

Abstract
Generalized arterial calcification of infancy (GACI), an autosomal recessive disorder caused by mutations in the ENPP1 gene, manifests with extensive mineralization of the cardiovascular system. The affected individuals in most cases die within the first year of life, and there is currently no effective treatment for this disorder. In this study, we characterized a spontaneous mutant mouse, asj-2J, as a model for GACI. These mice were identified as part of a phenotypic deviant search in a large-scale production colony of BALB/cJ mice at The Jackson Laboratory. They demonstrated a characteristic gait due to stiffening of the joints, with phenotypic similarity to a previously characterized asj (“ages with stiffened joints”) mouse, caused by a missense mutation in the Enpp1 gene. Complementation testing indicated that asj-2J and asj were allelic. PCR-based mutation detection strategy revealed in asj-2J mice a large, 40,035 bp, deletion spanning from intron 1 to the 3′-untranslated region of the Enpp1 gene, coupled with a 74 bp insertion. This was accompanied with a significant reduction in the plasma PPi concentration and reduced PPi/Pi ratio. As a consequence, extensive aberrant mineralization affecting the arterial vasculature, a number of internal organs, and the dermal sheath of vibrissae, a progressive biomarker of the ectopic mineralization process, was demonstrated by a combination of micro computed tomography, histopathology with calcium-specific stains, and direct chemical assay of calcium. Comparison of the asj and asj-2J mice demonstrated that the latter ones, particularly when placed on an acceleration diet high in phosphate and low in magnesium, had more extensive mineralization. Thus, the asj-2J mouse serves as a novel model for GACI, a currently intractable disorder.
Introduction
Generalized arterial calcification of infancy (GACI) is a severe ectopic mineralization disorder affecting primarily the arterial blood vessels in humans. The disease is often diagnosed by prenatal ultrasound, and the affected individuals in most cases die within the first year of life from cardiovascular complications [1], [2]. GACI is inherited in an autosomal recessive fashion, and most cases are due to mutations in the ENPP1 gene, which encodes ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1), an enzyme that hydrolyses ATP to AMP and inorganic pyrophosphate (PPi) [3]. Under physiological conditions, PPi serves as a powerful anti-mineralization factor, and with reduced ENPP1 activity in GACI, the ratio of inorganic phosphate (Pi) to PPi increases creating a pro-mineralization environment and allowing ectopic tissue mineralization to ensue [4]. There is currently no effective treatment for GACI.
A number of mouse models recapitulating the clinical features of human diseases with vascular mineralization have been described [5], [6]. One of them, the asj mouse, was recently identified as a result of ENU treatment in The Jackson Laboratory Neuromutagenesis Program [7]. These mice were originally noted to demonstrate a stiff posture, abnormalities in the front legs, and a progressive, age-associated stiffening of the joints. Pathological examination at seven months of age revealed very stiff and unbendable joints with severe osteoarthritis and mineralization, and consequently, this mutant mouse was designated as “ages with stiffened joints (asj)”. Molecular characterization of these mice identified a missense mutation (p.V246D) in the Enpp1 gene which resulted in markedly reduced ENPP1 enzymatic activity, ∼24% of the level in control mice, accompanied with a marked, >80% reduction in plasma PPi concentration [7]. Necropsy of the homozygous asj mice revealed extensive mineralization affecting dermal sheath of vibrissae, an observation we have previously made in Abcc6tm1Jfk knockout mice (designated hereafter as the Abcc6−/− mouse), a model for another ectopic mineralization disorder, pseudoxanthoma elasticum (PXE) [8]. The asj mouse also demonstrated extensive vascular mineralization, particularly when placed on a so-called “acceleration diet”, enriched in phosphate and low in magnesium content. We have also shown that this diet markedly enhances the vascular mineralization in Abcc6−/− mice [9], [10].
More recently, a spontaneous mouse phenotype with similarity to that of asj mouse was noted as part of the phenotypic deviant search in The Jackson Laboratory BALB/cJ large-scale production colony. In this study, we have characterized the phenotypic and histopathologic features of this spontaneous mutant mouse, designated as asj-2J, and we have identified a large deletion/insertion mutation in the Enpp1 gene.
Materials and Methods
Mouse maintenance and diets
The spontaneous mutant mouse (asj-2J) was discovered in 2011 at The Jackson Laboratory (JAX; Bar Harbor, ME) in a large-scale production colony of BALB/cJ mice. Two female mutant mice were initially identified by their slow, hobbling gate due to stiffened joints that worsens as they age, as well as their overall sickly appearance (Figure 1). The deviant mice were transferred to the Mouse Mutant Resource (MMR) at JAX for further study. Upon arrival in the MMR, ovarian transplants were performed on the two affected mice to facilitate breeding. Subsequently, mating of the ovarian transplant recipients to a BALB/cJ wild-type male, followed by random intercrossing of F1 mice revealed an autosomal recessive mode of inheritance. These procedures were approved by The Jackson Laboratory’s Institutional Animal Care and Use Committee and performed in accordance with National Institutes of Health guidelines for the care and use of animals in research.
Figure 1. Phenotypic presentation and extensive ectopic mineralization of asj-2J mice.

