

Abstract
The major challenge of photocatalytic water splitting, the prototypical reaction for the direct production of hydrogen by using solar energy, is to develop low-cost yet highly efficient and stable semiconductor photocatalysts. Herein, an effective strategy for synthesizing extremely active graphitic carbon nitride (g-C3N4) from a low-cost precursor, urea, is reported. The g-C3N4 exhibits an extraordinary hydrogen-evolution rate (ca. 20 000 μmol h−1 g−1 under full arc), which leads to a high turnover number (TON) of over 641 after 6 h. The reaction proceeds for more than 30 h without activity loss and results in an internal quantum yield of 26.5 % under visible light, which is nearly an order of magnitude higher than that observed for any other existing g-C3N4 photocatalysts. Furthermore, it was found by experimental analysis and DFT calculations that as the degree of polymerization increases and the proton concentration decreases, the hydrogen-evolution rate is significantly enhanced.
Keywords: graphitic carbon nitride, hydrogen production, polymerization, protonation, water splitting
Inorganic semiconductors have been at the forefront of photocatalytic water splitting for the synthesis of renewable fuels ever since the discovery of the phenomenon in the late 20th century.1 Advances towards an efficient photocatalyst suitable for hydrogen and oxygen evolution under visible light notably include: metal and nonmetal mixed oxides,2 sulfides and nitrides,3 doped perovskites,4 and nitrided pyrochlores.5 These successes have drawn considerable interest in the field, but are still far from meeting industrial requirements of efficiency and stability. The major challenge is to develop a highly efficient, low-cost, and robust photocatalyst that can successfully serve practical needs. A stable, organic photocatalyst, graphitic carbon nitride (g-C3N4), was found to show sufficient redox power to dissociate water under visible light in a suspension (see Figure S1 in the Supporting Information).6 However, the latest documented quantum yields for H2 production from water with g-C3N4 (excluding dye-sensitized systems) do not exceed 4 %,6, 7 which is still unsatisfactory for industrial applications.8 Graphitic carbon nitride is composed of extremely abundant elements and is nontoxic with proven stability, both thermally and in solutions of pH 1–14. Therefore, if an effective and facile strategy is devised to improve its energy-conversion efficiency, it can meet all the aforementioned three requirements for a practical photocatalyst. Although there has been extensive research on g-C3N4-based photocatalysts,7f,h,i little attention has been paid to the effects of the protonation and polymerization of pristine g-C3N4.
Herein, we report a g-C3N4 photocatalyst, synthesized from low-cost and abundant urea, that shows an extremely high quantum yield of 26.5 % under visible light: nearly an order of magnitude higher than the previous record of approximately 4 % for hydrogen evolution with g-C3N4.7 To the best of our knowledge, this value is much greater than that of most semiconductor photocatalysts, except sulfides (i.e. CdS and ZnS), which are highly efficient but inherently unstable.1 More importantly, we have determined a protonation mechanism from both experimental and computational findings to explain the extraordinary photocatalytic activity of g-C3N4 derived from a tailored polymerization route.
For comparison, samples of g-C3N4 were successfully prepared by using different precursors (urea, dicyandiamide (DCDA), and thiourea) under identical conditions and characterized by X-ray diffraction (XRD), attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy, transmission electron microscopy (TEM), and UV/Vis and Raman spectroscopy (see Figures S2 and S3 in the Supporting Information for details of characterization and analysis). Graphitic carbon nitride synthesized from different precursors (600 °C, 5 °C min−1 ramp rate) was tested for hydrogen evolution in an aqueous sacrificial solution containing triethanolamine (TEOA) at room temperature and atmospheric pressure, in a procedure similar to a previously reported method.6, 7f The fully optimized results are shown in Figure 1 and further summarized in Table 1. The urea-derived g-C3N4 exhibited superior hydrogen evolution in comparison to either the widely used DCDA- or thiourea-derived g-C3N4 under both full arc and visible-light irradiation (Figure 1 a,b). The urea-derived g-C3N4 evolved hydrogen at approximately 20 000 μmol h−1 g−1, 15 times faster than DCDA-derived and 8 times higher than thiourea-derived g-C3N4, as reflected in the turnover number (TON, over a platinum cocatalyst; see the Supporting Information for the calculation): Urea-derived g-C3N4 had a TON of 641.1, which is much higher than that of the other samples.
