

Abstract
The objective was to describe the pharmacokinetics of vortioxetine and evaluate the effect of intrinsic and extrinsic factors in the healthy population. Data from 26 clinical pharmacology studies were pooled. A total of 21,758 vortioxetine quantifiable plasma concentrations were collected from 887 subjects with corresponding demography. The doses ranged from 2.5 to 75 mg (single dose) and 2.5–60 mg (multiple QD doses). The pharmacokinetics of vortioxetine was best characterised by a two-compartment model with first-order absorption, lag-time and linear elimination, with interindividual error terms for absorption rate constant, oral clearance and central volume of distribution. The population mean was 32.7 L/hr for oral clearance and 1.97·103 L for the central volume of distribution. The average elimination half-life was 65.8 hr. CYP2D6 inferred metabolic status (ultra, extensive, intermediate or poor metabolisers) and age on oral clearance and height on central volume of distribution were identified as statistically significant covariate–parameter relationships. For CYP2D6 poor metabolisers, CL/F was approximately 50% to that seen in CYP2D6 extensive metabolisers. The impact of height on V2/F and age on CL/F was low and not considered to be clinically relevant. The final model was found to be reliable, stable and predictive. A reliable, stable and predictive pharmacokinetic model was developed to characterise pharmacokinetics of vortioxetine in the healthy population.
Major depressive disorder (MDD) is a leading cause of disability and is associated with significant reductions in quality of life, impaired productivity, reduced overall health and substantial economic costs 1. Vortioxetine (1-[2-(2,4-dimethyl-phenylsulfanyl)- phenyl]-piperazine hydrobromide) is a novel antidepressant with a multi-modal mechanism of action approved in the USA and EU for the treatment of major depressive disorder at doses of 5, 10, 15 and 20 mg. It works through a combination of two pharmacological modes of action: inhibition of the serotonin transporter and direct modulation of receptor activity. In vitro studies indicate that vortioxetine is a 5-HT3, 5-HT1D and 5-HT7 receptor antagonist, 5-HT1B receptor partial agonist, 5-HT1A receptor agonist, and inhibitor of the 5-HT transporter 2. In vivo non-clinical studies have demonstrated that vortioxetine increases the levels of the neurotransmitters serotonin, noradrenaline, dopamine, acetylcholine and histamine in specific brain areas 2. Because depression is a multi-factorial disorder, agents such as vortioxetine with multiple therapeutic targets may have advantages over ‘selective’ agents 3.
The clinical pharmacokinetics of vortioxetine, as determined by non-compartmental analysis (NCA), is characterised by a prolonged absorption, a medium clearance (47 L/hr) and a large volume of distribution (3.5 × 103 L) 4. The absolute bioavailability for oral administration was found to be 75% 4. Vortioxetine undergoes extensive metabolism, primarily via oxidation and subsequent glucuronic acid conjugation. In vitro data suggest that several CYP isoenzymes are involved in the oxidative metabolism of vortioxetine, including CYP2D6, CYP3A4/5, CYP2C9, CYP2C19, CYP2A6, CYP2C8 and CYP2B6 5. The major metabolite is the benzoic acid of vortioxetine (3-methyl-4-(2-piperazine-1-yl-phenylsulfanyl)-benzoic acid) Lu AA34443 5, which is considered to be much less pharmacologically active and incapable of crossing the blood–brain barrier (unpublished data). Clinical drug–drug interaction studies in healthy individuals have shown that co-administration of omeprazole (a CYP2C19 substrate and inhibitor) had no effect on the pharmacokinetics of vortioxetine, co-administration of bupropion (CYP2D6 inhibitor) increased the exposure of vortioxetine approximately 2-fold and co-administration of rifampicin (a broad CYP inducer) decreased the area under the curve (AUC) of vortioxetine by 72% 6. During the clinical development of vortioxetine, genotype data for CYP2D6, CYP2C19 and CYP2C9 have been collected from healthy individuals participating in clinical pharmacology studies.
Non-linear mixed effect modelling quantitatively estimates the magnitude of unexplained variability in the population of interest. Identification of factors contributing to this variability and exploration of their relationship to exposure is an important component of the population pharmacokinetic approach and can be used to support dosing recommendations.
A large number of clinical pharmacology studies with vortioxetine have been performed. The aim of the present meta-analysis was to pool subject-level data from a majority of these studies and perform a population pharmacokinetic analysis of vortioxetine in healthy individuals.
Methods
Studies analysed
Data from 26 clinical pharmacology single- and multiple-dose studies, performed in Europe, the USA or Japan, with rich PK sampling were pooled. Data obtained following oral administration of vortioxetine in the dose ranges of 2.5–75 mg for single dose and 2.5–60 mg/day for multiple doses were included in the analyses. For drug–drug interaction studies where vortioxetine was investigated with other compounds, only data for vortioxetine administered alone were included in the data set. For the renal and hepatic impairment studies, only data from the healthy control subjects were included. Study details are summarised in Table 1.
Table 1.