A: Note the appearance of stiffened front and hind feet in asj-2J mice (arrows, lower panel) in comparison to a wild-type littermate. B: Note the extensive mineralization (red color) in the dermal sheath of vibrissae, arterial blood vessels, and various internal organs in asj-2J mice, as visualized by special stain (Alizarin Red).
The asj-2J mice were transferred to the Animal Facility of Thomas Jefferson University where they were maintained in a climate-controlled environment. Enpp1 wild type mice as well as heterozygous and homozygous asj-2J mutant mice were generated from heterozygous matings. Mice were maintained either on standard laboratory diet (Laboratory Autoclavable Rodent Diet 5010; PMI Nutritional International) or fed an ‘acceleration diet’ (Harlan Teklad, Rodent diet TD.00442, Madison, WI), which we have previously shown to accelerate the ectopic mineralization in Abcc6−/− mice [9], [10]; this diet is enriched in phosphorus and has reduced magnesium content. The mice were euthanized by CO2 asphyxiation with the use of methods approved by the American Veterinary Medical Association and subjected to necropsy, as described previously in detail [11]. Mouse handling and care were followed according to animal welfare policies of the U.S. Public Health Service. All protocols were approved by the Institutional Animal Care and Use Committee of Thomas Jefferson University.
Genotyping and gene sequencing
Genomic DNA was isolated in tail clips from mice using DNeasy Blood & Tissue kit (Qiagen Inc., Valencia, CA). The entire coding region consisting of 25 exons and intron/exon boundaries of Enpp1 sequences were amplified using PCR primers (available upon request). The asj-2J mice were found to have a large deletion of ∼40 kb extending from intron 1 up to 3′UTR of the Enpp1 gene, with a 74 bp insertion (see Results). Three primers were designed for amplification of the wild-type, mutant, or both alleles in the same reaction. The primers were the following: p1, 5′-TCAGTGATTGGTCAACAGACACCT-3′ (in intron 1); p2, 5′-GGAAGACATGAATAGCAACTACCTG-3′ (in intron 1); and p3, 5′-CTTTGGTTATTGGAGGAGACAGAAA-3′ (in 3′UTR); for their positions along the Enpp1 gene, see Fig. 2. The primer pair p1/p2 produced a 1,042-bp wild-type allele, while primers p1/p3 resulted in a 654-bp mutant allele. Gene sequencing was performed at the Kimmel Cancer Center Nucleic Acid Facility at Thomas Jefferson University using the BigDye Terminator v.3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA). Products were analyzed on the 3730 DNA Analyzer (Applied Biosystems) and the results were visualized with Chromas software (Technelysium, South Brisbane, Australia). Repetitive sequences were analyzed by RepeatMasker program (http://www.repeatmasker.org/cgi-bin/WEBRepat Masker).
Figure 2. Schematic presentation of the large deletion/insertion mutation in the Enpp1 gene in asj-2J mice, and PCR-based genotyping.

A: Note the 40,035 bp deletion extending from intron 1 to the 3′-untranslated region of the gene. The deleted segment is replaced by insertion of a 74 bp fragment which is derived from the 3′ UTR of the gene. B: Development of specific primers (p1/p2/p3), with positions shown in A, allowed genotyping of the wild-type (WT) and mutant alleles and identification of wild-type, heterozygous and mutant homozygous mice. nc: negative control without DNA.
Small-animal computed tomography (micro CT scan)
Enpp1 wild-type and asj-2J mice were examined for mineralization at 12 weeks of age by CT, as described previously [7], [12]. Briefly, mice were anesthetized with a xylazine-ketamineacetopromazine cocktail (160 µl per 25 g body weight of 10 mg/kg xylazine, 200 mg/kg ketamine, 2 mg/kg acetopromazine) and then scanned with a MicroCAT II (ImTek Inc., Oak Ridge, TN). A 3-dimensional facial rendering was created for each mouse using Amira software, version 3.1 (Visualization Sciences Group, Burlington, MA).
Histopathological analysis
Muzzle skin (left side) and internal organs from euthanized mice were fixed in 10% phosphate-buffered formalin, routinely processed, and embedded in paraffin. Tissues were sectioned (6 µm) and stained with hematoxylin and eosin (H&E) and Alizarin red using standard procedures. Slides were examined under light microscopy for tissue mineralization [13].
Quantification of calcium and phosphate
To quantify the calcium deposition in various mouse tissues, muzzle skin (right side), abdominal aorta, right carotid artery and right kidney were harvested and decalcified with 0.15 N HCl for 48 hours (skin, aorta and carotid artery) or with 0.6 N HCl for 1 week (kidney) at room temperature. The calcium content in these samples as well as in serum was determined colorimetrically by the o-cresolphthalein complexone method [Calcium (CPC) Liquicolor; Stanbio Laboratory, Boerne, TX]. The values for calcium were normalized to tissue weight and in case of carotid artery to its total length. Serum calcium concentrations were measured using the same assay. The phosphate concentration of serum was determined with Malachite Green Phosphate Assay kit (BioAssay Systems, Hayward, CA).