Figure 1.
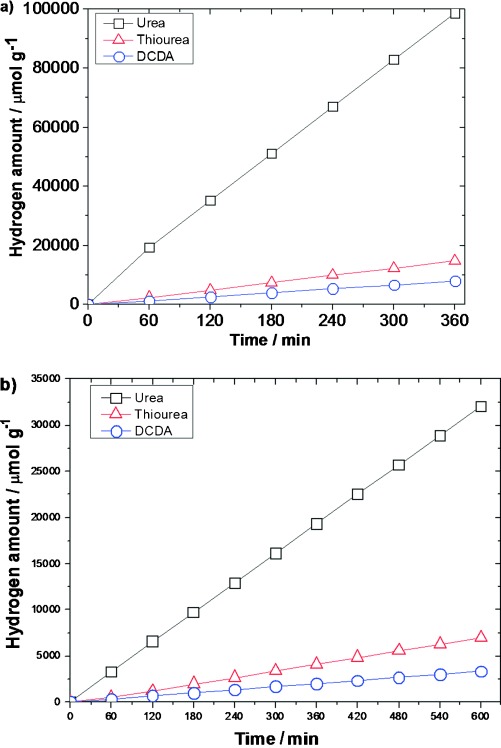
Hydrogen evolution with a 300 W Xe lamp, 3 wt % Pt, and TEOA as a hole scavenger: a) full arc, b) λ≥395 nm.
Table 1.
Summary of the properties of g-C3N4 synthesized from different precursors. The H2-evolution rate was measured by using a 300 W Xe lamp, 3 wt % Pt, and TEOA as a hole scavenger. All samples were synthesized at 600 °C in air. The TON value was calculated with respect to the Pt catalyst.
| Sample | HER rate [μmol h−1 g−1] | TON (6 h) | Band edge [nm] | SSA [m2 g−1] |
|---|---|---|---|---|
| urea | 19 412 | 641.1 | 415 | 43.8 |
| DCDA | 1350 | 52.5 | 451 | 12.8 |
| thiourea | 2470 | 96.4 | 453 | 18.5 |
Even under irradiation with visible light (λ≥395 nm, Figure 1 b), the urea-derived g-C3N4 evolved H2 at 3300 μmol h−1 g−1, more than 10 times faster than the DCDA-derived g-C3N4 at 300 μmol h−1 g−1 and nearly 7 times faster than thiourea-derived g-C3N4 at 500 μmol h−1 g−1. It may be tempting to conclude that the activity difference is due to differences in surface area; however, the urea-derived g-C3N4 has a specific surface area (SSA) only 3.4 times greater than that of DCDA-derived g-C3N4 and 2.4 times that of the sample of thiourea-derived g-C3N4 (see Figure S4). Meanwhile, the activity was over 15 times and 8 times higher than that of the samples synthesized by using DCDA and thiourea, respectively. Interestingly, urea-derived g-C3N4 calcined at 550 °C showed a surface area of 83.5 m2 g−1, double that of the sample calcined at 600 °C. However, the activity of the former sample is only a third that of the latter (see Figure S5 a). Zhang et al.9b prepared g-C3N4 at 550 °C (3 h) from a urea precursor and reported a hydrogen-evolution rate (HER) of 625 μmol h−1 g−1, which is similar to that of the sample prepared in this study. The difference in activity is due to the shorter calcination time employed during sample preparation, which probably affects the level of polymerization of the compound. Curiously, despite being able to absorb less light, the urea-derived carbon nitride still outperforms both DCDA- and thiourea-derived counterparts (Table 1). Therefore, the activity cannot be directly attributed to either surface area or optical absorption.
Extensive tests were carried out on the urea-derived sample owing to its excellent performance in evolving hydrogen from water. The synthesis parameters were tailored to augment hydrogen production from water, including the reaction temperature, ramp rate, cocatalyst species, and loading of the cocatalyst (see Figure S5).