Study subjects and drug administrations
| Study | Main purpose | # subjects1 | Doses (mg) |
|---|---|---|---|
| 1 | PK | 35 (35) | 10, 20, 30, 50 and 75 (SD) |
| 2 | PK | 56 (20) | 2.5, 5, 10, 20, 40 and 60 (MD) |
| 3 | AME | 6 (6) | 50 (SD) |
| 4 | PET | 16 (16) | 2.5, 10, 30 and 60 (MD) |
| 5 | DDI | 18 (9) | 10 (MD) |
| 6 | PET | 38 (38) | 2.5, 5 and 20 (MD) |
| 7 | PK | 60 (48) | 2.5, 5, 10, 20 and 40 (SD): 5, 10 and 20 per day (MD, men), 5 and 10 per day (MD, women) |
| 8 | DDI | 14 (8) | 20 (SD) |
| 9 | DDI | 24 (12) | 10 (MD) |
| 10 | BA | 24 (15) | 10 (SD) |
| 11 | DDI | 36 (18) | 10 (SD) |
| 12 | TQT | 164 (164) | 10 and 40 (MD) |
| 13 | DDI | 34 (26) | 10 (MD) |
| 14 | DDI | 28 (15) | 10 (MD) |
| 15 | DDI | 28 (17) | 10 (MD) |
| 16 | DDI | 66 (51) | 20 (SD) and 40 (SD) |
| 17 | SP | 48 (24) | 10 (SD) and 10 (MD) |
| 18 | SP | 17 (10) | 10 (SD) |
| 19 | SP | 25 (16) | 10 (SD) |
| 20 | BA | 22 (11) | 20 (SD) |
| 21 | BA | 23 (11) | 20 and 30 (SD) |
| 22 | BA | 18 (10) | 20 (SD and MD) and 30 (MD) |
| 23 | DDI | 26 (12) | 10 (MD) |
| 24 | BA | 21 (7) | 10/20 (MD) and 20/30 (MD) |
| 25 | SP | 16 (8) | 10 (SD) |
| 26 | BA | 24 (20) | 20 (SD) |
AME, Absorption, metabolism and excretion; BA, Relative or absolute bioavailability; DDI, Drug–drug interaction; PET, Positron emission tomography (imaging); PK, Pharmacokinetic and tolerability; TQT, Thoroughly QTc; SP, special population; SD, single dosing; MD, multiple dosing.
Number of subjects on vortioxetine treatment with number of men in parentheses.
Subjects
A total of 21,758 quantifiable plasma concentrations of vortioxetine collected from 887 healthy individuals were included in the analysis data set. The demographic characteristics of the healthy subject population are given in Table 2. All subjects gave written informed consent prior to undergoing pharmacokinetic evaluation and genotyping, and all studies were approved by local ethics committees. In addition, the studies were conducted in accordance with the revised Declaration of Helsinki for biomedical research involving human subjects and the rules of Good Clinical Practice.
Table 2.
Characteristics of the healthy individuals included in the vortioxetine pooled population pharmacokinetic analysis
| N | Mean | S.D. | Minimum | Median | Maximum | |
|---|---|---|---|---|---|---|
| Age (year) | 887 | 33.7 | 13.3 | 18 | 30 | 78 |
| Weight (kg) | 887 | 73.6 | 12.3 | 45.2 | 73.3 | 112 |
| Height (cm) | 887 | 172 | 9 | 144 | 173 | 203 |
| BMI (kg/m2) | 887 | 24.8 | 3.3 | 15.1 | 24.8 | 35.5 |
| Lean body-weight (kg) | 887 | 55.8 | 8.8 | 34.8 | 56.3 | 84.5 |
| Albumin (g/L) | 887 | 46.0 | 3.0 | 36 | 46 | 55 |
| ALAT (IU/L) | 881 | 23.4 | 12.4 | 5 | 20 | 128 |
| ASAT (IU/L) | 881 | 21.8 | 7.5 | 10 | 21 | 145 |
| Bilirubin (μmol/L) | 869 | 12.2 | 5.7 | 3 | 11 | 52 |
| Creatinine clearance (mL/min.) | 887 | 122 | 28 | 41.9 | 120 | 223 |
| Counts (frequency) | |
|---|---|
| Males:Females | 627:260 |
| Phenotype CYP2D6 UM:EM:IM:PM | 21:540:276:38 |
| Missing phenotype CYP2D6 data | 12 |
| Phenotype CYP2C19 EM:IM:PM | 548:282:44 |
| Missing phenotype CYP2C19 data | 13 |
| Phenotype CYP2C9 – EM:IM:PM | 786:17:0 |
| Missing phenotype CYP2C9 data | 84 |
| White | 657 |
| Black or African American | 98 |
| Asian | 129 |
| Native Hawaiian or Other Pacific Islander | 1 |
| American Indian or Alaska Native | 1 |
| Missing race | 1 |
| Non-Hispanic:Hispanic | 366:298 |
| Missing ethnicity data | 223 |
| Region EU:US:Japan | 327:444:116 |
S.D., standard deviation; UM, ultra metaboliser; EM, extensive metaboliser; IM, intermediate metaboliser; PM, poor metaboliser.
