing of the title compound, with hydrogen bonds shown as dashed lines (see Table 1 ▶ for details).
Figure 3.

A partial view of the crystal packing of the title compound. Blue dashed lines represent the intermolecular hydrogen bonds within two-molecule-thick chains and the green dashed lines represent the weak intermolecular C—H⋯π interactions (see Table 1 ▶ for details).
Synthesis and crystallization
The title compound was synthesized by a literature procedure (Rahman et al., 2012 ▶). The diazonuim salt was prepared with sodium nitrite and subsequent coupling with phenol to afforded the ethyl 4-[(4-hydroxyphenyl)diazenyl]benzoate, which was purified by crystallization and recrystallization from methanol. The azobenzene compound was alkylated with allyl bromide to give ethyl 4-{[4-(allyloxy)phenyl]diazenyl}benzoate, which was purified by crystallization from methanol/chloroform. The terminal double bonds-containing azobenzene compound was hydrolysed under basic conditions to yield the title compound. Red plate-like crystals were obtained by crystallization from an ethanol–ethyl acetate mixture (1:1); m.p. 494 K. 1H NMR (CDCl3): δ 8.18 (d, 2H, J = 8.2 Hz), 7.94 (d, 2H, J = 7.1 Hz), 7.93 (d, 2H, J = 6.7 Hz), 7.05 (d, 2H, J = 8.9 Hz), 6.04 (m, 1H, CH=), 5.45 (d, 1H, J = 16.6 Hz, =CH2), 5.31 (d, 1H, J = 10.2 Hz, =CH2), 4.60 (d, 2H, J = 4.1 Hz, OCH2).
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 2 ▶. Atoms O2 and O3 of the carboxylic acid group are each disordered over two positions and were refined with half occupancy each. The position of the O-bound H atom was located in a difference Fourier map and refined as a riding atom: O—H = 0.82 Å with U iso(H) = 1.5 U eq(O). The C-bound H atoms were positioned geometrically and refined using a riding model: C—H = 0.93–0.97 Å with U iso(H) = 1.2U eq(C). Two outlier reflections, 341 and 309, were omitted from the refinement.
Table 2. Experimental details.
| Crystal data | |
| Chemical formula | C16H14N2O3 |
| M r | 282.29 |
| Crystal system, space group | Triclinic, P
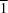
|
| Temperature (K) | 294 |
| a, b, c () | 5.0279(4), 8.9678(7), 15.9913(13) |
| , , () | 80.571(2), 83.874(2), 88.371(2) |
| V (3) | 707.19(10) |
| Z | 2 |
| Radiation type | Mo K |
| (mm1) | 0.09 |
| Crystal size (mm) | 0.78 0.22 0.09 |
| Data collection | |
| Diffractometer | Bruker APEX DUO CCD area detector |
| Absorption correction | Multi-scan (SADABS; Bruker, 2009 ▶) |
| T min, T max | 0.931, 0.992 |
| No. of measured, independent and observed [I > 2(I)] reflections | 12171, 3276, 2344 |
| R int | 0.023 |
| (sin /)max (1) | 0.651 |
| Refinement | |
| R[F 2 > 2(F 2)], wR(F 2), S | 0.043, 0.138, 1.04 |
| No. of reflections | 3276 |
| No. of parameters | 211 |
| H-atom treatment | H-atom parameters constrained |
| max, min (e 3) | 0.24, 0.16 |
Supplementary Material
Crystal structure: contains datablock(s) I. DOI: 10.1107/S1600536814023745/su2790sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536814023745/su2790Isup2.hkl
Supporting information file. DOI: 10.1107/S1600536814023745/su2790Isup3.cml
CCDC reference: 1031374
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
This research was supported by a PRGS Research Grant (No. RDU 130803).