Plasma collection and inorganic pyrophosphate assay
Whole blood was collected by cardiac puncture into heparin coated blood collection tubes and kept on ice until separation of plasma and erythrocytes by centrifugation. The plasma was collected, depleted of platelets by filtration (2,200×g at 4°C for 20 min) through a Centrisart I 300-kDa mass cutoff filter (Sartorius, New York, NY, USA), and stored at −80°C until further processing. PPi in plasma was measured by an enzymatic assay using uridine-diphosphoglucose (UDPG) pyrophosphorylase, with modifications, as described previously [4], [14], [15].
Statistical analysis
The comparisons in different groups of mice were completed using two-sided Kruskal-Wallis nonparametric tests which is comparable to one-way analysis of variance, but without the parametric assumptions. Fisher’s exact test was used to determine the difference between proportions in mineralization phenotypes in mice. Chi-squared test was used to compare observed data with expected data according to Mendelian inheritance. All statistical computations were completed using SPSS version 15.0 software.
Results
Characterization of asj-2J mice
The asj-2J mutant mice were identified at The Jackson Laboratory as part of the phenotypic deviant search in a large-scale production colony of BALB/cJ mice, as described in Materials and Methods. Putative homozygous mice, when maintained on standard mouse diet, looked hunched at 8 weeks, and the whole body appeared stiffened at about 12 weeks of age. In particular, stiffening of the front paws was noted, similar to previously published asj mice [7] (Fig. 1A). The distribution of the mutant genotype and the gender of the pups were subsequently determined in 86 mice representing a total of 19 litters as a result of heterozygous matings. The distribution of the genotype for the homozygous wild-type, heterozygous, and homozygous mutant mice, 23∶37∶26, did not statistically differ from the expected 1∶2∶1 ratio (Chi-squared test; p>0.01), and the male/female ratio, 41∶45, was approximately 1∶1, all consistent with autosomal recessive inheritance without gender preference.
Complementation testing was undertaken using the previously identified mutant mouse strain, C57BL/6J-Enpp1asj/GrsrJ, designated as the asj mouse, with similar phenotype. Heterozygous asj females were mated to a heterozygous male asj-2J mouse resulting in one affected pup in the first litter of six pups, thus asj-2J was suggested to be a re-mutation in the Enpp1 gene. The strain was officially named BALB/cJ-Enpp1asj-2J/GrsrJ.
Identification of a deletion/insertion mutation in Enpp1
Based on phenotypic similarities and the complementation tests between the asj and asj-2J mice, it was hypothesized that these mice might be allelic and harbor mutations in the Enpp1 gene. To search for mutations in the Enpp1 gene, primer pairs were developed for amplification of all 25 exons of the Enpp1 gene, together with flanking intronic sequences. PCR amplification of exon 1, but none of the remaining exons 2–25, resulted in a PCR product in homozygous mice, whereas products with correct Enpp1 sequences were obtained in heterozygous and wild-type mice. This result suggested the presence of a large deletion spanning most of the Enpp1 gene. In attempts to identify the break point downstream from the Enpp1 gene, the next gene, ∼44 kb downstream from Enpp1, was identified as Ctgf in reverse strand, and the last exon, no. 5, of this gene was subjected to PCR amplification. This approach clearly demonstrated the presence of Ctgf in homozygous asj-2J mice, indicating that the breakpoint of the large deletion resides somewhere between the end of exon 25 of Enpp1 and upstream from the end of Ctgf. To precisely map the boarders of the deletion, primer pairs at 5 kb interval were designed to span this critical region and PCR amplifications were performed. This approach mapped the 5′ end of the deletion within intron 1 at 2,376 bp upstream of exon 2 of Enpp1 and the 3′ end of the deletion at nucleotide position 199 downstream from the stop codon (TGA) in exon 25 of Enpp1 within the 3′-untranslated region of the gene (Fig. 2A). To verify the precise breakpoints of the deletion, primer pairs within intron 1 of Enpp1 (forward primer) and downstream from the identified breakpoint at the 3′ end (reverse primer) were generated. Sequencing of this region indicated that the mutation consisted of a large deletion of 40,035 bp, but also harbored an insertion of a 74 bp DNA fragment (Fig. 2A). A 74 bp segment with identical sequence was identified in the 3′-untranslated region of the Enpp1 gene in a region of mouse chromosome 10qA4 as part of the Long Interspersed Nuclear Element (LINE) L1MDa.
Analysis of the sequences spanning the deletion breakpoints was performed to detect possible repetitive elements using the RepeatMasker program. The breakpoint in intron 1 of the Enpp1 gene was adjacent to ∼5 kb Long Terminal Repeat (LTR) with a high content of repetitive elements (99.3%). The breakpoint at 3′-untranslated region of the Enpp1 gene was adjacent to a ∼500 bp LINE, specifically L1MDa element (70.1% repetitive elements), where the 74 bp insertion resides. These repeats may have played a role in mediating the large deletion in the Enpp1 gene in the asj-2J mice.
Precise knowledge of the breakpoints in the mutant mouse, in comparison to the wild-type sequence, allowed us to develop a PCR based genotyping strategy utilizing a common forward primer and separate reverse primers for the mutant and the wild-type allele. In the case of wild-type allele, a 1,042 bp PCR product results, while mutant allele yields a 654 bp band (Fig. 2B). This rapid genotyping allows us to routinely identify homozygous mutant, heterozygous and wild-type littermates in heterozygous crossings.