Bioanalytical methods
The analyses of plasma concentrations of vortioxetine were performed with a validated internal method (LC-MS/MS) according to 7. Details of the bioanalytical method used are given elsewhere 6.
Covariates
The following continuous covariates were included in the analysis: age, weight, height, body mass index (BMI), lean body mass (LBM), albumin, ALAT (SGPT), ASAT (SGOT), bilirubin and creatinine clearance. The following categorical covariates were included: sex, race, ethnicity (Hispanic or non-Hispanic), region (EU, USA or Japan) and inferred metabolic status for CYP2C9, CYP2C19 and CYP2D6. Each categorical covariate had to include at least 10 subjects; otherwise, the covariate was recategorised (if more than two categories) or omitted from the analysis.
Creatinine clearance (CrCL) was estimated from serum creatinine using the Cockroft–Gault formula. For race, the categories were White (Caucasian), Black or African American, Asian, native Hawaiian or other Pacific Islander and American Indian or Alaska Native, while the categories for ethnicity were non-Hispanic and Hispanic.
The CYP2D6, CYP2C9 and CYP2C19 genes display allelic heterogeneity and different alleles have been identified (see for example www.cypalleles.ki.se). Functionality (associated enzyme activity) tested in vivo can be divided into full functionality (FF), decreased functionality (DF) and non-functionality (NF). Categorisation of CYP enzymes genotype into inferred metabolic status was based on the ‘gene dose’ method proposed by Steimer et al. 8. Alleles with full functionality were assigned a gene dose of 1 and alleles with reduced functionality a value of 0.5, while alleles with no functionality were assigned a value of zero. If the sum of the gene dose for the alleles was zero, the inferred metabolic status was categorised as poor metaboliser (PM), if the sum was 0.5 or 1, the category was intermediate metaboliser (IM), if the sum was 1.5 or 2, the category was extensive metaboliser (EM) and for a sum over 2, the category was set to ultra metaboliser (UM).
Population pharmacokinetic analysis
Population pharmacokinetics analysis was performed by means of non-linear mixed effect modelling. Based on internal knowledge previously acquired during the development of the compound, the chosen initial structural model for vortioxetine was a two-compartment model with lag-time and first-order absorption and elimination (fig. 1). However, one- and three-compartment models were also evaluated. The two-compartment model was parameterised in terms of ka (absorption rate constant), CL/F (oral clearance), V2/F (volume of distribution for central compartment), Q/F (intercompartmental clearance), V3/F (volume of distribution for peripheral compartment) and ALAG1 (lag-time). Interindividual variability (IIV) was modelled as exponential terms for ka, CL/F and V2/F and a covariance term between CL/F and V2/F was included. Residual error was modelled as proportional. Plasma-concentration data were log-transformed (natural logarithm) prior to analysis in order to have a more robust optimisation and to increase the possibility of convergence. Individual elimination half-lives (t1/2) were estimated based on the empirical Bayes estimates of CL/F, V2/F, Q/F and V3/F. The half-life was estimated as ln(2)/beta, where
Fig 1.
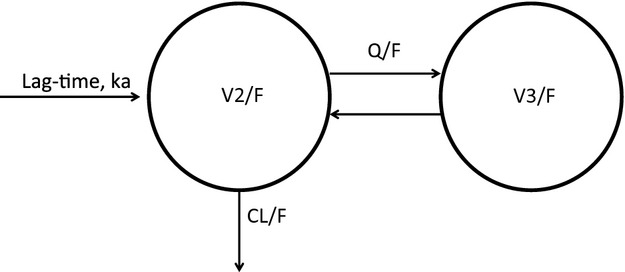
Structural model used for the population pharmacokinetic analysis of vortioxetine.
The influence of subject-specific covariates was assessed by the forward-inclusion and backward-deletion method. Missing continuous covariates were imputed with the median value for the population while missing categorical covariates were imputed with the most frequent category. Plots, physiologically based expectations and methods such as the GAM (generalised additive model) were used for selecting the covariate–parameter relationships to be tested in the forward-inclusion part. Discrimination between two nested models was based on the objective function value (OFV) and interindividual variability. A covariate relationship was only implemented in the model if
The OFV decreased with by least 6.64 points (p < 0.01) and
The interindividual variability for the pharmacokinetic parameter affected by the covariate was lower compared to the model without the covariate relationship.
The stepwise forward addition of covariates into the model continued until a decrease of 6.64 could no longer be reached. To determine whether all the covariates included in the fully parameterised population pharmacokinetic model continued to provide a significant influence on the population model, the covariates included in the full model were stepwise removed and the resulting reduced model evaluated to determine whether there was a significant degradation. The significance of each covariate was tested using the nested model criteria at a more stringent p-value of 0.005, resulting in a decrease in OFV of 7.88, to avoid false positives. Continuous covariates were entered into the model in a centred manner, while categorical covariates entered the model using dummy variables (e.g. 0 or 1 for sex). Since no non-linear trends could be found, all relationships were assumed to be linear.