X-ray photoelectron spectroscopy (XPS) was undertaken to accurately determine the specific bonding and structure of the samples. In all samples, the typical C 1s and N 1s peaks were observed, as in previous studies.9 A residual O 1s peak, probably due to calcination in air, was also present. XPS spectra of the three samples (prepared from urea, thiourea, and DCDA) are shown in Figures S6 (N 1s XPS) and S7 (C 1s and O 1s XPS spectra) of the Supporting Information. The C 1s spectra show C—C, C—N—C, and a trace amount of C—O bonding at 285.1, 288.2, and 289.2 eV, respectively. The N 1s spectra can be fitted to elucidate four separate signals, and provides a better idea of the bonding structure, since carbon spectra are susceptible to contamination. The N 1s core levels at 398.7, 399.7, and 400.9 eV correspond to sp2 C—N—C, sp3 H—N—[C]3, and C—NHx (amino functional groups), respectively.10 The weak peak at 404.4 eV can be attributed to terminal nitrate groups, charging effects, or π excitations.9a, 11 Hybridized sp3 nitrogen not only has three chemical bonds to carbon, but because of hybridization, can also be bonded to hydrogen perpendicular to the direction of the graphitic layer.
Figure 2 shows the distinctive trend in bonding ratios versus activity between samples (see also Table S1). The ratio of sp2 C—N—C bonds to the sum of sp3 H—N—[C]3 and C—NHx bonds (the latter represents the total amount of protons) is 2.83 in urea, 2.7 for thiourea, and only 2.31 in DCDA. As it is part of the heptazine ring, linked by a double and a single bond to two opposing carbon atoms, the sp2-bonded nitrogen atom is the principle participant that contributes to band-gap absorption and therefore is an extremely important part of the structure. Both hybridized sp3 nitrogen atoms and surface functional amino groups (C—NHx) are also key features when considering bulk and surface properties. Along with C—NHx bonding, graphitic carbon nitride possesses a positively charged, acidic surface, as confirmed by ζ-potential measurements (see Figure S8). Elemental analysis (EA) further confirmed the trend shown in Figure 2: As bulk H (atom %) increases, the HER per SSA (μmol m−2 h−1) decreases (see Figure S9). From these results and in combination with XPS analysis, we could conclude that a lower proton concentration leads to a larger HER.
Figure 2.
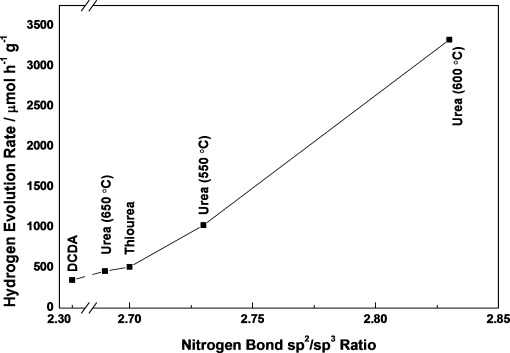
Ratios of bonds within the N 1s core-level peak in different samples and their comparison to the hydrogen-evolution rate (HER) under visible light (λ>395 nm): Decreasing proton concentration leads to a dramatic increase in photocatalytic activity.
The increase in proton concentration (from both sp3 nitrogen atoms and C—NHx) most likely stems from the route of condensation, and even though all three samples were synthesized under identical conditions, the extent of polymerization varied owing to the different precursors, which is consistent with the trends in XRD and FTIR measurements (see Figure S2). In particular, only urea-derived g-C3N4 loses hydrogen in the form of formaldehyde, owing to the presence of oxygen in the precursor (see Figures S10 and S11 for thermogravimetic analysis–differential scanning calorimetry–mass spectroscopy analysis). This trend not only applies to different precursors, but urea-derived g-C3N4 synthesized at different temperatures (550, 650 °C) also follows suit (Figure 2; see also Table S1): As protonation increases, activity decreases. Therefore, both the precursors and the synthetic parameters can control the protonation and polymerization, thus leading to varying activity. A small change in the preparation method can have a huge impact on photocatalytic hydrogen production (such as a change from 550 to 600 or 650 °C; see Figure S5 a and ramp-rate changes in Figure S5 b).