Demonstration of aberrant mineralization in asj-2J mice
Considering the notion that asj-2J mice are allelic to asj mice, which demonstrate considerable mineralization of soft connective tissues, the asj-2J mice were examined for mineralization in the muzzle skin containing the dermal sheath of vibrissae, the first site of mineralization in asj as well as Abcc6−/− mice [7], [8]. This was first done non-invasively in 3 month old mice by micro CT analysis, and the results demonstrated extensive mineralization corresponding to the muzzle skin as well as in juxta-articular connective tissues, spinal and intercostal tissues, and in various arteries (Fig. 3). The mineralization of the dermal sheath of vibrissae was subsequently confirmed by histopathologic examination of the muzzle skin. Histopathology of skin sections with Alizarin red stain demonstrated extensive calcification in asj-2J mice at 3 months of age when kept on control diet (Fig. 1B). Complete necropsies of the mice also revealed extensive arterial mineralization in descending thoracic aorta, carotid artery, and in a number of internal organs, including the heart, spleen, lung, and the kidney (Fig. 1B).
Figure 3. Non-invasive demonstration of ectopic mineralization in asj-2J mice by micro CT.
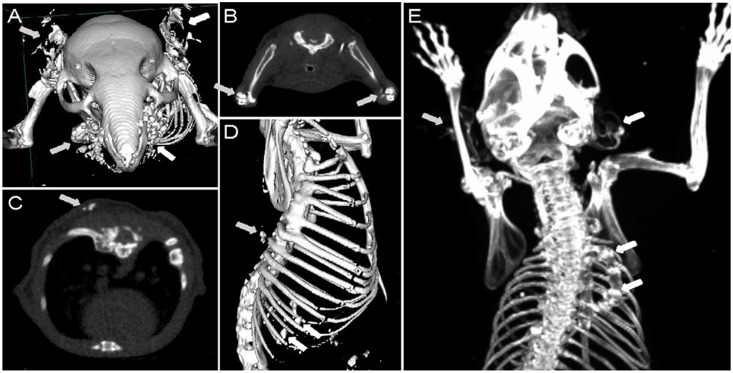
Note the extensive mineralization in the muzzle skin containing the dermal sheath of vibrissae and in the ears (A, arrows); in the juxta-articular connective tissue in the elbows (B, arrows); axial view of dorsal artery (C, arrow); spinal and intercostal tissues (D, arrows); and in the right rib fusion leading to scoliosis as well as in the ears (E, arrows).
We have previously demonstrated that a special diet, so-called “acceleration diet”, low in magnesium and high in phosphorus, facilitates the mineralization process in asj and Abcc6−/− mice [9], [10]. In this study, the asj-2J mice were placed on the acceleration diet at 4 weeks of age, at the time of weaning, and they were necropsied at 12 weeks of age and analyzed for mineralization and compared to that in the same mice kept on control diet. Histopathologic examination of the muzzle skin indicated extensive mineralization that appeared semi-quantitatively to be more abundant in asj-2J mice on the acceleration diet than in mice on the control diet (compare Fig. 1 and Fig. 4). To quantitate the amount of mineralization, a piece of muzzle skin, the abdominal aorta, carotid artery, and kidney were dissected and calcium deposits were solubilized, after which the calcium content was measured by a colorimetric chemical assay. The results indicated that tissue mineralization in homozygous mice placed on acceleration diet was higher than in the corresponding mice kept on control diet (Fig. 5 and Table 1). It should be noted that the heterozygous Enpp1+/asj-2J mice when on control diet did not show any evidence of aberrant tissue mineralization; however, the same mice, when placed on acceleration diet, demonstrated increased mineralization in the kidney (Table 1 and Fig. 5). This observation may reflect reduced capacity of the kidney to respond to the diet rich in phosphate, as noted in our previous studies [16]. Finally, the calcium contents in these tissues in males (n = 4) and females (n = 8), when analysed separately, were not statistically different (Kruskal-Wallis test; p = 0.9).
Figure 4. Enhanced mineralization in asj-2J mice placed on “acceleration diet”, rich in phosphate and low in magnesium.

The mice were placed on this diet at 4 weeks of age and necropsy was performed at 12 weeks. Note extensive mineralization as visualized by special stain (Alizarin Red). Assessment of the histopathology suggested that the mineralization was more extensive in mice kept on acceleration diet compared to the same mice kept on control diet (compare mineralization in Fig. 1B).
Figure 5. Quantitation of ectopic mineralization by chemical assay of calcium content in the muzzle skin (A), aorta (B), carotid artery (C), and kidney (D) in 12-week old asj-2J mice kept either on normal mouse diet or placed on acceleration diet at 4 weeks of age.

The statistical analysis was performed with Kruskal-Wallis nonparametric test; *, p<0.01 in comparison to wild-type Enpp1+/+ mice; +p<0.01 as compared with asj-2J mice kept on regular diet. Values are mean ± SE, n = 6–12 per group.