Model validation
The stability of the parameter estimates in the final model was assessed using the condition number and nonparametric bootstrap analysis. The condition number was computed as the square root of the ratio of the largest eigenvalue to the smallest eigenvalue of the covariance matrix. Condition numbers less than 1000 indicate acceptable stability of the parameter estimates 9. For the bootstrap analysis, 200 data sets were generated by repeated random sampling with replacement from the NONMEM input data file, and the final NONMEM model was fitted to the bootstrap data sets. The mean and 95% confidence intervals for the population PK parameters were derived and compared with the estimates for the original data set. The bootstrap 95% confidence intervals were estimated by ranking the parameters from the bootstrap runs. Convergence of bootstrap and set success rate percent were estimated. Visual predictive check (VPC) plots were constructed to evaluate the model predictability. The median and 95% prediction intervals (2.5 and 97.5% percentiles) of simulated concentrations were plotted together with dose-normalised (to 10 mg) observed concentrations after single dosing. In addition, the normalised prediction distribution errors (NPDE) were estimated and used for assessment of predictability. For a model with good predictability, the NPDE should follow an N(0,1) distribution 10. Also the shrinkage for the individually estimated concentrations (epsilon shrinkage) and parameters (eta shrinkage) were calculated 11. Although no formal criterion exists, a level of 0.2 was used to conclude ‘negligible shrinkage’.
Simulations
Using the parameter estimates from the final model, steady-state plasma-concentration profiles were simulated for the doses 5, 10, 15 and 20 mg, using the same healthy population as included in the population pharmacokinetic modelling regarding covariate distributions. One thousand plasma-concentration profiles per dose at steady state were simulated. From the plasma-concentration profiles and estimated CL/F values, the pharmacokinetic parameters AUC0–24 (area under the plasma-concentration curve from dosing to 24 hr post-dose) was estimated as dose/(CL/F) and Cmax (maximal plasma concentration) was read directly from the plasma-concentration profiles. The arithmetic mean, median, standard deviation and the 2.5% and 97.5% percentiles for AUC0–24 and Cmax were estimated.
Softwares
The non-linear mixed effect modelling was performed with the software NONMEM® version 7.1 (ICON Development Solutions, Ellicott City, MD, USA), compiled using the Intel Visual FORTRAN Compiler (Santa Clara, CA, USA) (standard edition version 9.1). Data management, running and post-processing of NONMEM® were done with an in-house system based on Perl and S-PLUS®.
Results
For 6 of the 26 studies included in the data set, no information about ethnicity was available. Therefore, for the 223 subjects from these six studies, the imputed ethnicity was ‘non-Hispanic’ (the most frequent value among the subjects with ethnicity information). The CYP2C9 genotype was missing for 84 of the subjects and was imputed as ‘extensive metaboliser’ (EM). For CYP2C19 and CYP2D6, 13 and 12 subjects had missing data, respectively; these were imputed as ‘extensive metaboliser’ (EM). For the continuous covariates, the following were missing: ALAT (6 subjects), ASAT (6 subjects) and bilirubin (18 subjects). All missing continuous covariates were imputed using the median value.
The initial two-compartment structural model described the data well. A 1-compartment model was significantly worse, while a three-compartment model did not further improve the fit. Interindividual variability was modelled as exponential terms on ka, CL/F and V2/F and with a covariance term between CL/F and V2/F. Residual error was expressed as proportional.
In the forward-inclusion covariate model building, age and CYP2D6 inferred metabolic status on CL/F and height on central volume of distribution were found to be significant covariate–parameter relationships (full model). Backward deletion revealed that all covariates in the full model were retained in the final model, that is, OFV increased by more than 7.88 points by deleting any of them, and the final model was accordingly identical to the full model. The forward-inclusion steps are summarised in Table 3. CYP2C19-inferred metabolic status was highly correlated with CL/F in the first forward-inclusion step, with a drop of 72 points in OFV; but this relationship was not further evaluated in the next covariate model step due to the over-parameterisation that resulted from having both CYP2D6- and CYP2C19-inferred metabolic status in the same model. All tested models for continuous covariates were linear, since plots of pharmacokinetic parameter values versus covariates did not indicate any non-linearity. In addition, power covariate models for the age-CL/F and height-V2/F relationships were tested for the final NONMEM model, but these were not better.
Table 3.
Summary of the covariate model steps
| Step | Covariate–parameter relationship | ΔOFV1 |
|---|---|---|
| 1 (forward inclusion)2 | CYP2D6-inferred metabolic status on CL/F | −148 |
| 2 (forward inclusion) | Age on CL/F | −75 |
| 3 (forward inclusion) | Height on V2/F | −63 |
| 4 (backward deletion) | >7.88 |
Change in objective function value (OFV) value compared to previous step.
CYP2C19-inferred metabolic status was highly correlated with CL/F in the first forward-inclusion step, with a drop of 72 points in OFV; but this relationship was not further evaluated in the next covariate model step due to over-parameterisation.