To determine exactly why polymerization and protonation status influences H2-production rates, we modeled protonation by DFT simulations using periodic supercells. Time-dependent DFT (TDDFT) simulations were performed on cluster models to determine the effects of hydrogen on excited-state properties. The density of states (DOS) is shown in Figure 3 c. It can be clearly seen that the conduction-band edge (CBE) of the protonated system is shifted down in energy (towards more positive values with respect to the normal hydrogen electrode, NHE) by 0.34 eV. This shift significantly modifies the electrochemical properties, as it provides a lower overpotential for reduction reactions, as also shown in the UV/Vis absorption spectra (see Figure S2 c). The reason behind the drop in the position of the CBE can be clearly seen in the site-decomposed DOS (see discussion in the Supporting Information and Figure S12). The effects of protonation on excited-state properties of a molecular model were also calculated. The lowest energy vibrationally stable structure involves strong distortions from planarity of all three heptazine rings. The onset of optical absorption on protonated g-C3N4 occurs at a lower energy (more positive with respect to the NHE) than for deprotonated g-C3N4. Indeed, two absorption peaks of the C18N28H13 model occur at lower energies than that of the initial absorption peak of the C18N28H12 model, in qualitative agreement with the DFT DOS in Figure 3. This result verifies our DFT-based electronic-structure analysis with TDDFT.
Figure 3.
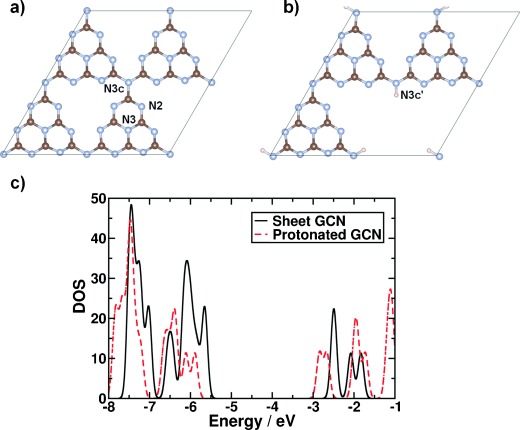
Geometric and electronic structure of g-C3N4. a) Supercell model of sheet carbon nitride; b) supercell model of protonated carbon nitride. Nitrogen is denoted by light-blue spheres, carbon by red-gray spheres, and hydrogen by white spheres. c) Total density of states for sheet carbon nitride (black line) and protonated carbon nitride (red dashed line). Energy is given with respect to the zero of the simulation for sheet carbon nitride. The DOS of the protonated carbon nitride has been shifted so that the corresponding zero points align.
We also plotted the distribution of the lowest-energy exciton for both carbon nitride models (see Figure S13 b,c). For deprotonated C3N4, the exciton is distributed relatively homogeneously over the cluster, with transitions from occupied N pz orbitals to empty C pz* orbitals. For protonated C3N4, the exciton is more heterogeneous; the photohole on the protonated heptazine ring and the photoelectron are distributed evenly on the other two heptazine rings. Although there is a better spatial separation between photohole and photoelectron, both charge carriers are more localized around the central N3 site, and thus are not as available to participate in the photochemical reactions. Moreover, this localization will act to increase the exciton-recombination rate, thus hindering the efficient utilization of charge carriers.
A relatively small change in protonation has a two-pronged detrimental effect on the reduction ability of g-C3N4. Protonation significantly reduces the reduction potential and also localizes excitons around a central nitrogen N3 site, thus hindering migration to active sites. In this material, protonation is essentially controlled through the degree of polymerization, but also coupled with the degree of condensation. Graphitic carbon nitride, if “underpolymerized” (at low temperatures), has incomplete heptazine coupling, which results in excess hydrogen-passivating N3c′ nitrogen sites, thus hampering activity. If “overpolymerized” (at about 650 °C), the structure of g-C3N4 tends to overly condense into buckled multilayered crystals of reduced surface area; as a result, the density of active sites is reduced, and photocatalytic ability is adversely affected. Moreover, the buckling also distorts the sp2 planar geometry, thus leading to charge-trapping states at nitrogen sites and hence reduced activity.