Table 1. Aberrant tissue mineralization in Enpp1+/+, Enpp1+/asj-2J and Enpp1asj-2J mice on normal and acceleration diet compared with Enpp1asj mice on acceleration diet.
| Mouse | Number ofmice examined | Soft tissue mineralization (%) | ||||||||
| Vibrissae | Liver | Kidney | Heart | Aorta | Eyes | Carotidartery | Lung | Spleen | ||
| Normal diet: | ||||||||||
| Enpp1+/+ | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Enpp1+/asj-2J | 11 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Enpp1asj-2J | 12 | 100 | 8 | 100 | 100 | 67 | 33 | 83 | 92 | 67 |
| Acceleration diet: | ||||||||||
| Enpp1+/+ | 8 | 0 | 0 | 88 | 0 | 0 | 0 | 0 | 0 | 0 |
| Enpp1+/asj-2J | 8 | 0 | 0 | 100 | 0 | 0 | 0 | 0 | 0 | 0 |
| Enpp1asj-2J | 6 | 100 | 100** | 100 | 100 | 100 | 100* | 100 | 100 | 100 |
| Acceleration diet: | ||||||||||
| Enpp1asj | 9 | 100 | 56 | 100 | 67 | 56 | 44+ | 78 | N/A | N/A |
Mice were placed on either normal diet or acceleration diet at 4 weeks of age, and tissues were collected for histopathology at 3 months of age. The values represent the number of mice, as percent of all animals examined, affected by mineralization as examined by hematoxylin and eosin stain on one section. Statistical analyses were performed with Fisher’s Exact test. Comparison to Enpp1asj-2J mice on normal diet: *P<0.05; **P<0.01. Comparison to Enpp1asj-2J mice on acceleration diet: + P<0.05.
Comparison of mineralization in asj and asj-2J mice
As indicated above, C57BL/6J-Enpp1asj and BALB/cJ-Enpp1asj-2J mice are allelic in Enpp1 gene with similar phenotypic features. The mutation in asj mice has previously shown to be a missense mutation, p.V246D [7], while the asj-2J mice have a large deletion eliminating 24 out of 25 exons, i.e., 57% of the entire gene and 93% of the coding region. To examine whether the differences in the mutations might lead to different degrees of ectopic mineralization, these two mutant mouse strains were placed on acceleration diet at 4 weeks of age, and complete necropsies were performed at 12 weeks, i.e., after 8 weeks on special diet. Examination of the internal organs, including arterial blood vessels, demonstrated considerable mineralization in both mouse strains - however, the mineralization was more frequent and more extensive in asj-2J mice as compared to that in asj mice (Table 1). The degree of mineralization was also quantitated by chemical assay of calcium in muzzle skin in asj and asj-2J mice when placed on acceleration diet. The asj-2J mice showed significantly higher level of calcium than that in asj mice; asj: 126.3±5.8 (n = 9) and asj-2J: 176.8±4.8 (n = 6) µmol Ca/g tissue (p<0.01).
Chemical assays in plasma/serum
The asj-2J mice were further analyzed by determining the calcium, phosphorus, and inorganic pyrophosphate concentrations in serum/plasma when kept on regular control diet. The results indicated normal levels of serum calcium and phosphate, and consequently the Ca/P ratio was not altered. However, there was a significant reduction, ∼80% in the inorganic pyrophosphate levels in the plasma of asj-2J mice in comparison to the wild-type littermates (Table 2). As a consequence, the PPi/Pi ratio was significantly reduced from 4.2 to 0.88 (×1,000) (p<0.01).
Table 2. Calcium, phosphorus and pyrophosphate concentrations in serum/plasma of mice maintained on a regular rodent diet.
| Parameter | Concentration (mean ± S.E.) | ||
| Enpp1+/+ (n = 8) | Enpp1+/asj-2J (n = 11) | Enpp1asj-2J (n = 12) | |
| Calcium (mg/dL) | 9.38±0.18 | 9.52±0.15 | 9.41±0.17 |
| Phosphorus (mg/dL) | 9.32±0.32 | 9.30±0.31 | 9.37±0.24 |
| Ca/P ratio | 1.01±0.03 | 1.03±0.03 | 1.01±0.03 |
| PPi (µM) | 2.53±0.44 | 1.78±0.23 | 0.52±0.08* |
| PPi/Pi ratio (×1,000) | 4.22±0.74 | 2.96±0.38 | 0.88±0.12* |
Blood samples were collected by cardiac puncture, Ca and P concentrations were determined in serum, and PPi levels were measured in heparinized plasma after removal of platelets. Statistical significance in comparison to Enpp1+/+ mice: *P<0.01.