Table 4 gives the parameter values for the final model, while fig. 2 shows the goodness-of-fit plots. The mean group CL/F values for the four different categories of CYP2D6-inferred metabolic status were 53 L/hr (UM), 34 L/hr (EM), 27 L/hr (IM) and 18 L/hr (PM). A box plot of CYP2D6-inferred metabolic status versus empirical Bayesian estimated CL/F values are shown in fig. 3. For CYP2C19, the CL/F values were 36 L/hr (EM), 29 L/hr (IM) and 25 L/hr (PM) [taken from the model with CYP2C19 on CL/F only]. CL/F decreased with age by 0.28 L/hr for every year. The interindividual variability for CL/F and V2/F decreased from 33% to 31% (V2/F) and from 46% to 42% (CL/F) compared to the base model. The covariance matrix for random effects (etas) for the final model is given in Table 5.
Table 4.
Parameter values for the final population pharmacokinetic model of vortioxetine in healthy individuals
| Model parameters | Parameter estimate |
|---|---|
| Absorption rate constant (ka) | |
| Estimate (1/hr) | 0.142 |
| RSE1 (%) | 2.7 |
| IIV2 (%) | 51 |
| RSE1 (%) | 7.0 |
| Volume of distribution, central comp (V2/F) | |
| Estimate (L) | 1.97 × 103 |
| RSE1 (%) | 1.6 |
| IIV2 (%) | 31 |
| RSE1 (%) | 7.7 |
| Height on V2/F (L/cm) | 17.4 |
| RSE1 (%) | 12 |
| Oral clearance (CL/F) | |
| Estimate (L/hr) | 52.9 (UM4), 34.1 (EM4), 26.6 (IM4), 18.1 (PM4) |
| RSE1 (%) | 6.0 (UM4), 1.7 (EM4), 2.5 (IM4), 6.6 (PM4) |
| IIV2 (%) | 42 |
| RSE1 (%) | 7.0 |
| Correlation CL/F – V2/F | 0.61 |
| Age on CL/F (L/hr/yr) | 0.277 |
| RSE1 (%) | 8.6 |
| Volume of distribution, peripheral comp (V3/F) | |
| Estimate (L) | 6.61 × 102 |
| RSE1 (%) | 3.8 |
| IIV2 (%) | – |
| Intercompartmental clearance (Q/F) | |
| Estimate (L/hr) | 22.5 |
| RSE1 (%) | 4.6 |
| IIV2 (%) | – |
| Lag-time (ALAG) | |
| Estimate (hr) | 0.781 |
| RSE1 (%) | 0.8 |
| IIV2 (%) | – |
| Residual error | |
| Estimate | 0.0654 |
| Estimate3 (%) | 26 |
| RSE1 (%) | 4.4 |
UM, ultra metaboliser; EM, extensive metaboliser; IM, intermediate metaboliser; PM, poor metaboliser.
Relative standard error (RSE) was calculated as Standard Error/Estimate*100 from NONMEM® results.
Interindividual variability.
Percentage standard deviation calculated as 100*sqrt(Estimate).
CYP2D6-inferred metabolic status.
Fig 2.

Goodness-of-fit plots for the final population pharmacokinetic model for vortioxetine in healthy individuals.
Fig 3.
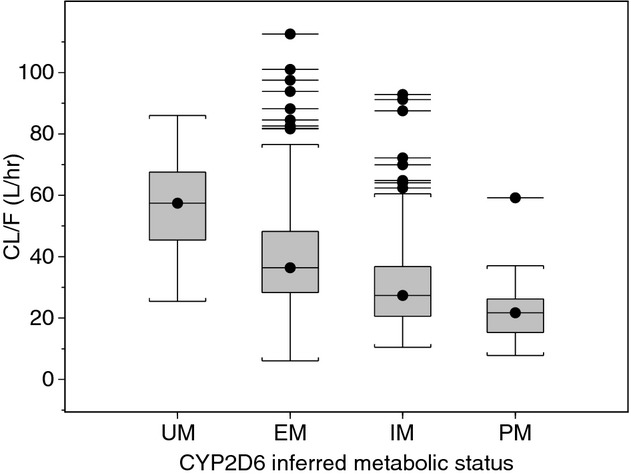
CYP2D6-inferred metabolic status (UM, ultra metaboliser; EM, extensive metaboliser; IM, intermediate metaboliser; PM, poor metaboliser) versus oral clearance (CL/F) for vortioxetine. Two subjects with CL/F values over 150 L/hr are excluded from the plot in order to improve readability.
Table 5.
Covariance matrix for random effects (etas) for the final population pharmacokinetic model of vortioxetine in healthy individuals
| Parameter | ka | V2/F | CL/F |
|---|---|---|---|
| ka | 0.26 | – | – |
| V2/F | – | 0.094 | – |
| CL/F | – | 0.078 | 0.18 |
The mean elimination half-life (t1/2) of vortioxetine for all subjects, estimated using empirical Bayesian estimates of the final model parameters, was 65.8 hr with a standard deviation of 26.8 hr and a median of 61.3 hr. The estimated mean t1/2 values were 42, 60, 73 and 104 hr for CYP2D6-inferred metabolic status of UM, EM, IM and PM, respectively. By multiplying the t1/2 values by 3.3, one obtains the time when 90% steady state is reached. In fig. 4, the fraction of subjects reaching 90% steady state is plotted against days of vortioxetine dosing.