supplementary crystallographic information
Crystal data
| C16H14N2O3 | Z = 2 |
| Mr = 282.29 | F(000) = 296 |
| Triclinic, P1 | Dx = 1.326 Mg m−3 |
| Hall symbol: -P 1 | Melting point: 494 K |
| a = 5.0279 (4) Å | Mo Kα radiation, λ = 0.71073 Å |
| b = 8.9678 (7) Å | Cell parameters from 3848 reflections |
| c = 15.9913 (13) Å | θ = 2.5–27.4° |
| α = 80.571 (2)° | µ = 0.09 mm−1 |
| β = 83.874 (2)° | T = 294 K |
| γ = 88.371 (2)° | Plate, red |
| V = 707.19 (10) Å3 | 0.78 × 0.22 × 0.09 mm |
Data collection
| Bruker APEX DUO CCD area-detector diffractometer | 3276 independent reflections |
| Radiation source: fine-focus sealed tube | 2344 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.023 |
| φ and ω scans | θmax = 27.6°, θmin = 1.3° |
| Absorption correction: multi-scan (SADABS; Bruker, 2009) | h = −6→6 |
| Tmin = 0.931, Tmax = 0.992 | k = −11→11 |
| 12171 measured reflections | l = −20→20 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.043 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.138 | H-atom parameters constrained |
| S = 1.04 | w = 1/[σ2(Fo2) + (0.0696P)2 + 0.0848P] where P = (Fo2 + 2Fc2)/3 |
| 3276 reflections | (Δ/σ)max < 0.001 |
| 211 parameters | Δρmax = 0.24 e Å−3 |
| 0 restraints | Δρmin = −0.16 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R-factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | Occ. (<1) | |
| O1 | 1.26119 (18) | −0.00858 (10) | 0.39744 (5) | 0.0464 (3) | |
| O2 | −0.234 (5) | 0.8701 (17) | −0.0212 (8) | 0.068 (3) | 0.50 |
| O3 | −0.424 (5) | 0.940 (2) | 0.1072 (12) | 0.0525 (17) | 0.50 |
| H3 | −0.5052 | 1.0045 | 0.0769 | 0.079* | 0.50 |
| O2X | −0.392 (5) | 0.943 (2) | 0.0945 (13) | 0.062 (3) | 0.50 |
| O3X | −0.267 (5) | 0.8749 (16) | −0.0231 (8) | 0.065 (3) | 0.50 |
| H3X | −0.3393 | 0.9554 | −0.0404 | 0.098* | 0.50 |
| N1 | 0.6027 (2) | 0.37818 (12) | 0.20184 (7) | 0.0442 (3) | |
| N2 | 0.4446 (2) | 0.45802 (12) | 0.24161 (7) | 0.0444 (3) | |
| C1 | 1.6508 (4) | −0.33243 (19) | 0.43299 (12) | 0.0807 (6) | |
| H1A | 1.5953 | −0.3863 | 0.3931 | 0.097* | |
| H1B | 1.7412 | −0.3818 | 0.4775 | 0.097* | |
| C2 | 1.6019 (3) | −0.18848 (16) | 0.42651 (9) | 0.0538 (4) | |
| H2A | 1.6599 | −0.1378 | 0.4674 | 0.065* | |
| C3 | 1.4597 (3) | −0.09989 (14) | 0.35815 (8) | 0.0437 (3) | |
| H3A | 1.5844 | −0.0364 | 0.3182 | 0.052* | |