Discussion
GACI is an autosomal recessive disorder that manifests with profound arterial mineralization often diagnosed by prenatal ultrasound. The affected children are born with cardiovascular complications and the majority of them die within the first year of life [1], [2]. GACI in most cases is caused by mutations in the ENPP1 gene encoding an enzyme, ENPP1, which catalyzes the hydrolysis of ATP to form AMP and PPi. The extracellular PPi is a powerful anti-mineralization factor while Pi acts as a pro-mineralization factor, and consequently, a physiological PPi/Pi ratio is critical in preventing ectopic mineralization under normal homeostasis [4]. There are a number of enzymes and transport systems that contribute to the extracellular PPi/Pi ratio. In addition to ENPP1, these include CD73, a plasma membrane linked enzyme that breaks down AMP to adenosine and Pi, and tissue non-specific alkaline phosphatase (TNAP) that converts PPi to Pi [5], [17]. The activity of TNAP is inhibited by adenosine, and reduced CD73 activity results in reduction of adenosine concentration, thus allowing accelerated conversion of PPi to Pi by TNAP, and therefore promotes ectopic mineralization [18]. In addition, a number of transmembrane protein systems have been shown to facilitate transport of PPi and Pi from intracellular milieu to the extracellular space, including ankylosis protein (ANK), and type I–III sodium-dependent Pi co-transporters, SLC17A1, SLC34, and SLC20 [19], [20], [21]. Finally, recent studies have suggested that ABCC6, a transmembrane transporter, which is nonfunctional in patients with PXE, is required for release of ATP to the extracellular milieu [22]. Thus, in the absence of functional ABCC6, the extracellular concentration of ATP is reduced, and consequently, there is less ATP to serve as a substrate for ENPP1 to generate extracellular PPi. Collectively, under normal physiologic conditions, there is a complex pro-mineralization/anti-mineralization network that is required to maintain the normal homeostatic ratio of PPi/Pi [2], [5]. Mutations in many of the genes controlling this ratio have been shown to result in ectopic mineralization of the soft connective tissues, particularly in the skin and the arterial blood vessels. For example, mutations in the ENPP1 gene result in GACI, mutations in the ABCC6 gene underlie PXE, and patients with mutations in the NT5E gene, which encodes CD73, develop arterial calcification due to CD73 deficiency (ACDC) [3], [23], [24].
A number of animal models, particularly targeted and spontaneous mutant mice, have been extremely helpful in providing pathomechanistic information on ectopic mineralization in human diseases [5], [6]. In this study, we describe a novel mutant mouse, asj-2J, which was identified in the colony breeding program of The Jackson Laboratory. This mouse was noted to have extensive mineralization of the dermal sheath of vibrissae as well as arterial blood vessels, and the mice developed a phenotypic gait due to periarticular mineral deposits. The mineralization phenotype could be significantly accelerated by placing the mice on “acceleration diet”, enriched in phosphate and low in magnesium. The phenotypic similarity of these mutant mice with a previously described asj mouse prompted us to test the hypothesis that asj-2J mice were allelic, and complementation studies supported the notion that both mice had mutations in the same gene, Enpp1. Previous studies have demonstrated that asj mice harbor a homozygous missense mutation, p.V246D, reflecting T-to-A transversion in Enpp1 as a result of ENU treatment [7]. The mutation in asj-2J mice was determined to consist of a large deletion spanning from intron 1 all the way to the end of Enpp1, thus completely eliminating functional gene in these mice. As a consequence, these mice were shown to have markedly reduced PPi plasma concentrations leading to reduced PPi/Pi ratio which allows ectopic mineralization to ensue. It should be noted that the asj-2J mice developed more extensive mineralization than asj mice when placed on the acceleration diet. This may reflect the differences in the type of mutations, the asj-2J having a large deletion, essentially comparable to a complete ablation of the gene, while asj mice have a missense mutation allowing residual level of ENPP1 activity [5]. In fact, measurements of ENPP1 in the liver of asj mice revealed low levels of activity as compared to Enpp1 wild-type mice, yet there was clearly measurable activity above the background [5]. It should be noted, however, that these two mice were on different inbred strain backgrounds, asj on C57BL/6J and asj-2J on BALB/cJ, respectively. We have previously demonstrated that different strain backgrounds can influence the degree of mineralization [10], [13], an observation that may impact on our comparison of differences in asj and asj-2J mice. Nevertheless, since the mutations in the ENPP1 gene in patients with GACI result in loss-of-function, the asj-2J mouse with complete ablation of the gene appears to be a suitable model system to study this disease [3], [25].
In addition to asj and asj-2J mice, four other mutations in the mouse Enpp1 gene have previously been described. One of them, so called “tip toe walking” (Enpp1ttw/ttw) mouse, has been shown to harbor a stop codon mutation, p.G568X, in Enpp1 [26], while a mouse with similar phenotype (Enpp1ttw-Ham) was shown to harbor a splice-site mutation c.259+1G>T in Enpp1 [27]. A genetically engineered knock-out mouse, Enpp1tm1Gdg, was shown to exhibit phenotypic similarities to Enpp1ttw/ttw mice [28], while another mutant mouse, Enpp1m1Amgn, with a p.C397S mutation showed reduced long bone mineral density and crystal-related arthropathy [29]. While the previously published Enpp1 mutant mice were shown to develop ectopic mineralization, their characterization was largely focused on skeletal alterations and perispinal soft tissue mineralization. Also, while arterial calcification was noted in some of these previously described mice, the histologic lesions were not evident until ∼16–22 weeks of age. In asj-2J mice, when kept on either standard mouse diet or the “acceleration diet”, mineralization of the dermal sheath of vibrissae was noted at ∼4 weeks of age. This was accompanied with early demise of asj-2J mice, frequently at the age of 12–16 weeks of life. This is likely due to cardiovascular involvement, particular occlusion of small and medium sized arteries. Thus, this mouse displays features of GACI, and can serve as a platform to test various anti-mineralization compounds that could be used for treatment of ectopic mineralization disorders. It is also of interest that a novel in vivo model mimicking features of GACI, a zebrafish enpp1 mutant dragonfish, has recently been described [30]. This model was also treated with bisphosphonate etidronate which rescued aspects of the mineralization phenotype in this fish.