Fig 4.
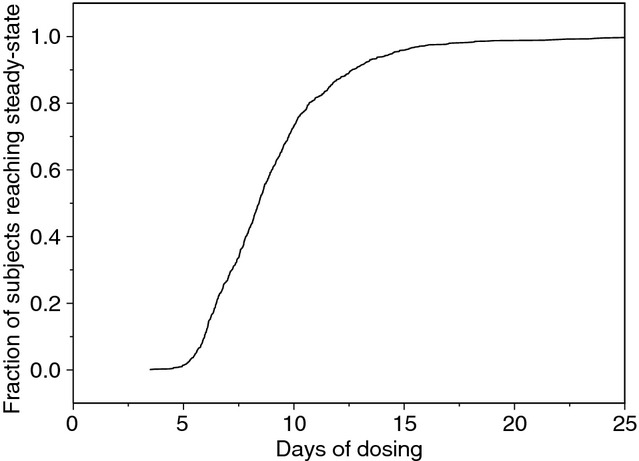
Fraction of healthy individuals reaching 90% steady state versus days of dosing of vortioxetine.
Model validation
Of the 200 bootstrap runs, 199 converged, giving a convergence rate of 99.5%. The differences between the parameter values estimated with the bootstrap replicated data sets, and the original NONMEM input data file were all within ±6%. The condition numbers for the base and the final models were 5.1 and 5.4, respectively. In both cases, these were well below the criterion of 1000 for a stable non-linear model, indicating very stable models, as confirmed by the high convergence rate for the bootstrap analysis of the final model.
The eta shrinkages for the final model were 22% (ka), 16% (V2/F) and 2% (CL/F). Epsilon shrinkage was 4%. According to the rule-of-thumb of 20–30% 12, a small degree of shrinkage was only found for ka. The VPC plot (fig. 5) showed that the observed values lay within the 95% confidence intervals. The estimated NPDE values followed an N(0,1) distribution.
Fig 5.
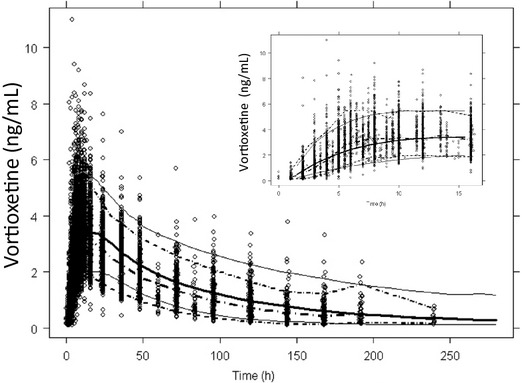
Visual predictive check (VPC) plot for the final population pharmacokinetic model for vortioxetine in healthy individuals. Dose-normalised observed data (dots) and median, 2.5% and 97.5% percentiles for simulated data (solid lines) and dose-normalised observed data (dotted lines) are shown. The insert shows the first 20 hr after dosing.
Simulations
Using the final model, simulations of steady-state plasma-concentration profiles were performed for the approved doses 5, 10, 15 and 20 mg. Descriptive statistics of AUC0–24 and Cmax are given in tables6 and 7, respectively.
Table 6.
Simulated AUC0–24 values at steady state in the healthy population after multiple administrations of 5–20 mg vortioxetine
| Dose (mg/day) | Median (ng h/mL) | Mean (ng h/mL) | S.D. (ng h/mL) | 95% PI (ng h/mL) |
|---|---|---|---|---|
| 5 | 161 | 163 | 101 | 68;406 |
| 10 | 323 | 326 | 202 | 137;812 |
| 15 | 484 | 490 | 303 | 205;1219 |
| 20 | 645 | 653 | 404 | 273;1625 |
S.D., standard deviation; PI, prediction interval.
Table 7.
Simulated Cmax values at steady state in the healthy population after multiple administrations of 5–20 mg vortioxetine
| Dose (mg/day) | Mean (ng/mL) | Median (ng/mL) | S.D. (ng/mL) | 95% PI (ng/mL) |
|---|---|---|---|---|
| 5 | 7.2 | 7.1 | 4.3 | 3.1;17.4 |
| 10 | 14.4 | 14.2 | 8.5 | 6.1;34.9 |
| 15 | 21.6 | 21.4 | 12.8 | 9.2;52.3 |
| 20 | 28.8 | 28.5 | 17.0 | 12.3;69.8 |
S.D., standard deviation; PI, prediction interval.