| H3B | 1.3758 | −0.1671 | 0.3273 | 0.052* | |
| C4 | 1.1092 (2) | 0.08615 (13) | 0.34590 (7) | 0.0373 (3) | |
| C5 | 1.1259 (3) | 0.09639 (14) | 0.25801 (7) | 0.0421 (3) | |
| H5A | 1.2504 | 0.0384 | 0.2297 | 0.051* | |
| C6 | 0.9539 (3) | 0.19454 (14) | 0.21297 (8) | 0.0438 (3) | |
| H6A | 0.9626 | 0.2013 | 0.1541 | 0.053* | |
| C7 | 0.7698 (3) | 0.28242 (13) | 0.25406 (8) | 0.0395 (3) | |
| C8 | 0.7558 (3) | 0.27159 (14) | 0.34268 (8) | 0.0434 (3) | |
| H8A | 0.6323 | 0.3302 | 0.3709 | 0.052* | |
| C9 | 0.9240 (3) | 0.17465 (14) | 0.38793 (8) | 0.0436 (3) | |
| H9A | 0.9147 | 0.1678 | 0.4468 | 0.052* | |
| C10 | 0.2754 (3) | 0.55478 (13) | 0.18998 (8) | 0.0412 (3) | |
| C11 | 0.2795 (3) | 0.56604 (17) | 0.10240 (9) | 0.0572 (4) | |
| H11A | 0.3976 | 0.5065 | 0.0729 | 0.069* | |
| C12 | 0.1078 (3) | 0.66589 (17) | 0.05898 (9) | 0.0579 (4) | |
| H12A | 0.1103 | 0.6733 | 0.0002 | 0.069* | |
| C13 | −0.0685 (3) | 0.75530 (14) | 0.10282 (8) | 0.0427 (3) | |
| C14 | −0.0730 (3) | 0.74195 (15) | 0.19032 (8) | 0.0463 (3) | |
| H14A | −0.1914 | 0.8010 | 0.2201 | 0.056* | |
| C15 | 0.0973 (3) | 0.64144 (15) | 0.23369 (8) | 0.0455 (3) | |
| H15A | 0.0919 | 0.6322 | 0.2926 | 0.055* | |
| C16 | −0.2538 (3) | 0.86292 (15) | 0.05719 (8) | 0.0461 (3) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0458 (5) | 0.0547 (5) | 0.0370 (4) | 0.0192 (4) | −0.0066 (4) | −0.0037 (4) |
| O2 | 0.062 (4) | 0.097 (5) | 0.039 (3) | 0.042 (3) | −0.008 (2) | 0.009 (3) |
| O3 | 0.052 (3) | 0.062 (3) | 0.041 (5) | 0.029 (2) | 0.000 (3) | −0.009 (3) |
| O2X | 0.067 (7) | 0.076 (3) | 0.041 (4) | 0.039 (4) | −0.006 (4) | −0.009 (3) |
| O3X | 0.071 (6) | 0.079 (4) | 0.045 (3) | 0.045 (3) | −0.012 (2) | −0.012 (3) |
| N1 | 0.0475 (6) | 0.0424 (6) | 0.0412 (6) | 0.0075 (5) | −0.0088 (5) | −0.0013 (4) |
| N2 | 0.0459 (6) | 0.0442 (6) | 0.0423 (6) | 0.0092 (5) | −0.0089 (5) | −0.0032 (4) |
| C1 | 0.1015 (15) | 0.0604 (10) | 0.0731 (11) | 0.0305 (10) | −0.0072 (10) | 0.0038 (8) |
| C2 | 0.0522 (8) | 0.0555 (8) | 0.0526 (8) | 0.0157 (7) | −0.0097 (6) | −0.0056 (6) |
| C3 | 0.0425 (7) | 0.0438 (7) | 0.0446 (6) | 0.0098 (5) | −0.0055 (5) | −0.0081 (5) |
| C4 | 0.0359 (6) | 0.0389 (6) | 0.0359 (6) | 0.0044 (5) | −0.0062 (5) | −0.0020 (5) |
| C5 | 0.0418 (7) | 0.0462 (7) | 0.0372 (6) | 0.0100 (5) | −0.0021 (5) | −0.0067 (5) |
| C6 | 0.0482 (8) | 0.0497 (7) | 0.0324 (6) | 0.0071 (6) | −0.0062 (5) | −0.0031 (5) |
| C7 | 0.0411 (7) | 0.0372 (6) | 0.0391 (6) | 0.0038 (5) | −0.0081 (5) | −0.0011 (5) |
| C8 | 0.0464 (7) | 0.0426 (6) | 0.0410 (6) | 0.0123 (5) | −0.0040 (5) | −0.0088 (5) |
| C9 | 0.0472 (7) | 0.0497 (7) | 0.0339 (6) | 0.0105 (6) | −0.0060 (5) | −0.0072 (5) |
| C10 | 0.0414 (7) | 0.0389 (6) | 0.0425 (6) | 0.0045 (5) | −0.0097 (5) | −0.0014 (5) |
| C11 | 0.0620 (9) | 0.0640 (9) | 0.0432 (7) | 0.0305 (7) | −0.0043 (6) | −0.0079 (6) |
| C12 | 0.0668 (10) | 0.0678 (9) | 0.0360 (6) | 0.0298 (8) | −0.0064 (6) | −0.0039 (6) |
| C13 | 0.0418 (7) | 0.0426 (7) | 0.0421 (6) | 0.0094 (5) | −0.0067 (5) | −0.0028 (5) |
| C14 | 0.0465 (8) | 0.0483 (7) | 0.0448 (7) | 0.0132 (6) | −0.0061 (6) | −0.0110 (5) |
| C15 | 0.0492 (8) | 0.0498 (7) | 0.0383 (6) | 0.0073 (6) | −0.0093 (5) | −0.0076 (5) |
| C16 | 0.0440 (8) | 0.0490 (7) | 0.0436 (7) | 0.0149 (6) | −0.0046 (6) | −0.0048 (6) |
Geometric parameters (Å, º)
| O1—C4 | 1.3613 (14) | C5—C6 | 1.3868 (17) |
| O1—C3 | 1.4317 (15) | C5—H5A | 0.9300 |
| O2—C16 | 1.238 (13) | C6—C7 | 1.3812 (18) |
| O3—C16 | 1.36 (2) | C6—H6A | 0.9300 |
| O3—H3 | 0.8200 | C7—C8 | 1.3989 (17) |
| O2X—C16 | 1.18 (2) | C8—C9 | 1.3700 (18) |
| O3X—C16 | 1.280 (14) | C8—H8A | 0.9300 |
| O3X—H3X | 0.8200 | C9—H9A | 0.9300 |
| N1—N2 | 1.2481 (16) | C10—C15 | 1.3781 (18) |
| N1—C7 | 1.4195 (16) | C10—C11 | 1.3855 (19) |
| N2—C10 | 1.4254 (16) | C11—C12 | 1.3817 (19) |
| C1—C2 | 1.297 (2) | C11—H11A | 0.9300 |
| C1—H1A | 0.9300 | C12—C13 | 1.3899 (18) |
| C1—H1B | 0.9300 | C12—H12A | 0.9300 |
| C2—C3 | 1.4790 (19) | C13—C14 | 1.3825 (18) |
| C2—H2A | 0.9300 | C13—C16 | 1.4813 (18) |
| C3—H3A | 0.9700 | C14—C15 | 1.3801 (18) |
| C3—H3B | 0.9700 | C14—H14A | 0.9300 |
| C4—C5 | 1.3870 (16) | C15—H15A | 0.9300 |
| C4—C9 | 1.3956 (17) | ||
| C4—O1—C3 | 117.90 (9) | C8—C9—C4 | 120.20 (11) |
| C16—O3—H3 | 109.5 | C8—C9—H9A | 119.9 |
| C16—O3X—H3X | 109.5 | C4—C9—H9A | 119.9 |
| N2—N1—C7 | 114.00 (10) | C15—C10—C11 | 119.92 (12) |
| N1—N2—C10 | 114.63 (10) | C15—C10—N2 | 114.92 (11) |
| C2—C1—H1A | 120.0 | C11—C10—N2 | 125.15 (12) |
| C2—C1—H1B | 120.0 | C12—C11—C10 | 119.87 (12) |
| H1A—C1—H1B | 120.0 | C12—C11—H11A | 120.1 |
| C1—C2—C3 | 124.08 (15) | C10—C11—H11A | 120.1 |
| C1—C2—H2A | 118.0 | C11—C12—C13 | 120.24 (12) |
| C3—C2—H2A | 118.0 | C11—C12—H12A | 119.9 |
| O1—C3—C2 | 107.64 (10) | C13—C12—H12A | 119.9 |
| O1—C3—H3A | 110.2 | C14—C13—C12 | 119.40 (12) |
| C2—C3—H3A | 110.2 | C14—C13—C16 | 119.70 (11) |
| O1—C3—H3B | 110.2 | C12—C13—C16 | 120.89 (11) |
| C2—C3—H3B | 110.2 | C15—C14—C13 | 120.30 (12) |
| H3A—C3—H3B | 108.5 | C15—C14—H14A | 119.9 |
| O1—C4—C5 | 124.57 (11) | C13—C14—H14A | 119.9 |
| O1—C4—C9 | 115.09 (10) | C10—C15—C14 | 120.24 (12) |
| C5—C4—C9 | 120.33 (11) | C10—C15—H15A | 119.9 |
| C6—C5—C4 | 118.92 (11) | C14—C15—H15A | 119.9 |
| C6—C5—H5A | 120.5 | O2X—C16—O2 | 123.9 (14) |
| C4—C5—H5A | 120.5 | O2X—C16—O3X | 117.6 (15) |
| C7—C6—C5 | 121.13 (11) | O2—C16—O3X | 8 (2) |
| C7—C6—H6A | 119.4 | O2X—C16—O3 | 7.0 (19) |
| C5—C6—H6A | 119.4 | O2—C16—O3 | 128.6 (14) |
| C6—C7—C8 | 119.40 (11) | O3X—C16—O3 | 121.9 (13) |
| C6—C7—N1 | 116.43 (11) | O2X—C16—C13 | 120.1 (11) |
| C8—C7—N1 | 124.16 (11) | O2—C16—C13 | 115.8 (10) |
| C9—C8—C7 | 120.02 (11) | O3X—C16—C13 | 122.3 (9) |
| C9—C8—H8A | 120.0 | O3—C16—C13 | 115.6 (9) |
| C7—C8—H8A | 120.0 | ||
| C7—N1—N2—C10 | −179.99 (10) | C15—C10—C11—C12 | 1.0 (2) |
| C4—O1—C3—C2 | 178.33 (11) | N2—C10—C11—C12 | −179.04 (13) |
| C1—C2—C3—O1 | 132.68 (17) | C10—C11—C12—C13 | 0.1 (3) |
| C3—O1—C4—C5 | 2.60 (19) | C11—C12—C13—C14 | −0.9 (2) |
| C3—O1—C4—C9 | −178.51 (11) | C11—C12—C13—C16 | 179.98 (14) |
| O1—C4—C5—C6 | 178.13 (11) | C12—C13—C14—C15 | 0.4 (2) |
| C9—C4—C5—C6 | −0.70 (19) | C16—C13—C14—C15 | 179.59 (12) |
| C4—C5—C6—C7 | 0.6 (2) | C11—C10—C15—C14 | −1.5 (2) |
| C5—C6—C7—C8 | −0.3 (2) | N2—C10—C15—C14 | 178.60 (11) |
| C5—C6—C7—N1 | −179.63 (11) | C13—C14—C15—C10 | 0.7 (2) |
| N2—N1—C7—C6 | −178.28 (11) | C14—C13—C16—O2X | 4.5 (14) |
| N2—N1—C7—C8 | 2.47 (19) | C12—C13—C16—O2X | −176.3 (14) |
| C6—C7—C8—C9 | 0.1 (2) | C14—C13—C16—O2 | 178.8 (10) |
| N1—C7—C8—C9 | 179.33 (12) | C12—C13—C16—O2 | −2.0 (11) |
| C7—C8—C9—C4 | −0.2 (2) | C14—C13—C16—O3X | −176.7 (11) |
| O1—C4—C9—C8 | −178.46 (11) | C12—C13—C16—O3X | 2.5 (11) |
| C5—C4—C9—C8 | 0.5 (2) | C14—C13—C16—O3 | −1.5 (10) |
| N1—N2—C10—C15 | 178.98 (11) | C12—C13—C16—O3 | 177.7 (10) |
| N1—N2—C10—C11 | −1.0 (2) |
Hydrogen-bond geometry (Å, º)
Cg1 is the centroid of the C4–C9 ring.
| D—H···A | D—H | H···A | D···A | D—H···A |
| O3—H3···O2i | 0.82 | 1.90 | 2.71 (3) | 166 |
| C6—H6A···O2ii | 0.93 | 2.59 | 3.367 (15) | 145 |
| C3—H3A···Cg1iii | 0.97 | 2.66 | 3.504 (2) | 145 |
Symmetry codes: (i) −x−1, −y+2, −z; (ii) −x+1, −y+1, −z; (iii) x+1, y, z.
References
- Bai, B. L., Wang, H., Xin, H., Long, B. & Li, M. (2007). Liq. Cryst. 34, 659–665.
- Bruker (2009). APEX2, SAINT and SADABS. Bruker AXS Inc., Madison, Wisconsin, USA.
- Ikeda, T. & Tsutsumi, O. (1995). Science, 268, 1873–1875. [DOI] [PubMed]
- Jayalakshmi, V., Hegde, G., Nair, G. & Prasad, S. K. (2009). Phys. Chem. Chem. Phys. 11, 6450–6454. [DOI] [PubMed]
- Kang, S. K. & Samulski, E. T. (2000). Liq. Cryst. 27, 371–376.
- Lai, L.-L., Su, F.-Y., Lin, Y.-J., Ho, C.-H., Wang, E., Hung, C.-H., Liu, Y.-H. & Wang, Y. (2002). Helv. Chim. Acta, 85, 1517–1522.
- Lee, J. W., Jin, J. I., Achard, M. F. & Hardouin, F. (2001). Liq. Cryst. 28, 663–671.
- Lutfor, M. R., Hegde, G., Pour, M. A., Yusoff, M. M. & Kumar, S. (2013a). J. Fluorine Chem. 156, 230–235.
- Lutfor, M. R., Yusoff, M. M., Srinivasa, H. T., Samah, N. A., Malek, N. F. M. A. & Kumar, S. (2013b). New J. Chem. 37, 2460–2467.
- Macrae, C. F., Bruno, I. J., Chisholm, J. A., Edgington, P. R., McCabe, P., Pidcock, E., Rodriguez-Monge, L., Taylor, R., van de Streek, J. & Wood, P. A. (2008). J. Appl. Cryst. 41, 466–470.
- Paleos, C. M. & Tsiourvas, D. (2001). Liq. Cryst. 28, 1127–1161.
- Rahman, M. L., Kwong, H. C., Mohd. Yusoff, M., Hegde, G., Mohamed Tahir, M. I. & Rahman, M. Z. A. (2012). Acta Cryst. E68, o2958. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Takahashi, A., Mallia, V. A. & Tamaoki, N. (2003). J. Mater. Chem. 13, 1582–1587.
- Yu, Q.-D. & Liu, Y.-Y. (2009). Acta Cryst. E65, o2326. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I. DOI: 10.1107/S1600536814023745/su2790sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536814023745/su2790Isup2.hkl
Supporting information file. DOI: 10.1107/S1600536814023745/su2790Isup3.cml
CCDC reference: 1031374
Additional supporting information: crystallographic information; 3D view; checkCIF report