A limited number of studies have been published testing the efficacy of bisphosphonates, stable non-hydrolyzable pyrophosphate analogues of pyrophosphate, to counteract the development of ectopic mineralization in patients with GACI [31], [32], [33], [34]. These case studies have yielded somewhat conflicting results; while improvement has been reported in some cases, in several studies the effects are not clear, and significant side effects from bisphosphonates have also been reported. The reasons for these diverse outcomes are not entirely clear but may relate to the fact that GACI is a complex clinical disorder with unpredictable progression, and there is no biomarker to measure the disease activity. The degree of disease progression is primarily assessed by occasional imaging studies of ectopic mineralization and the ultimate outcome of patient survival. Thus, the asj-2J mouse, genetically uniform on homogeneous strain background and under controlled environmental conditions, can serve as a platform to test pharmacological compounds with anti-mineralization properties, including various bisphosphonates.
In conclusion, the asj-2J mouse serves as a novel model to study molecular alterations in GACI, and it provides a platform to test various pharmacologic approaches for treatment of GACI and related, currently intractable, ectopic mineralization disorders.
Acknowledgments
The authors thank Joshua Kingman, Rachel Nordstrom and Dian Wang for laboratory assistance; Dr. Madhukar Thakur and Neil Mehta for micro CT scan; and Drs. Christine Feldmyer and Thea Price for interpretation of the images. Mark Pawlowski and Amy Terry assisted in processing histopathology; Jocelyn Andrel Sendecki in statistical analysis; and Carol Kelly in manuscript preparation.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper.
Funding Statement
This work was supported by National Institutes of Health (NIH)/National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) grants K01 AR064766 (to QL), R21 AR063781 (to JPS), and R01 AR28450 (to JU). This work was also supported by NIH/National Center for Research Resources/Office of Research Infrastructure Programs grants OK010972-35, OD011163-02, RR001183, and RR032339 (The Jackson Laboratory). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Nitschke Y, Rutsch F (2012) Generalized arterial calcification of infancy and pseudoxanthoma elasticum: two sides of the same coin. Front Genet 3:302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rutsch F, Nitschke Y, Terkeltaub R (2011) Genetics in arterial calcification: pieces of a puzzle and cogs in a wheel. Circ Res 109:578–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rutsch F, Ruf N, Vaingankar S, Toliat MR, Suk A, et al. (2003) Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nature Genet 34:379–381. [DOI] [PubMed] [Google Scholar]
- 4. O’Neill WC, Sigrist MK, McIntyre CW (2010) Plasma pyrophosphate and vascular calcification in chronic kidney disease. Nephrol Dial Transplant 25:187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li Q, Uitto J (2013) Mineralization/anti-mineralization networks in the skin and vascular connective tissues. Am J Pathol 183:10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mackenzie NC, Huesa C, Rutsch F, MacRae VE (2012) New insights into NPP1 function: lessons from clinical and animal studies. Bone 51:961–968. [DOI] [PubMed] [Google Scholar]
- 7. Li Q, Guo H, Chou DW, Berndt A, Sundberg JP, et al. (2013) Mutant Enpp1asj mouse as a model for generalized arterial calcification of infancy. Dis Model Mech 6:1227–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Klement JF, Matsuzaki Y, Jiang QJ, Terlizzi J, Choi HY, et al. (2005) Targeted ablation of the Abcc6 gene results in ectopic mineralization of connective tissues. Mol Cell Biol 25:8299–8310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jiang Q, Uitto J (2012) Restricting dietary magnesium accelerates ectopic connective tissue mineralization in a mouse model of pseudoxanthoma elasticum (Abcc6−/−) . Exp Dermatol 21:694–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li Q, Uitto J (2010) The mineralization phenotype in Abcc6−/− mice is affected by Ggcx gene deficiency and genetic background–a model for pseudoxanthoma elasticum. J Mol Med (Berl) 88:173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Silva KA, Sundberg JP (2012) Necropsy methods. In: Hedrich HJeditor. The Laboratory Mouse. 2nd ed. London: Academic Press. pp. 779–806. [Google Scholar]
- 12. Le Corre Y, Le Saux O, Froeliger F, Libouban H, Kauffenstein G, et al. (2012) Quantification of the calcification phenotype of Abcc6-deficient mice with microcomputed tomography. Am J Pathol 180:2208–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Berndt A, Li Q, Potter CS, Liang Y, Silva KA, et al. (2013) A single-nucleotide polymorphism in the Abcc6 gene associates with connective tissue mineralization in mice similar to targeted models for pseudoxanthoma elasticum. J Invest Dermatol 133:833–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tolouian R, Connery SM, O’Neill WC, Gupta A (2012) Using a filtration technique to isolate platelet free plasma for assaying pyrophosphate. Clin Lab 58:1129–1134. [PMC free article] [PubMed] [Google Scholar]