Cmax and AUC0–24 at steady state for 20 mg were simulated for a typical subject with a height of 175 cm and age of 40 years, a tall person (187 cm), a short person (155 cm), an old person (75 years) and a young person (18 years) to assess the impact of height and age on exposure. The short person (155 cm) had 5% higher Cmax and AUC0–24 values than the average subject (175 cm). The tall person (187 cm) had 4% lower Cmax and AUC0–24 values than the average subject. The young person (18 years) had a 14% lower Cmax and a 15% lower AUC0–24 than the average subject, while the old person (75 years) had a 38% higher Cmax and a 40% higher AUC0–24 than the average subject.
Discussion
A reliable, stable and predictive population pharmacokinetic model for vortioxetine in healthy subjects has been developed on the basis of data collected from 887 subjects in 26 studies. Based on the parameter estimates, general goodness-of-fit and NPDE plots, a two-compartment model with first-order absorption and elimination described the data well. Also the predictability of the model was good, based on the VPC plot.
Vortioxetine is a compound with a medium clearance and a large volume of distribution. The oral clearance (47 L/hr), volume of distribution (3.7 × 103 L) and elimination half-life (57 hr) of vortioxetine in healthy young subjects have been estimated from non-compartmental analysis 4. These values can be compared with the values obtained in the present population pharmacokinetic analysis of 33 L/hr for CL/F (from the base model), 2.6 × 103 L for the sum of the central and peripheral volumes of distribution and 66 hr for the elimination half-life.
With an absolute bioavailability of 75% and with a population mean oral clearance of 33 L/hr (from the base model), the average systemic clearance is around 25 L/hr. The sum of the population mean of the central and peripheral volumes of distribution after oral administration, that is, V2/F + V3/F, is 2.60 × 103 L, which gives a volume of distribution of 1.9 × 103 L assuming a bioavailability of 75%.
All body size measures, except BMI, were strongly correlated with the central volume of distribution, although only height remained in the final model. Weight, LBM and height are all heavily correlated, especially for subjects participating in clinical pharmacology studies with narrow inclusion ranges; hence, inclusion of the most significant of them in the model often excludes the other ones. The small effect of height, as a measure of body size, on the simulated vortioxetine exposure (±5% for tall/short subjects compared to average height) is not considered of clinical relevance.
CYP2D6-inferred metabolic status was shown to have a significant impact on CL/F, where extensive metabolisers in general had 1.9 times the CL/F of poor metabolisers. This is in line with a drug–drug interaction study with vortioxetine and bupropion (a CYP2D6 inhibitor), in which the CL/F of vortioxetine was reduced approximately by half when co-administered with bupropion 6. For CYP2C19, EMs had on average 1.4 times the CL/F of PMs. However, in a drug–drug interaction study, no effects on vortioxetine steady-state exposure were observed with co-administration of a single dose of omeprazole (a CYP2C19 inhibitor) 12.
Age was found to impact the oral clearance to a statistically significant degree. CL/F decreased by 0.28 L/hr per year of increasing age, corresponding up to a 40% higher exposure in a 75-year-old subject compared with the population mean of 44 years. The age effect on CL/F cannot be explained by the fact that renal clearance decreases with age, since the renal clearance of vortioxetine is negligible. Vortioxetine is mainly metabolised through CYP2D6 and to a lesser degree through CYP2C19, but we are not aware of any age dependency (in the adult population) of CYP2D6 or CYP2C19 human activity in the literature. A relationship between creatinine clearance and clearance of many drugs eliminated via non-renal routes has previously been reported 13,14. A recent review of all new drug applications approved by the US Food and Drug Administration between 2003 and 2007 showed that of 23 non-renally excreted molecules whose pharmacokinetics had been studied in subjects with impaired kidney function, 13 displayed altered pharmacokinetics and six had alterations important enough to justify dose adjustments 15,16. In addition, a review by Touchette and Slaughter 13 reported that there exists some evidence, although it is inconclusive that the activity of the CYP2D6 isozyme is reduced with renal impairment. In the present population pharmacokinetic analysis, creatinine clearance was not a significant covariate in the final model, but could be because the inclusion criteria for the studies limit the range of creatinine clearance. The apparent age dependency of oral clearance may in fact be an effect of decreasing kidney function with age. However, the modest impact of age on the exposure of vortioxetine is not considered to be clinically relevant.
In the population studied, men had a larger volume of distribution and a higher oral clearance compared to women, and sex was by itself a significant covariate for both volume of distribution and oral clearance. Population mean values for men were 1.94 × 103 L (V2/F) and 34 L/hr (CL/F), while for women, the values were 1.81 × 103 L (V2/F) and 30 L/hr (CL/F). However, when the effect of height on V2/F was incorporated in the model, sex was no longer a significant covariate, that is, body size (in this case, height) explains the difference between the sexes in V2/F and CL/F.
The population pharmacokinetic meta-analysis performed was in healthy individuals. It should be noted that due to sometimes narrow inclusion criteria in clinical pharmacology studies, the intrinsic and extrinsic factors of the population studied do not always fully reflect the ones in the patient population.
In summary, we have shown that vortioxetine is a compound with a large volume of distribution, a medium clearance and a relatively long terminal half-life. The oral clearance is significantly related to age and CYP2D6-inferred and CYP2C19-inferred metabolic status, while the central volume of distribution is significantly related to body size (height). The impact of these covariates is of limited clinical relevance, and except for strong CYP2D6 inhibitors and broad CYP450 inducers, no dose adjustment is needed 17.
Conflict of interest
All authors are employed by H. Lundbeck A/S or Takeda Pharmaceuticals Ltd, the sponsors of the original studies.
References
- 1.World Health Organization. Mental health: depression. http://www.who.int/mediacentre/factsheets/fs369/en/. (last accessed on 21 January 2014)
- 2.Bang-Andersen B, Ruhland T, Jørgensen M, Smith G, Frederiksen K, Jensen KG, et al. Discovery of 1-[2-(2,4-dimethylphenylsulfanyl)phenyl]piperazine (Lu AA21004): a novel compound for the treatment of mood and anxiety disorders. J Med Chem. 2011;54:3206–21. doi: 10.1021/jm101459g. [DOI] [PubMed] [Google Scholar]
- 3.Millan MJ. Multi-target strategies for the improved treatment of depressive states: conceptual foundations and neuronal substrates, drug discovery and therapeutic application. Pharmacol Ther. 2006;110:135–370. doi: 10.1016/j.pharmthera.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 4.Areberg J, Søgaard B, Højer A-M. The clinical pharmacokinetics of Lu AA21004 and its major metabolite in healthy young volunteers. Basic Clin Pharmacol Toxicol. 2012;111:198–205. doi: 10.1111/j.1742-7843.2012.00886.x. [DOI] [PubMed] [Google Scholar]
- 5.Hvenegaard MG, Bang-Andersen B, Pedersen H, Jørgensen M, Püschl A, Dalgaard L. Identification of the cytochrome P450 and other enzymes involved in the in vitro oxidative metabolism of a novel antidepressant, Lu AA21004. Drug Metab Dispos. 2012;40:1357–65. doi: 10.1124/dmd.112.044610. [DOI] [PubMed] [Google Scholar]
- 6.Chen G, Lee R, Højer A-M, Buchbjerg JK, Serenko M, Zhao Z. Pharmacokinetic drug interactions involving vortioxetine (Lu AA21004), a multimodal antidepressant. Clin Drug Investig. 2013;33:727–36. doi: 10.1007/s40261-013-0117-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.U.S. Department of Health and Human Services, Food and Drug Administration. Guidance for industry – bioanalytical method validation. 2001. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm368107.pdf (last accessed on 21 January 2014)
- 8.Steimer W, Zöpf K, von Amelunxen S, Pfeiffer H, Bachofer J, Popp J, et al. Allele-specific change of concentration and functional gene dose for the prediction of steady-state serum concentrations of amitriptyline and nortriptyline in CYP2C19 and CYP2D6 extensive and intermediate metabolizers. Clin Chem. 2004;50:1623–33. doi: 10.1373/clinchem.2003.030825. [DOI] [PubMed] [Google Scholar]
- 9.Bonate PL. Pharmacokinetic-Pharmacodynamic Modeling and Simulation. New York: Springer Science + Business Media Inc; 2006. [Google Scholar]
- 10.Brendel K, Comets E, Laffont C, Laveille C, Mentré F. Metrics for external model evaluation with an application to the population pharmacokinetics of gliclazide. Pharm Res. 2006;23:2036–49. doi: 10.1007/s11095-006-9067-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karlsson MO, Savic RM. Diagnosing model diagnostics. Clin Pharmacol Ther. 2007;82:17–20. doi: 10.1038/sj.clpt.6100241. [DOI] [PubMed] [Google Scholar]
- 12.Savic RM, Karlsson MO. Importance of shrinkage in empirical Bayes estimates for diagnostics: problems and solutions. AAPS J. 2009;11:558–69. doi: 10.1208/s12248-009-9133-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Touchette MA, Slaughter RL. The effect of renal failure on hepatic clearance. DICP. 1991;25:1214–24. doi: 10.1177/106002809102501111. [DOI] [PubMed] [Google Scholar]
- 14.Nolin TD, Naud J, Leblond FA, Pichette V. Emerging evidence of the impact of kidney disease on drug metabolism and transport. Clin Pharmacol Ther. 2008;83:898–903. doi: 10.1038/clpt.2008.59. [DOI] [PubMed] [Google Scholar]
- 15.Naud J, Nolin TD, Leblond FA, Pichette V. Current understanding of drug disposition in kidney disease. J Clin Pharmacol. 2012;52:10S–22S. doi: 10.1177/0091270011413588. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Zhang L, Abraham S, Apparaju S, Wu TC, Strong JM, et al. Assessment of the impact of renal impairment on systemic exposure of new molecular entities: evaluation of recent new drug application. Clin Pharmacol Ther. 2009;85:305–11. doi: 10.1038/clpt.2008.208. [DOI] [PubMed] [Google Scholar]
- 17.Brintellex SmPC. Summary of product characteristics. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002717/WC500159449.pdf. 2014 (last accessed on 21 January 2014)