As mentioned previously, apart from the need for a photocatalyst to be cheap and robust, it must exhibit a high quantum yield for hydrogen production from water if it is to be considered commercially viable. Compounds that traditionally exhibit high efficiencies either suffer from instability (e.g. sulfides13) or are made of relatively expensive metals (e.g. GaAs–GaInP214). The cheap and stable urea-derived g-C3N4 in this study has a peak internal quantum yield of 28.4 % at 365 nm (Figure 4 a). Even under irradiation with visible light at λ=400 nm, the quantum yield is 26.5 %, nearly an order of magnitude greater than the highest reported (3.75 % at 420 nm,7f obtained by liquid exfoliation). To ensure the reliability of our measurement, we examined as a reference a benchmark cyanamide-derived g-C3N4, which showed comparable activity (Table 2; the small difference is due to the use of a 395 nm long-pass filter instead of a 420 nm filter). As proposed previously, the huge enhancement in hydrogen-evolution rate of our urea-derived sample (3327.5 vs. 142.3 μmol h−1 g−1) can be attributed to a lower protonation status and the condensation state. Recently, a facile synthetic method for three-dimensional porous g-C3N4 was introduced by using aggregates of melamine and cyanuric acid (MCA) co-crystals in dimethyl sulfoxide (DMSO, sample denoted MCA_DMSO) as precursors.12 It was reported that the quantum yield of g-C3N4_MCA_DMSO at λ=420 nm was 2.3 % (Table 2), much higher than that of melamine-derived bulk g-C3N4 (0.26 %) under the same conditions. We repeated this study and observed very similar morphologies and optical properties to those reported (see Figure S14). Correspondingly, a similar quantum yield of 3.1 % at λ=400 nm was obtained (Table 2; the difference is due to the wavelength of the band-pass filter). Since MCA_DMSO is another oxygen-containing precursor, the rise in the quantum yield as compared to that of a melamine sample further supports our proposed protonation mechanism. Furthermore, the reason why our optimized urea-derived g-C3N4 is more than 10 times more efficient than MCA_DMSO g-C3N4, in terms of quantum yield at 400 nm, given the very similar specific surface area of these two samples, is because of the much higher oxygen concentration in the urea precursor, which helps to passivate protonation sites and polymerize g-C3N4 without structural instability/buckling.
Figure 4.
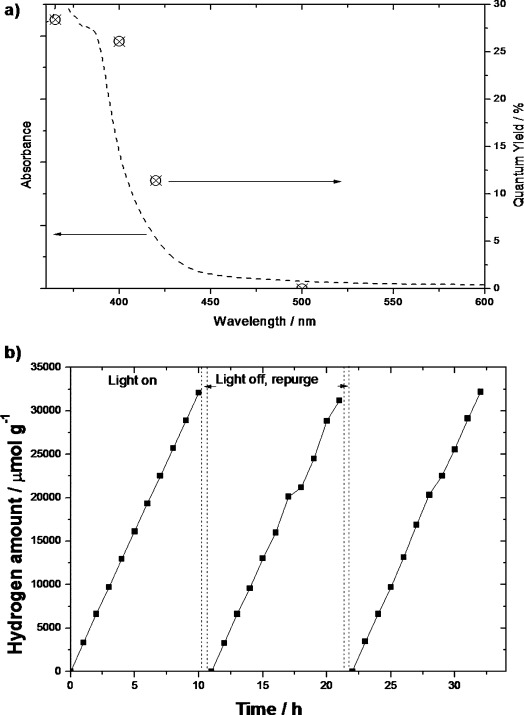
a) Quantum yield of urea-based g-C3N4, as measured by using band-pass filters at specific wavelengths (absorbance is shown by the black dashed line, internal quantum yield by circles with crosses). b) Stability test of the urea-derived g-C3N4 under irradiation with visible light (λ≥395 nm).
Table 2.
Comparison of typical g-C3N4 photocatalysts reported for hydrogen production and the corresponding quantum yields.
| Photocatalyst[a] | Band gap [eV] | HER rate under visible light [μmol h−1 g−1]b | QY [%]c | Ref. |
|---|---|---|---|---|
| g-C3N4 (cyanamide) | 2.7 | 106.9 | ca. 0.1 % (420–460 nm) | 7e |
| mpg-C3N4 | 2.7 | 1490 | N/A | 7c |
| g-C3N4 (S-doped) | 2.85 | 800 | N/A | 7a |
| g-C3N4 nanosheets | 2.35 | 1860 | 3.75 (420 nm) | 7f |
| g-C3N4 (MCA_DMSO) | 2.83 | N/A | 2.3 (420 nm) | 12 |
| our g-C3N4 (cyanamide) | 2.7 | 142.3 | N/A | this study |
| our g-C3N4 (MCA_DMSO) | 2.8 | 261.3 | 3.1 (400 nm) | this study |
| our g-C3N4 (urea) | 2.85 | 3327.5 | 26.5 (400 nm) | this study |
All photocatalysts listed were loaded with 3 wt % Pt cocatalyst, except g-C3N4 (S-doped), which was loaded with 6 wt % Pt cocatalyst. mpg-C3N4=mesoporous carbon nitride.
HER rates for external samples were measured by using a λ≥420 nm long-pass (LP) filter either, anda λ≥395 nm LP filter for our g-C3N4 samples (cyanamide and urea). The small differences observed are therefore due to the long-pass filters used.
The band-pass value for the filter used for measurement of the quantum yield is indicated by the value in brackets. N/A=not applicable.
The stability of the optimized photocatalyst was also tested in an extended experiment (Figure 4 b). The high activity was reproducible, and the material showed excellent stability during a period of 30 h. Attributes for a high quantum yield commonly include good absorption, efficient charge separation, and rapid carrier transfer to the surface for redox reactions. Even though the overall band gap of urea-based carbon nitride is larger than that of both the DCDA- and thiourea-derived carbon nitrides, it produces more hydrogen; therefore, the bulk absorption and band gap are not the determining factor in overall activity. Owing to the fewer protons in g-C3N4 (urea), the band-gap and therefore the overpotential is larger, which also results in better separation of charge and consequently causes better migration of charger carriers to active sites. The trend of the proton concentration and the trend of polymerization for the sample set are in very good agreement with the corresponding activity, which contributes to charge transport. Therefore, for the first time, it has been demonstrated that the protonation and degree of polymerization determines hydrogen-evolution rates from water, whereby the surface area plays a minor role.
To conclude, we have presented a novel strategy for the production of a structure-controlled graphitic carbon nitride, which acts as a highly efficient photocatalyst for hydrogen synthesis from water by using solar energy. The internal quantum yield in the visible region is 26.5 %, nearly an order of magnitude higher than reported previously. Under full-arc irradiation, the optimized g-C3N4 can be tailored to produce approximately 20 000 μmol g−1 of hydrogen per hour from water. The optimized g-C3N4 photocatalyst is very stable. It exhibited a near-linear profile of H2 production from water for 30 h and resulted in a TON of over 641 in a 6 h test under irradiation with a 300 W Xe lamp. Data from XPS, FTIR spectroscopy, ζ-potential measurements, and XRD show that both the protonation status and the degree of polymerization can influence the g-C3N4 hydrogen-evolution rate. In other words, it has been experimentally proven that as the degree of polymerization increases and the protonation status is reduced, the hydrogen-evolution rate can be significantly enhanced. By using two different lines of computational evidence (DFT and TDDFT), we showed that this enhancement is because of a shift in the position of the conduction-band edge, thus increasing the overpotential for reduction reactions at the surface. There is a significant shift in the CBE position even for limited proton concentrations that are less than experimental values. Furthermore, excess protonation localizes photoelectrons at non-active redox sites. It was also shown that both a high degree of polymerization and a low level of protonation can be achieved by means of an oxygen-containing precursor, such as urea in this study, as further verified by another recently reported oxygen-containing g-C3N4 precursor.12
Supporting Information
Supporting information for this article, including experimental details, is available on the WWW under http://dx.doi.org/10.1002/anie.201403375.
References
- 1.Fujishima A, Honda K, Kohayakawa K. Nature. 1972;238:37–6570. doi: 10.1038/238037a0. [DOI] [PubMed] [Google Scholar]
- 1b.Chen X, Shen S, Guo L, Mao SS. Chem. Rev. 2010;110 doi: 10.1021/cr1001645. [DOI] [PubMed] [Google Scholar]
- 2.Kato H, Kobayashi H, Kudo A. J. Phys. Chem. B. 2002;106:12441–12447. [Google Scholar]
- 2b.Martin DJ, Umezawa N, Chen X, Ye J, Tang J. Energy Environ. Sci. 2013;6 [Google Scholar]
- 3.Darwent JR, Porter G. J. Chem. Soc. Chem. Commun. 1981:145–146. [Google Scholar]
- 3b.Hitoki G, Takata T, Kondo JN, Hara M, Kobayashi H, Domen K. Chem. Commun. 2002 doi: 10.1039/b202393h. [DOI] [PubMed] [Google Scholar]
- 4.Kato H, Kudo A. J. Phys. Chem. B. 2002;106:5029–5034. [Google Scholar]
- 5.Liu M, You W, Lei Z, Zhou G, Yang J, Wu G, Ma G, Luan G, Takata T, Hara M, Domen K, Li C. Chem. Commun. 2004:2192–2193. doi: 10.1039/b407892f. [DOI] [PubMed] [Google Scholar]
- 6.Wang X, Maeda K, Thomas A, Takanabe K, Xin G, Carlsson JM, Domen K, Antonietti M. Nat. Mater. 2008;8:76–80. doi: 10.1038/nmat2317. [DOI] [PubMed] [Google Scholar]
- 7.Yue B, Li Q, Iwai H, Kako T, Ye J. Sci. Technol. Adv. Mater. 12 doi: 10.1088/1468-6996/12/3/034401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7a.Jorge AB, Martin DJ, Dhanoa MTS, Rahman AS, Makwana N, Tang J, Sella A, Corà F, Firth S, Darr JA, McMillan PF. J. Phys. Chem. C. 2011;117 [Google Scholar]
- 7b.Ge L, Han C, Xiao X, Guo L, Li Y. Mater. Res. Bull. 2013;48 [Google Scholar]
- 7c.Schwinghammer K, Tuffy B, Mesch MB, Wirnhier E, Martineau C, Taulelle F, Schnick W, Senker J, Lotsch BV. Angew. Chem. 2013;125 doi: 10.1002/anie.201206817. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2013;52 [Google Scholar]
- 7d.Xiang Q, Yu J, Jaroniec M. J. Phys. Chem. C. 2013;115 [Google Scholar]
- 7e.Wang X, Maeda K, Chen X, Takanabe K, Domen K, Hou Y, Fu X, Antonietti M. J. Am. Chem. Soc. 2011;131 doi: 10.1021/ja809307s. [DOI] [PubMed] [Google Scholar]
- 7f.Xu J, Li Y, Peng S, Lu G, Li S. Phys. Chem. Chem. Phys. 2009;15 doi: 10.1039/c3cp44687e. [DOI] [PubMed] [Google Scholar]
- 7g.Liu G, Niu P, Sun C, Smith SC, Chen Z, Lu GQ, Cheng H-M. J. Am. Chem. Soc. 2013;132 doi: 10.1021/ja103798k. [DOI] [PubMed] [Google Scholar]
- 7h.Wang Y, Zhang J, Wang X, Antonietti M, Li H. Angew. Chem. 2010;122 doi: 10.1002/anie.201000120. [DOI] [PubMed] [Google Scholar]
- 7i.Angew. Chem. Int. Ed. 2010;49 [Google Scholar]
- 8.Li KF, Martin D, Tang JW. Chin. J. Catal. 2011;32:879–890. [Google Scholar]
- 9a.Thomas A, Fischer A, Goettmann F, Antonietti M, Müller J-O, Schlögl R, Carlsson JM. J. Mater. Chem. 2008;18:4893–4908. [Google Scholar]
- 9b.Zhang Y, Liu J, Wu G, Chen W. Nanoscale. 2012;4 doi: 10.1039/c2nr30948c. [DOI] [PubMed] [Google Scholar]
- 10.Li J, Shen B, Hong Z, Lin B, Gao B, Chen Y. Chem. Commun. 2012;48:12017–12019. doi: 10.1039/c2cc35862j. [DOI] [PubMed] [Google Scholar]
- 11.Dong F, Sun Y, Wu L, Fu M, Wu Z. Catal. Sci. Technol. 2012;2:1332–1335. [Google Scholar]
- 12.Jun Y-S, Park J, Lee SU, Thomas A, Hong WH, Stucky GD. Angew. Chem. 2013;125:11289–11293. doi: 10.1002/anie.201304034. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2013;52 [Google Scholar]
- 13.Matsumura M, Saho Y, Tsubomura H. J. Phys. Chem. 1983;87:3807. [Google Scholar]
- 13b.Reber JF, Meier K. J. Phys. Chem. 1984;88 [Google Scholar]
- 14.Khaselev O, Turner JA. Science. 1998;280:425–427. doi: 10.1126/science.280.5362.425. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.