- 15. Li Q, Price TP, Sundberg JP, Uitto J (2014) Juxta-articular joint-capsule mineralization in CD73 deficient mice: Similarities to patients with NT5E mutations. Cell cycle 13:2609–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li Q, Chou DW, Price TP, Sundberg JP, Uitto J (2014) Genetic modulation of nephrocalcinosis in mouse models of ectopic mineralization: the Abcc6(tm1Jfk) and Enpp1(asj) mutant mice. Lab Invest 94:623–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nitschke Y, Rutsch F (2012) Genetics in arterial calcification: lessons learned from rare diseases. Trends Cardiovasc Med 22:145–149. [DOI] [PubMed] [Google Scholar]
- 18. Markello TC, Pak LK, St Hilaire C, Dorward H, Ziegler SG, et al. (2011) Vascular pathology of medial arterial calcifications in NT5E deficiency: Implications for the role of adenosine in pseudoxanthoma elasticum. Mol Genet Metab 103:44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zaka R, Williams CJ (2006) Role of the progressive ankylosis gene in cartilage mineralization. Curr Opin Rheumatol 18:181–186. [DOI] [PubMed] [Google Scholar]
- 20. Bottger P, Hede SE, Grunnet M, Hoyer B, Klaerke DA, et al. (2006) Characterization of transport mechanisms and determinants critical for Na+-dependent Pi symport of the PiT family paralogs human PiT1 and PiT2. Am J Physiol Cell Physiol 291:C1377–1387. [DOI] [PubMed] [Google Scholar]
- 21. Miyamoto K, Haito-Sugino S, Kuwahara S, Ohi A, Nomura K, et al. (2011) Sodium-dependent phosphate cotransporters: lessons from gene knockout and mutation studies. J Pharm Sci 100:3719–3730. [DOI] [PubMed] [Google Scholar]
- 22. Jansen RS, Duijst S, Mahakena S, Sommer D, Szeri F, et al. (2014) ABCC6-mediated ATP secretion by the liver Is the main source of the mineralization inhibitor inorganic pyrophosphate in the systemic circulation-brief report. Arterioscler Thromb Vasc Biol 34:1985–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li Q, Jiang Q, Uitto J (2008) Pseudoxanthoma elasticum: oxidative stress and antioxidant diet in a mouse model (Abcc6−/−). J Invest Dermatol 128:1160–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. St Hilaire C, Ziegler SG, Markello TC, Brusco A, Groden C, et al. (2011) NT5E mutations and arterial calcifications. N Engl J Med 364:432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lorenz-Depiereux B, Schnabel D, Tiosano D, Hausler G, Strom TM (2010) Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. Am J Hum Genet 86:267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Okawa A, Nakamura I, Goto S, Moriya H, Nakamura Y, et al. (1998) Mutation in Npps in a mouse model of ossification of the posterior longitudinal ligament of the spine. Nature Genet 19:271–273. [DOI] [PubMed] [Google Scholar]
- 27. Takabayashi S, Seto S, Katoh H (2014) A new Enpp1 allele, Enpp1(ttw-Ham), identified in an ICR closed colony. Exp Anim 63:193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sali A, Favaloro JM, Terkeltaub R, Goding JW (1999) Germline deletion of the nucleoside triphosphate pyrophosphohydrolase (NTPPPH) plasma cell membrane glycoprotein-1 (PC-1) produces abnormal calcification of periarticular tissues. In: Vanduffel L LReditor. Ecto-ATPases and related ectoenzymes. Maastricht, The Netherlands: Shaker Publishing. pp. 267–282. [Google Scholar]
- 29. Babij P, Roudier M, Graves T, Han CY, Chhoa M, et al. (2009) New variants in the Enpp1 and Ptpn6 genes cause low BMD, crystal-related arthropathy, and vascular calcification. J Bone Min Res 24:1552–1564. [DOI] [PubMed] [Google Scholar]
- 30. Apschner A, Huitema LF, Ponsioen B, Peterson-Maduro J, Schulte-Merker S (2014) Zebrafish enpp1 mutants exhibit pathological mineralization, mimicking features of generalized arterial calcification of infancy (GACI) and pseudoxanthoma elasticum (PXE). Dis Model Mech 7:811–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ramjan KA, Roscioli T, Rutsch F, Sillence D, Munns CF (2009) Generalized arterial calcification of infancy: treatment with bisphosphonates. Nat Clin Pract Endocrinol Metab 5:167–172. [DOI] [PubMed] [Google Scholar]
- 32. Rutsch F, Boyer P, Nitschke Y, Ruf N, Lorenz-Depierieux B, et al. (2008) Hypophosphatemia, hyperphosphaturia, and bisphosphonate treatment are associated with survival beyond infancy in generalized arterial calcification of infancy. Circ Cardiovasc Genet 1:133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Edouard T, Chabot G, Miro J, Buhas DC, Nitschke Y, et al. (2011) Efficacy and safety of 2-year etidronate treatment in a child with generalized arterial calcification of infancy. Eur J Pediatr 170:1585–1590. [DOI] [PubMed] [Google Scholar]
- 34. Otero JE, Gottesman GS, McAlister WH, Mumm S, Madson KL, et al. (2013) Severe skeletal toxicity from protracted etidronate therapy for generalized arterial calcification of infancy. J Bone Miner Res 28:419–430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper.