

Abstract
To catalyze pre-mRNA splicing, U6 snRNA positions two metals that interact directly with the scissile phosphates. The U6 metal ligands correspond stereospecifically to metal ligands within the catalytic domain V of a group II self-splicing intron. In domain V, the ligands are organized by base-triple interactions, which also juxtapose the 3′ splice site with the catalytic metals. However, in the spliceosome, the mechanism for organizing catalytic metals and recruiting the substrate has remained unclear. Here we show by genetics, crosslinking, and biochemistry in yeast that analogous triples form in U6 and promote catalytic metal binding and both chemical steps of splicing. Because the triples include an element that defines the 5′ splice site, the triples also provide a mechanism for juxtaposing the pre-mRNA substrate with the catalytic metals. Our data indicate that U6 adopts a group II intron-like tertiary conformation to catalyze splicing.
Introns are removed from pre-mRNAs by the spliceosome - a dynamic ribonucleoprotein (RNP) machine composed of 80 conserved proteins and five small nuclear RNAs (snRNAs; ref. 1). While proteins play key supporting roles in catalysis2,3, the catalytic core itself is composed of RNA (ref. 4). Indeed, this RNA-based core catalyzes two sequential phosphotransesterifications that are identical to the reactions performed by group II intron RNAs, which self-splice in the absence of proteins. Specifically, in both systems an intronic 2′ hydroxyl first attacks the 5′ splice site to form a branched lariat structure5, and then the 5′ exon attacks the 3′ splice site to form mRNA. These two reactions were proposed to be catalyzed by a general, two-metal mechanism6, in which one divalent metal stabilizes the nucleophile and the second divalent metal stabilizes the leaving group. Indeed, crystal structures of group II introns have revealed that ligands in the catalytic domain V position two divalent metals within 4 Å, the preferred distance for the two-metal mechanism, and that these metals interact with the 5′ splice site7,8 (c.f. 9,10). Supporting a catalytic role for these metals, divalent metals stabilize the leaving groups during group II intron splicing, thus promoting catalysis9,10. Indicating a two metal mechanism for pre-mRNA splicing as well, we have recently demonstrated that ligands in U6 snRNA (Fig. 1a) bind two distinct divalent metals that catalyze splicing by interacting with the leaving groups during both chemical steps4.
Figure 1. Base-triple interactions in the group II intron catalytic core and their proposed counterparts in the spliceosome.
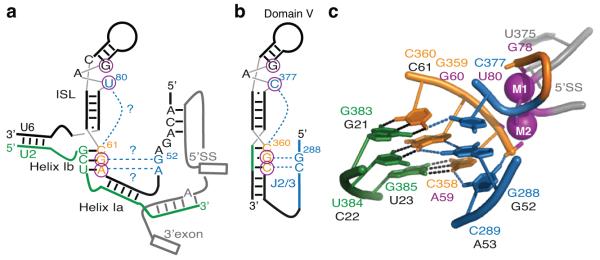
(a,b) Secondary structure model of key RNA structures present in the spliceosomal (a) and group II intron (b) catalytic cores. Residues in the catalytic triad are colored orange and their base-pairing partners green. Residues involved in base-triple interactions in domain V and their proposed counterparts in U6 are colored blue. The U6 and domain V residues that bind catalytic metals are circled4,7. The base-triple interactions tested in this study are shown in a as blue dashed lines highlighted with question marks. The pre-mRNA in a is shown in grey in a configuration before branching. c, Structure of the group II intron domain V, highlighting the catalytic triplex (PDB 4FAQ, ref. 8). Coloring is as in a. Residue numbers are shown in orange, green, and blue for the group II intron, with the proposed corresponding residues in the U6 snRNA denoted below in black or magenta (for catalytic metal ligands). Watson-Crick interactions are shown as black dashed lines and base-triple interactions are shown as blue dashed lines. The catalytic metals (M1 and M2), their non-bridging oxygen ligands, and the scissile phosphate at the 5′ splice site are colored magenta. Note that this particular class of group II introns contains an unusual CGC triad, rather than the canonical AGC.
In the group II intron catalytic core, the conserved AGC triad of domain V together with nucleotides in the upper portion of the stem-loop, including a conserved bulged position, bind two distinct metals (Fig. 1b,c; refs. 7,11). The ligands that form the two metal sites are brought together by base-triple interactions between the AGC triad and the bulge in a configuration stabilized by a conserved distal element, termed the J2/3 linker12,13 (Fig. 1b,c). By organizing domain V, the triple helix positions the two catalytic metals with the 4 Å spacing preferred for phosphoryl transfer catalysis6,8,10. Additionally, the J2/3 linker functions in both steps of splicing12,13 and recognizes the 3′ splice site14, thereby promoting docking of the 3′ splice site into the catalytic core. Thus, in the group II intron the triple helix effects catalysis both by positioning catalytic metal ligands and recruiting the 3′ splice site. In the spliceosome, however, the mechanism for catalytic metal positioning and substrate docking has remained unclear.
Nonetheless, the RNA structures at the heart of the spliceosome share several similarities to the catalytic core of group II introns. Like RNA domains of group II introns, the snRNAs define and juxtapose the chemically reactive sites in the substrate, through U2/U6 helix Ia and adjacent interactions15-17 (Fig. 1a). Additionally, similarly to the catalytic domain V of group II introns, U2/U6 helix Ib and the intramolecular stem-loop (ISL) of U6 adopt a secondary structure in which a conserved AGC triad is situated five base pairs away from a conserved bulge (Fig. 1a,b), both of which are important for each step in splicing15,17-20. Moreover, the U6 catalytic metal ligands, situated in the triad and the bulge, correspond directly and stereospecifically to the domain V metal ligands4,7,8.
The functional and structural similarities between group II introns and the spliceosome led to the prediction that a group II-like triple helix may form in U6 snRNA to similarly position analogous metal ligands. This spliceosomal triplex would join the AGC triad, the bulge, and the terminal GA of the conserved ACAGAGA sequence (Fig. 1a, ref. 11). Because the 5′ end of the ACAGAGA sequence base pairs with the 5′ splice site21,22, such a triple helix would also provide a structural mechanism for docking of the 5′ splice site, required for both transesterifications, into the catalytic RNA core. Recently, three-dimensional modeling of the spliceosomal active site revealed that such a triplex is consistent with current experimental data23. To determine if the U6 snRNA positions catalytic metals in a manner similar to domain V of group II introns, we therefore investigated whether a group II intron-like catalytic triplex forms in the U6 snRNA.
Through molecular genetics in vivo and in vitro, in combination with site-directed crosslinking, we present compelling evidence that a triple helix equivalent to that seen in domain V forms in U6 snRNA during catalytic activation of the spliceosome. Our data define a structural mechanism by which the spliceosome positions two metals in a catalytic configuration and juxtaposes these metals with the substrate reactive groups within a single catalytic core for both steps of splicing.
RESULTS
Genetic evidence for the U6 triplex
The AGC triad of U6 snRNA can base pair with U6 downstream, to form an extended ISL (refs. 16,24); with U4, to form U4/U6 stem I (ref. 25); and with U2, to form U2/U6 helix Ib (refs. 15,19), but genetics implicates a function for the AGC triad beyond these roles (refs. 15,19). For instance, a thorough in vivo compensatory analysis that repaired all of these predicted base pairing interactions was unable to alleviate the lethality of three mutations in the AGC triad19. To determine whether the residual function of the AGC triad reflects formation of a catalytic triplex (Fig. 1), we tested exhaustively whether the growth defects of the AGC triad could be suppressed in an allele- and position-specific manner by compensatory mutations in the predicted base-triple partners7,8,11 (Supplementary Note 1). A similar mutational approach has been applied previously in the study of triple helix interactions in other RNAs (refs. 26,27) and subsequently validated by direct structural observation using NMR or X-ray crystallography27,28.
The final position of the AGC triad, C61 (U6-C61) has been predicted to interact with the bulged U6 ISL residue, U80, that binds two catalytic metals (Fig. 1a, refs. 4,7,11). While C61 is tolerant to mutation15,19, the C61G mutation grows slowly, even when helix Ib is repaired by a compensatory mutation in U2 (Fig. 2a, ref. 19), allowing us to assay for suppression by U6-U80 mutations. Strikingly, U80C suppressed the growth defect of this repaired helix Ib double mutant (Fig. 2a). Suppression was both allele- and position-specific; other alleles at U80 did not improve growth of the helix Ib double mutant (Fig. 2a), and U80C did not suppress the growth defect of mutations at other positions in the AGC triad, such as A59U or G60U (Fig. 2b, Supplementary Fig. 1a), which were by contrast suppressed by mutations in their respective, predicted base-triple partners (see below); indeed, U80C exacerbated mutations at A59 and G60 (Fig. 2b, Supplementary Table 1), which would be expected for simultaneous disruption of two different base-triple interactions within a triple helix, as observed for the PAN RNA triplex27. Thus, the specific suppression by U80C provides evidence for a direct interaction between the bulged U6-U80 in the U6 ISL and U6-C61 of the AGC triad (Supplementary Note 2).
Figure 2. Genetic evidence for a base-triple interaction between U80 and C61 in the U6 snRNA.

(a,b) Spot assays showing growth on selective media of equivalent numbers of yeast cells containing combinations of alleles at U6-U80 and U6-C61 (a) or U6-A59 (b). The allele combinations for the U2/U6 helix Ib base pair mutated in each case are indicated above each panel. Alleles of U6-U80 present in each row are indicated on the left. c, Diagrams of isomorphic base-triple interactions: C*C-G (left, as in group II intron structure; ref. 11), U*C-G (middle, predicted for the spliceosome), and C*G-C (right, predicted for the spliceosome suppressor). The location of the glycosyl bond is highlighted with a circle - open black for the major groove interacting residue, filled black for the catalytic triad, and gray for U2 or equivalent residues in group II intron.
The other two positions of the AGC triad, U6-G60 and U6-A59, have been predicted to pair with U6-G52 and U6-A53, respectively, of the conserved ACAGAGA sequence. Importantly, these two residues fall between the 5′ splice site binding site of U6 and U2/U6 helix Ia, which is immediately adjacent to the catalytic core, such that triplex formation would promote docking of the 5′ splice site into the catalytic core (Fig. 1a; ref. 11). All three point mutations at the central position of the AGC triad, G60, are lethal and only the conservative substitution G60A can be suppressed by restoring base-pairing in U2/U6 helix Ib (refs. 15,19). Nevertheless, we found that a mutation of the predicted base-triple residue, G52, suppressed G60U, albeit mildly (Fig. 3a). This marks the first observed suppression of G60U (Supplementary Note 3). Remarkably, suppression of G60U did not require restoration of base-pairing in U2/U6 helix Ib (Fig. 3a). As we observed for mutations at U6-C61 (Fig. 2), suppression of G60U was allele- and position-specific (Fig. 3a-b, Supplementary Fig. 1b, Supplementary Note 4), thus providing compelling evidence for an interaction between G52 of the ACAGAGA sequence and G60 of the AGC triad, in the context of a base triple that includes helix Ib base pairing.
Figure 3. Genetic evidence for base-triple interactions between the AGC triad and the ACAGAGA region of the U6 snRNA.

(a,b) Spot assays showing growth on selective media of equivalent numbers of yeast cells containing combinations of alleles at G52 and G60 (a) or C61 (b). (c,d) Spot assays showing growth on selective media of equivalent numbers of yeast cells containing combinations of alleles at A53 and A59 (c) or C61 (d). Matrices are presented as in Figs. 2a,b. Note that in a yeast also contained a mutation at position 59 in U4 to repair U4/U6 stem I (ref. 19). For additional positional specificity controls see Supplementary Fig. 1. (e,f) Diagrams of observed (group II intron) and predicted (spliceosome) isomorphic base-triple interactions11 involving the first (f) and second (e) residues of the catalytic triad. Diagrams are as in Fig. 2c.
Mutations at U6-A59 showed a wider range of phenotypes, allowing for multiple tests of suppression at this position. Although restoring U2/U6 helix Ib suppresses A59C and A59G almost completely, A59U is not suppressed at all by restoring U2/U6 helix Ib, U4/U6 stem I, or both (Fig. 3c, ref. 19). Remarkably, a mutation at the predicted base-triple partner, U6-A53C, suppressed not only the sick phenotype of A59C and the lethality of A59G but also the lethality of A59U - all without U2/U6 helix Ib repair (Fig. 3c, Supplementary Fig. 1c). This marks the first observed suppression of A59U (Supplementary Note 4). While A53C suppressed all three alleles at position A59, suppression was position-specific (Fig. 3d, Supplementary Fig. 1d). Additionally, only the A53C allele suppressed all three A59 point mutations (Fig. 3c), suggesting a specific mechanism of suppression (see below). Together, these data, especially the positional specificity, provide evidence for a base-triple interaction between A53 of the ACAGAGA sequence and A59 of the AGC triad.
If the suppression of AGC triad mutations is direct, via formation of compensatory base-triple interactions, as the allele- and position-specificity of our data strongly implies, then suppressor combinations should be capable of forming similar base-triple interactions. Consequently, we assessed the structural similarity between potential suppressor base triples and wild-type base triples, modeled based on the group II structure7,11. Where appropriate, to guide this comparison we utilized published matrices enumerating the many possible base-base interactions, observed crystallographically or predicted by modelling29. Note that two of the three group II intron triples are unusual11 and the third triple, while resembling analogous triples, is not strictly isosteric with any of these interactions29. Two base-base interactions are generally considered similar if the configurations conserve the distance between their linkages to the sugar-phosphate backbone. We focused specifically on the C1′-C1′ distance between the AGC triad residues and their predicted base-triple partners (Figs. 2c, 3e,f) because this distance likely influences catalytic metal positioning by the AGC triad4.
For each of the predicted base triples we discovered a suppressor base triple that could form interactions that resembled the configuration of the wild-type base triple, predicted from the group II structure (Figs. 2c, 3e-f, Supplementary Note 5). For example, with suppression of the U6-C61G U2-G21C double mutant by U6-U80C (Fig. 2a), O6 of C61G could interact with N4 of U80C to form a similar base triple (Fig. 2c). Similarly, for the base triple involving G52 and G60, the C1′-C1′ distance for our proposed suppressor base-triple is remarkably similar to that of the predicted wild-type base triple (within 0.4 Å, Supplementary Table 2). For the final base triple, we can in some cases model a plausible suppressor base-triple (e.g. Fig. 3f), but the capacity of the A53C mutation to suppress generally suggests that it might do so by forming a base-neutral interaction with the phosphate backbone of A59, just as the equivalent residue of the group II intron interacts with the backbone11 (Fig. 1c, Supplementary Notes 5, 6). This analysis suggests that the base-triple suppressor combinations likely act directly by restoring a configuration of the bases similar to that observed in the wild-type context.
Overall, our analysis is consistent with a mechanism of suppression in which the novel suppressor combinations permit growth by forming base-triple interactions structurally similar to those predicted for the wild-type context. Further, as was observed previously for other triple helix interactions27, we observed that a mutation that suppressed disruption of one base-triple exacerbated disruption of another base-triple (in 75% of the tests, Supplementary Table 1) - a result that is consistent with triplex formation, because these tests would disrupt two, rather than one, of the three triples. Thus, while our data are not sufficient to define base-triple interactions at atomic resolution, which is currently beyond state-of-the-art, our data establish compelling evidence that the residual function of the AGC triad, not accounted for by pairing with U2, U4, or U6, is to form a triplex interaction, in the context of helix Ib, with the bulge of the U6 ISL and the ACAGAGA region of U6, which binds the 5′ splice site.
Physical evidence for the U6 triplex
As a result of triple helix formation in domain V, two of the base-triple partners (C377 and G288) stack through their base rings (Fig. 4a). To investigate whether the corresponding positions in U6 (U80 and G52) form a similar stacking interaction, we designed a crosslinking assay. We reconstituted U6-depleted extract with a synthetic U6 (U6-4SU80) containing 4-thio-uridine at U80 and a single radioactive label at G52 (Fig. 4a; Supplementary Fig. 2a). If these two bases stack, UV irradiation should induce formation of a covalent linkage between U80 and G52 (ref. 30), with the site-specific radiolabel facilitating identification of such a linkage.
Figure 4. The U6 triplex forms in vitro.

a, Stacking interaction between C377 and G288, as observed in the group II intron crystal structure (PDB 4FAQ, ref. 8); left, side view; right, top view; equivalent spliceosome residues are indicated in parentheses. The positions of the 4-thio-uridine (4SU) and of the radioactive label (32p) are indicated in red. b, Denaturing PAGE analysis of U6-4SU80 recovered from in vitro splicing reactions after UV irradiation. Where indicated, unlabeled ACT1 pre-mRNA was present in the reactions. The U6 crosslinks (X1, X2, and X3) are indicated to the right. The efficiency of X1 formation is quantified below the gel; error bars represent s.d. from three technical replicates. c, Predicted products for P1 nuclease and NaOH digestion; note that P1 nuclease is able to cleave 5′ of UV-crosslinked RNA residues46. d, Denaturing PAGE analysis of RNA products following P1 nuclease digestion of un-crosslinked U6 or X1 excised from a gel like that shown in b. A 5′-32pGpG dinucleotide was also digested with P1 as a size marker. Curvy blue arrow signifies a crosslink. For full gels see Supplementary Fig. 8a,b.
We detected three major crosslinks that were dependent on both UV irradiation and 4-thio-uridine (Fig. 4b, Supplementary Fig. 2b), including an extract-independent crosslink (X2) and the previously reported U4/U6 crosslink involving U80 (X3, Fig. 4b; ref. 31; see Supplementary Fig. 2c-e for characterization of X2 and X3). Most importantly, we observed extract-dependent and pre-mRNA stimulated formation of a crosslink migrating closely to uncrosslinked U6 (X1, Fig. 4b, Supplementary Fig. 2c). To determine if this reflected intramolecular crosslinking, we performed P1 nuclease digestion, which degrades RNA to single nucleotides with 5′-monophosphates (ref. 32, Fig. 4c). Whereas uncrosslinked U6 digested to radiolabeled mononucleotide, reflecting formation of 5′-pG (Fig. 4c,d), X1 digested to a species migrating slower than the mononucleotide and close to a dinucleotide standard (Fig. 4d), providing evidence that G52 crosslinked to U80. In contrast, when we performed RNA hydrolysis, which yields single nucleotides with cyclic 3′-monophosphates (Fig. 4c), both uncrosslinked U6 and X1 yielded radiolabeled mononucleotide, reflecting formation of Ap-3′ (Supplementary Note 7, Supplementary Fig. 2f,g), thereby ruling out that U80 crosslinked to A51. We conclude that X1 results from crosslinking between G52 and U80. Given an analogous crosslink between equivalent positions in the group II intron, which stack in the domain V triplex13, and the chemical mechanism of 4-thio-uridine crosslinking (Supplementary Note 8), the simplest explanation for our observed crosslink is that U80 and G52 stack in the context of a triplex structure, thus providing physical evidence for a group II intron-like U6 triplex in the spliceosome.
The NTC promotes U6 triplex formation
The crosslinking of U80 to G52 allowed us to investigate when the U6 triplex forms during the splicing cycle. To do so we induced crosslinking of 4SU80-U6 spliceosomes after stalling at defined stages through the use of dominant negative mutations of DEAH-box ATPases known to promote conformational rearrangements during in vitro splicing1. Unexpectedly, we detected X1 at very high levels in Bact spliceosomes, stalled by a dominant-negative Prp2p mutant (rPrp2p-K252A) at the final ATP-dependent step in spliceosome activation (Fig. 5a, Supplementary Fig. 3), providing evidence that the U6 triplex forms before the final stage in catalytic activation. Strikingly, immunoprecipitation via Prp19p revealed that more than 50% of U6 in Bact spliceosomes formed the X1 crosslink in extract (Fig. 5a). Such high crosslinking efficiency requires stacking of the residues involved33-35 and suggests that the U6 triplex is a defining feature of spliceosomes poised for final activation.
Figure 5. The NTC promotes formation of the U6 triplex.
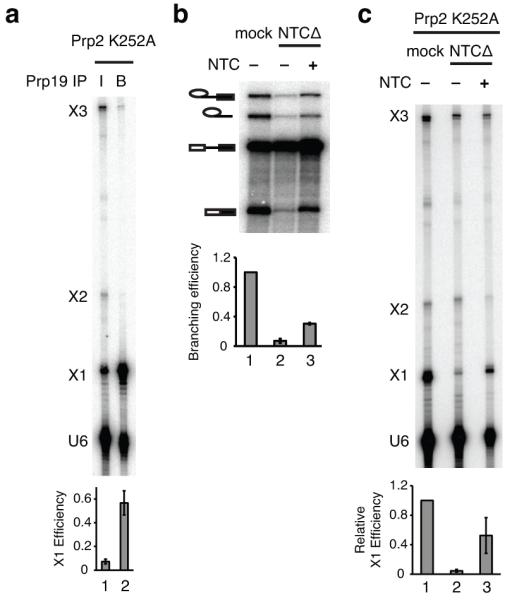
a, Denaturing PAGE analysis of radiolabeled U6-4SU80 extracted from a splicing reaction following UV irradiation and immunoprecipitation via Prp19p. 5% of the input (I) and all of the beads (B) were analyzed. b, Denaturing PAGE analysis of radiolabeled ACT1 pre-mRNA following in vitro splicing in extracts depleted of the NTC (NTCΔ) with or without addition of purified yeast NTC (ref. 36). c, Denaturing PAGE analysis of U6-4SU80 recovered from in vitro splicing reactions after UV irradiation. Splicing of unlabeled ACT1 pre-mRNA was performed in extracts depleted of the NTC (NTCΔ) and reconstituted with radiolabeled U6-4SU80. In panels a and c, rPrp2p-K252A was added to stall spliceosomes immediately after NTC binding and before Prp2p-dependent activation. In panels a and c, the efficiency of X1 formation is quantified below the gel; in panel c the X1 efficiency was normalized to the mock-depleted reactions for individual experiments. In panel b the splicing efficiency, normalized to the mock-depleted reaction, is quantified below the gel. Error bars represent s.d. from three technical replicates. For full gels see Supplementary Fig. 8c,d,e.
Given triplex formation in Bact spliceosomes, we asked whether triplex formation required the Prp19p-associated complex (NTC), which is necessary for stabilization of Bact spliceosomes after release of U4 snRNA (ref. 36). Depletion of the NTC from splicing extracts resulted in a strong reduction in X1 and purified NTC rescued the crosslink (Fig. 5b-c), defining a role for the NTC in stabilizing the U6 base-triple interactions (see also Supplementary Fig. 4 and Supplementary Note 9). Further, given the involvement of the ACAGAGA sequence in the U6 triplex, our data indicate that the 5′ splice site docks into the catalytic core at the stage of NTC binding (see Discussion).
The U6 triplex is present at branching and exon ligation
Biochemical data and crystal structures of the group II intron imply a role for the domain V triplex in both catalytic steps7,8,13,20. Further, we have shown in the spliceosome that catalytic metal ligands that would be organized by the triplex function in both catalytic steps4. Consequently, we tested whether the U6 triplex is present during each catalytic step.
To test for the triplex at the branching stage, we stalled spliceosomes just after branching by addition of a dominant negative Prp16p mutant that remains bound to the spliceosome (rPrp16p-K379A) and isolated from glycerol gradients the resulting spliceosome complexes, which had catalyzed branching but had not undergone the Prp16p-dependent rearrangement (denoted B*(prp16)) (Fig. 6a,b). UV irradiation of these spliceosomes induced high levels of X1 (Fig. 6c, lane 1, top), suggesting the presence of the triplex at the branching stage. To test this interpretation further, we utilized Prp16p as a specific immuno-affinity handle for X1 in genuine B*(prp16) spliceosomes, since Prp16p binds stably to this complex in a Prp2p-dependent manner just before branching37 and dissociates just after branching38. X1 was efficiently immunoprecipitated from the B*(prp16) peak (Fig. 6c, lane 4; see also Supplementary Fig. 5). By contrast, when we stalled spliceosomes before branching at the Prp2p stage, X1 did not efficiently immunoprecipitate from the Bact peak, indicating that the immunoprecipitation of X1 via Prp16p was specific for the B*(prp16) complex (Fig. 6c, lane 6, Supplementary Note 10).
Figure 6. The U6 triplex is present at the branching and exon ligation stages of splicing.

a, General splicing pathway highlighting the complexes used to probe for the triplex at the catalytic stages. Individual snRNPs are shown as ovals. The relevant complexes are labeled; the dominant negative ATPase mutant that was added to induce accumulation of specific complexes is indicated in parentheses. Note that here we designate as B*(prp16) those spliceosomes that have catalyzed branching but have not undergone the Prp16p-dependent rearrangement. b, Glycerol gradient sedimentation profile of the complexes used for crosslinking. Spliceosomes containing U6-4SU80 were assembled on Cy3-labeled UBC4 pre-mRNA and then sedimented. For each gradient fraction, the Cy3-labeled pre-mRNA, lariat intermediate, or excised intron, corresponding to Bact, B*(prp16), or P complex, respectively, is quantified as a fraction of the total Cy3 signal. (c,d), Denaturing PAGE analysis of radiolabeled U6-4SU80 recovered after complex isolation, UV irradiation, and immunoprecipitation via Prp16p (c) or Prp22p (d). The indicated spliceosomal complexes were UV-irradiated and immunoprecipitated after isolation from peak glycerol gradient fractions, as illustrated in b. Representative gels are shown for the glycerol gradient fractionated spliceosomes (GG spliceosome) before (input) and after (αPrp16 or αPrp22) immunoprecipitation. The lower panels in c and d show the UBC4 pre-mRNA substrate present in the fractions used for immunoprecipitation, detected by Cy3. The X1 immunoprecipitation efficiency (relative to X1 present in the input) is quantified in each panel. Error bars represent s.d. from three technical replicates. For full gels see Supplementary Fig. 8f,g,h.
To test for the triplex at the exon ligation stage, we stalled spliceosomes just after exon ligation by addition of rPrp22p-K512A and isolated the resulting P complexes from glycerol gradients (P peak; Fig. 6a,b). UV irradiation of these complexes induced the X1 crosslink (Fig. 6d, lane 1, top), suggesting the presence of the triplex at the exon ligation stage. To test this implication further, we used Prp22p as a specific immuno-affinity handle for P complex spliceosomes, because Prp22p only binds stably to this complex, in a manner dependent on Prp16p, Slu7p and Prp18p just before exon ligation37,39, and dissociates just after exon ligation40. When spliceosomes were stalled after exon ligation, we observed significant immunoprecipitation of X1 from fractions of the P peak, (Fig. 6d, lane 3). By contrast, when spliceosomes were stalled just after branching, X1 immunoprecipitated 3-fold less efficiently from equivalent fractions (Fig. 6d, lanes 3,4), even though the accumulating B*(prp16) complexes and associated X1 crosslink migrated in the same fractions (Fig. 6b); thus, the immunoprecipitation of X1 via Prp22p was specific for the P complex. We conclude that U80 and G52 also stack at the exon ligation stage. These results indicate that the U6 catalytic triplex is present during both catalytic steps of splicing and provides a structural framework for the organization of the U6 ligands for the catalytic metals that function during both reactions4.
The U6 triplex promotes catalytic metal binding
The U6 triplex provides a structural mechanism for positioning the two catalytic metals4 in a proper configuration for phosphoryl transfer (Fig. 1). To determine whether the U6 triplex promotes catalytic metal binding, we tested whether mutations predicted to disrupt the triplex impair catalytic metal binding. The mutations U6-G52A and U6-A53U confer lethality or a growth defect in vivo (Fig. 3) and have exon ligation defects in vitro (Supplementary Fig. 6, Supplementary Note 11), which we infer result from disruption of their base-triple interactions with the AGC triad. We assayed the impact of these mutations on catalytic metal binding during branching using a pre-mRNA substrate that bears sulfur substitutions at both the leaving group and the non-bridging pro-RP oxygen at the 5′ splice site (3′S-PS(RP)), which disrupt interactions with both catalytic Mg2+ ions (Fig. 7a, ref. 4). In a background of Mg2+, these interactions can be rescued by binding of two thiophilic Cd2+ ions at high concentrations4. In a background of Mn2+, which is more thiophilic than Mg2+, Cd2+ can rescue at lower concentrations, because Mn2+ can populate one site (M2) and enable high-affinity binding of Cd2+ to the other site, M1 (Fig. 7e, ref. 4). Thus, at limiting Cd2+ concentrations (e.g. 10 μM), Mn2+ significantly potentiates the efficiency of branching for the 3′S-PS(RP) substrate (compared to Mg2+, Fig. 7b,c). Remarkably, in a background of Mn2+, we found that mutations in U6 predicted to destabilize the base-triple interactions (G52A and A53U) compromised Cd2+-dependent rescue of the 3′S-PS(RP) substrate (Fig. 7b,c). Indeed, compared to U6 wild-type, U6-G52A increased the apparent transition midpoint for Cd2+ rescue of the 3′S-PS(RP) substrate by more than 10-fold (Fig. 7d). These data indicate that destabilization of the U6-G52*U6-G60/U2-C22 base-triple disrupted metal binding at the catalytic core (Fig. 7f). Consistent with this interpretation, the strong G52A-induced defect, relative to wild type, in branching at low Cd2+ concentrations was almost completely suppressed at high Cd2+ concentrations (Fig. 7d). In contrast, U6-U80C, predicted to maintain the proper configuration of the U6 triplex11, did not compromise Cd2+ rescue of the 3′S-PS(RP) substrate at limiting Cd2+ (Fig. 7c) and did not affect the transition midpoint for rescue by Cd2+ (Fig. 7d). Together, these results establish that the triplex promotes binding of catalytic metals and define an essential role for the U6 triplex during catalysis of branching.
Figure 7. The U6 triplex promotes catalytic metal binding during branching.

a, A diagram of the 3′S-PS(RP) pre-mRNA. b, Denaturing PAGE analysis of splicing of the 3′S-PS(RP) pre-mRNA catalyzed by affinity-purified spliceosomes in the presence of the indicated combinations of metals; WT, wild type. In b, the band marked * results from exonucleolytic degradation that stops at the sulfur. c, Quantification of Mn2+ potentiation of Cd2+-dependent rescue of branching of the 3′S-PS(RP) pre-mRNA by spliceosomes containing the indicated U6 variants. Mn2+ potentiation is quantitated relative to Mg2+. Error bars represent ranges of two technical replicates. d, Titration curves showing efficiency of Cd2+-mediated rescue of branching of the 3′S-PS(RP) pre-mRNA by affinity-purified spliceosomes reconstituted with the indicated U6 variants. Hill fits to the data, assuming one rescuing metal, are shown (solid lines). The apparent transition midpoints are indicated below the label of each U6 variant. Error bars represent s.d. of three independent replicates. (e, f) Diagrams showing predicted catalytic metal binding by spliceosomes containing U6 WT (e) or U6 G52A (f). Relevant U6 ligands and the nucleophile are colored red. Metals are colored magenta (Cd2+) and blue (Mn2+), and their interactions with specific U6 ligands are depicted as dashed lines, with differential shading intensity illustrating differences in the expected strength of interaction with oxygen vs. sulfur, as inferred from studies with model compounds47. Shading of metals bound at M1 and M2 is further adjusted to reflect experimental observations.
The U6 triplex promotes exon ligation
Because our crosslinking experiments implied that the triplex is present during exon ligation (Fig. 6d), we tested whether exon ligation required the triplex. Of the bases involved in the triplex, only mutations at A59 show a specific defect in exon ligation18. Our in vivo results suggest that A59 forms a base-triple interaction with A53, similar to that observed for the equivalent positions in the group II intron (Fig. 3c, Fig. 8a). Thus, we asked whether the exon ligation defects conferred by U6-A59 mutations could be suppressed by mutations at U6-A53. Strikingly, the A53C mutation strongly improved exon ligation efficiency for both A59U and A59G (Fig. 8b,c). Suppression was allele-specific because A59G was enhanced rather than suppressed by other A53 mutations and A59U was only mildly suppressed by other A53 mutations. Importantly, the observed suppression was also position-specific, as neither A59G nor A59U were substantially suppressed by a mutation at the neighboring G52 (Fig. 8b,c), which nevertheless suppressed the exon ligation defect conferred by a mutation at its base-triple partner G60 (Supplementary Fig. 7a,b). A53C also suppressed the exon ligation defect of compromised pre-mRNA reporters both in vitro and in vivo (Supplementary Fig. 7d,e). Overall the allele- and position-specific suppression of exon ligation defects we observed in vitro explicitly paralleled the suppression we observed in vivo (Fig. 3a,c). As in vivo, we infer that the in vitro suppression reflects the formation of a group II-like base-triple that includes an interaction between A53C and the backbone of A59 (Fig. 8a). Together, these results indicate that the U6 triplex not only forms at the exon ligation stage (Fig. 6d) but also functions at this catalytic stage both in vivo and in vitro.
Figure 8. The U6 triplex promotes exon ligation in vitro.

a, Configuration of the C288*C358-G385 base triple in the group II intron (PDB 4FAQ, ref. 8), equivalent to the spliceosomal U6-A53*U6-A59/U2-U23 base triple; left, side view; right, top view. b, Denaturing PAGE analysis of splicing of ACT1 pre-mRNA in extracts reconstituted with the indicated U6 variants. No dep., no depletion; no rec., no reconstitution. Upper case indicates wild-type allele. c, Quantification of exon ligation for the indicated U6 variants, normalized to wild-type U6; exon ligation was calculated as mRNA/lariat intermediate (ref. 19). Error bars represent s.d. of two independent experiments and two technical replicates for each experiment. The efficiency of branching was within 15% of wild type for all U6 variants (quantification not shown). For full gel see Supplementary Fig. 8i.
DISCUSSION
Here we provide compelling evidence for the formation of a catalytic triplex in the spliceosome, analogous to the metal binding catalytic triplex of group II introns (Fig. 1). Specifically, despite the dynamic nature of the AGC triad15, the role for the triad in catalysis4, and the unusual and discontinuous nature of the group II intron triplex7,8, U6 mutations within the ACAGAGA and ISL bulged elements suppressed growth defects of AGC triad mutants in the context of U2/U6 helix Ib in an allele- and position-specific manner at all three triples, providing independent and corroborative evidence for tertiary interactions between these highly conserved U6 residues (Figs. 2,3). By site-directed crosslinking we revealed physical evidence for the U6 triplex (Fig. 4) and showed that, strikingly, the triplex forms upon NTC binding (Fig. 5), before the last ATP-dependent activation step, suggesting that the catalytic core is formed already at the pre-catalytic stage. Finally, we established that the catalytic triplex is present at both catalytic stages of splicing (Fig. 6), promotes both branching and exon ligation, and plays an essential role in positioning catalytic metals (Figs. 7,8). These data establish clear evidence that a catalytic triplex forms at the heart of the active spliceosome. The triple helix provides a mechanism by which the spliceosome structures U6 snRNA in a conformation competent to bind the two divalent metals that catalyze splicing4. Furthermore, the triplex rationalizes how the spliceosome recruits the pre-mRNA reactive phosphates to this metal binding core for catalysis (see below). Finally, our results extend the deep mechanistic parallels between the spliceosome and group II introns and further support the likelihood of a common evolutionary precursor5.
The AGC triad was identified early on as a strictly conserved element highly sensitive to mutation15 and was later shown to base pair with U4 (ref. 25) in assembling spliceosomes and with U2 in activated spliceosomes15,17,19. With this work we show that in activated spliceosomes the triad not only interacts with U2 but also with distal regions in U6 to yield base-triples (Figs. 2, 3, 4). Indeed, the triplex, with the AGC triad at the backbone, rationalizes the asymmetric phenotypes in U2/U6 helix Ib - the strong phenotypes in U6 and the weak phenotypes in U2 (refs. 15,19), an asymmetry that is mirrored in the U6-U6 triple interactions.
Because the two metal ions required for splicing catalysis are bound by the AGC triad and the bulge of the U6 ISL (ref. 4), the catalytic triplex provides a mechanism to juxtapose these two metal ion-binding sites in U6 for two metal-ion catalysis. Indeed, our results indicate that the integrity of the U6 triplex is essential for proper metal positioning by the catalytic core, as observed during branching (Fig. 7).
The catalytic triplex also provides a mechanism for recruiting the 5′ splice site to the metal binding core. The 5′ splice site is defined through base-pairing interactions with the 5′ end of the ACAGAGA sequence of U6 (refs. 21,22). Because the 3′ end of the ACAGAGA sequence participates directly in the triplex (Fig. 1a), the triplex likely recruits the U6/5′ splice site interaction to the catalytic core for branching. The timing of triplex formation supports this hypothesis. By site-directed crosslinking we found that the triplex forms before the final catalytic activation step of the spliceosome, mediated by Prp2p; the triplex forms after the release of U4 snRNA from U6 and upon binding of the NTC and associated factors to the spliceosome (Fig. 5). Because the NTC consolidates interactions between the exonic region of the 5′ splice site and U5 and promotes correct pairing between the intronic region of the 5′ splice site and the ACAGAGA sequence of U6 (ref. 41), our results argue for coupling between binding of the 5′ splice site and formation of a catalytic configuration of the RNA core. Similarly, during the exon ligation stage the integrity of the triplex may be coupled to recruitment of the 3′ splice site (Supplementary Note 12). Such coupling highlights the dependence of the catalytic core on substrate binding and may allow sampling and proofreading of the substrate through inspection of the catalytic core at subsequent steps in the splicing pathway. Indeed, downstream of NTC binding, both Prp2p and Prp16p have been implicated in proofreading through, in part, destabilization of a weakly-formed catalytic core17,42,43.
Before branching, Prp2p has been implicated in destabilizing the catalytic core, including the triplex, to promote on-pathway rearrangements43. Our in vitro analysis indicates that the U6 triplex must restabilize to promote branching (Fig. 6c, Fig. 8, Supplementary Fig. 7). Cwc2p stabilizes the triplex before Prp2p activity (Supplementary Fig. 4), and following Prp2p action, when Cwc2p becomes essential for branching, Cwc2p interacts with the U6 ISL (ref. 2). Thus, Cwc2p may act to re-stabilize the catalytic core for branching catalysis.
Following branching, Prp16p has been implicated in destabilizing the catalytic core17, likely to accommodate substrate repositioning - the replacement of the branchsite with the 3′ splice site at the catalytic metal binding site4. Consistent with a role in destabilizing the helix Ib component of the catalytic core17, we found that mutations predicted to destabilize the U6 triplex (U6-A53C and U6-A53G) suppressed a hypomorphic allele of prp16 (Supplementary Fig. 7c). These data imply that Prp16p destabilizes the U6 triplex after the spliceosome has catalyzed branching. Interestingly, the transition from branching to exon ligation requires disruption of the U6/5′ splice site pairing44. Thus, our data also suggest that disruption of the U6/5′ splice site pairing after branching is coupled to destabilization of the U6 triplex.
As we have found for other elements of the catalytic core17,45, we found evidence that the triplex is only transiently destabilized during the catalytic stage; the triplex reforms at the exon ligation stage (Fig. 6d, Supplementary Fig. 7) and is necessary for efficient exon ligation (Fig. 8). Interestingly, while the catalytic triplex of the group II intron similarly appears to function in both catalytic conformations, the group II triplex has also been suggested to disrupt transiently between the two steps of splicing, to allow substrate repositioning8.
Overall our results indicate that the spliceosomal catalytic core shares remarkable structural and functional similarity, if not also evolutionary origins5, with the catalytic core of group II self-splicing introns. Thus, these data underscore evidence that splicing catalysis is performed by an RNA-based catalytic core4 stabilized by a complex protein scaffold2,3.
ONLINE METHODS
Yeast strains and plasmids
To test combinations of alleles in U2 and U6, we used strain yHM118, in which the endogenous U2 and U6 genes were deleted and viability rescued with the URA3 marked plasmid pU2U6U (ref. 45). To test, in addition, mutant versions of U4, we used strain yJPS628, containing pU2U6U4 (ref. 19). The HIS3 marked plasmid pJPS216 encodes U2 (ref. 48); the TRP1 marked plasmid pSX6 encodes U6 (ref. 15); and the ADE2 marked plasmid pJPS464 endcodes U4 (ref. 19). Mutated U2, U6, and U4 plasmids, used for in vivo assays, were described previously19 or generated for this study by QuikChange mutagenesis (Stratagene) and verified by sequencing. For in vitro transcription, mutant U6 templates were generated by QuikChange mutagenesis (Stratagene) from pJPS488, which encodes wild-type U6 downstream of a T7 promoter49.
In vitro splicing was performed using extracts prepared from the following strains: yJPS1405 (PRP19 with a C-terminal biotin tag, ref. 4); yJPS1489 (CWC2-TAP, Open Biosystems, ref. 50); yJPS1492 (CWC25-HA, same as ySCC25, ref. 51); yJPS1510 (PRP19P-HA, same as ySCC1, ref. 36).
Plasmid shuffle growth assays
Variants of pJPS216 and pSX6 were co-transformed into yJPS328 and selected on plates lacking histidine and tryptophan. To test for interactions with U4, variants of pJPS216, pSX6, and pJPS464 were co-transformed and selected on plates lacking histidine, tryptophan, and adenine. Duplicate colonies were grown in liquid media lacking the appropriate nutrients to an OD600 of approximately 0.8, and then spotted onto media containing 5-fluroorotic acid (5-FOA) to counter select for the wild-type plasmid52. Cells were grown at 30 °C for 3-10 days for phenotypic analysis.
To test for a genetic interaction between PRP16 and the U6 triplex, variants of pSX6 and wild-type pJPS216 were co-transformed into yHM187 (prp16-302; ref. 45) or yHM118 (PRP16) and selected on plates lacking histidine and tryptophan. Duplicate colonies were grown in liquid media lacking histidine and tryptophan to an OD600 of approximately 0.4, and then spotted onto media containing 5-fluroorotic acid (5-FOA) to counter select for the wild-type plasmid52. Cells were grown at 20 °C and photographed after 6 days.
Note that the growth spots presented reflect two related growth parameters. First, the spots reflect colony size and consequently colony growth. Second, the spots reflect the number of colonies growing in the spot. Before spotting equal numbers of cells onto 5-FOA to counter-select against cells that have retained the wild-type, URA3-marked plasmid, cultures were grown selecting for the mutated plasmids but not the wild-type plasmid. Phenotypically wild-type mutants consequently passively lost the wild-type plasmid (~1-5% of cells/cell division) and consequently yielded a large fraction of cells that were viable on 5-FOA. By contrast, phenotypically sick or lethal mutants died in the culture, if they passively lost the wild-type plasmid, so a large fraction of cells retained the wild-type, URA3-marked plasmid and consequently died on 5-FOA, leading to spots with decreased colony density.
Copper growth assays
yJPS1035 (ref. 45) was first transformed with ACT1-CUP1 reporter variants on LEU2 marked plasmids (derived from pJPS1920, ref. 45) and then co-transformed with pJPS216 and pSX6 variants and selected on plates lacking histidine, tryptophan, and leucine. Cotransformants were streaked to media lacking leucine and containing 5-flouroorotic acid (5-FOA) (ref. 52) and grown for 3 days at 30 °C. Colonies were then purified on minimal media lacking leucine. Duplicate colonies were grown in liquid media lacking leucine to an OD600 of approximately 0.2 and spotted on media lacking leucine and containing various concentrations of copper sulfate. Cells were grown at 30 °C and photographed after 3 days.
Splicing extracts, U6 depletion and reconstitution, and in vitro splicing
Splicing extracts were prepared from yJPS1405, unless otherwise noted. Preparation of splicing extracts, in vitro splicing, U6 depletion and reconstitution, affinity-purification via Prp19p, and incubation of affinity-purified spliceosomes were performed essentially as described4. For crosslinking experiments, U6 was added back to a final concentration of 2-4 nM. In vitro splicing in extract was carried out for 25 minutes at 20 °C, unless otherwise noted. Where noted, 20% of the standard splicing extract volume was replaced with recombinant, dominant-negative Prp2p-K252A, Prp16p-K379A, or Prp22p-K512A in buffer D, at a final concentration of approximately 40-80 ng/μL.
Radiolabeled ACT1 pre-mRNA was synthesized by in vitro transcription. Fluorescently labeled UBC4 pre-mRNA was synthesized by splint-mediated ligation, essentially as described53. Radiolabeled 3′S-PS(RP) and 3′O-PO pre-mRNAs were synthesized as described4.
In Figs. 7b-c, spliceosomes from extracts reconstituted with the indicated U6 variants were assembled on the 3′S-PS(RP) substrate, affinity-purified via Prp19p, and incubated in the absence of ATP in buffer PK (3% PEG, 60 mM KPO4, pH 7.0) with 1 mM MgCl2 or 1 mM MnCl2 in the presence of various amounts of CdCl2. In Fig. 7d, spliceosomes from extracts reconstituted with the indicated U6 variants were assembled on the 3′S-PS(RP) substrate, affinity-purified via Prp19p, and incubated in the absence of ATP in buffer PK (pH 7.0) with 1 mM MnCl2 in the presence of various amounts of CdCl2.
Preparation of recombinant proteins
Recombinant Prp16p, Prp22p, and Prp2p were expressed in E. coli Rosetta2 DE3pLysS cells (Novagen), essentially as described54. Recombinant Prp2p was expressed in the presence of 2% ethanol at 17 °C for 17 hours. All proteins were purified by Ni2+-NTA affinity chromatography, followed by glycerol gradient, and dialyzed against buffer D before use.
Synthesis of U6 snRNA for in vitro reconstitution
Synthetic U6, with modifications for crosslinking, was constructed by splint-mediated ligation using RNA oligonucleotides corresponding to the following residues: U6 1-51, U6 52-79, U6 80-112. Oligonucleotides U6 52-79 and U6 80-112 (containing U80 or 4SU80) were purchased from Dharmacon, deprotected, and used as supplied. U6 1-51 was generated from full length, in vitro transcribed U6 by DNAzyme-mediated cleavage after A51 using U6 A51 DNAzyme (5′-CTGATCATTCCGAGCCGGACGACTGTATTGT-3′, a modified 8-17 DNAzyme, ref. 55). For a typical cleavage reaction, 2-4 nmoles of gel-purified, full length, transcribed U6 was mixed with a 1.2-fold molar excess of U6 A51-Dzyme (IDT) in cleavage buffer (100 mM Tris-Cl, pH 7.4; 50 mM NaCl) and then annealed by heating to 90 °C followed by cooling to room temperature for 25 minutes. Following hybridization, cleavage was performed overnight at 37 °C in cleavage buffer supplemented with 10 mM MgCl2. U6 1-51 product was gel purified and the 3′-cyclic phosphate resulting from DNAzyme cleavage was removed by incubation with polynucleotide kinase (NEB) at 37 °C for 2-4 hrs. The U6 52-79 oligonucleotide was 5′ phosphorylated with γ-32P-ATP (Perkin Elmer, 6000 Ci/mmol) prior to ligation. Ligation of the three oligonucleotides and purification of ligated U6 was performed by splint-mediated ligation, as described4.
Wild-type and mutant U6 used for the in vitro splicing experiments were synthesized by in vitro transcription according to standard procedures using templates derived from linearization of pJPS488 with DraI, as described49.
UV crosslinking
For crosslinking, splicing reactions (20-40 μL) were transferred to a round-bottom 96-well plate (Fisher Scientific) on ice and irradiated using a UMP UV lamp (FBUVLS-80, Fisher Scientific) set at 365nm and placed at approximately 4 cm from the bottom of the well so that light would shine perpendicularly on the plane of the sample. Irradiation was performed under an aluminum foil tent at 4 °C for 90 min. Following crosslinking, RNA was analyzed by 8% denaturing PAGE.
Analytical digestions
For nuclease P1 digestion, gel purified X1 RNA (~3,000-10,000 cpm) was incubated with nuclease P1 (1U/μL; US Biological) in a 4 μL reaction at 37 °C for 18-20 hrs. For alkaline hydrolysis, gel purified RNA (~3,000-10,000 cpm) was incubated with 20 mM NaOH at 90 °C for 3 hrs. Following digestions, RNA was analyzed by 20% denaturing PAGE. For RNaseH digestion of X3, gel purified X3 RNA (~1,000 cpm) was hybridized to 10 pmoles of a DNA oligo complementary to U2 snRNA (nucleotides 35-62) or U4 snRNA (nucleotides 81-100) in buffer TEN (10 mM Tris, pH 7.4; 1 mM EDTA; 100 mM NaCl) in a 3 μL reaction volume on a thermal cycler. Cleavage was then performed by the addition of 1U RNase H (Fermentas) and incubation at 37 °C for 1 hour.
NTC Depletion and complementation
Splicing extracts were prepared from a strain containing Prp19p-HA (yJPS1510). For depletion, 200 μL of freshly prepared extract was incubated at 4 °C for 2 hours with 150 μL of protein A-sepharose slurry conjugated to anti-HA antibodies (12CA5, 2 mg/mL of bead slurry, ref. 56). After centrifugation at 800 × g for 4 minutes, the supernatant was used as NTC-depleted extract.
For complementation, the NTC complex was isolated from yeast splicing extracts prepared from yJPS1510, as follows. The splicing extract was precipitated with 40% ammonium sulfate to obtain the 40P fraction, as described57. The 40P fraction (280 mg) was resuspended in 1 mL of buffer D supplemented with 60 mM potassium phosphate, pH 7.0 (buffer DK) and was incubated at 4 °C for 2 hours with 200 μL slurry of protein A-sepharose conjugated to anti-HA antibodies (12CA5, 2 mg/mL of bead slurry; crosslinked with DMP (Sigma; ref. 58). Beads were then washed 3 times at 4 °C with 6 mL of buffer DK supplemented with 0.01% NP-40 substitute (Fluka), brought to room temperature and washed with an additional 6 mL of the same buffer. The bound NTC was eluted with 300 μL of 0.6 mM HA peptide at room temperature with rotation for 30 mins. The eluate was concentrated 10-fold using a Vivaspin-500 column (Sartorius), flash frozen, and stored at -80 °C. Typically 1-2 μL of the concentrated NTC were used for complementation of a 10 μL splicing reaction.
Preparation of Bact, B*(prp16), and P complexes
To isolate Bact or B*(prp16) complex spliceosomes, splicing reactions (80 μL) were performed using UBC4 pre-mRNA, labeled fluorescently with Cy3 at the seventh residue of the 3′ exon53, and extracts from yJPS1492. The splicing reactions were supplemented with rPrp2p-K252A, to accumulate Bact, or rPrp16p-K379A, to accumulate B*(prp16), and were loaded on 11 mL 15-40% glycerol gradients in buffer G50 (20 mM Hepes, pH 7.9; 50 mM KCl, 0.5 mM EDTA). The gradients were centrifuged at 234,325 × g in a Beckman SW 41 rotor for 13 hrs. Fractions were collected by hand from the top of the gradient; RNA was extracted from the fractions and analyzed by denaturing PAGE. The splicing species were visualized by detection of the Cy3 label using a Typhoon Trio phosphorimager (Amersham Biosciences). The lariat intermediate generally peaked in fractions 16-20, so fractions 16-18 were collected to enrich for the B*(prp16) complex; the pre-mRNA generally peaked in fractions 18-22, so fraction 18-20 were collected to enrich for the Bact complex.
To isolate P complex spliceosomes stalled after exon ligation, splicing reactions (80 μL) were performed with the fluorescently labeled UBC4 pre-mRNA and supplemented with rPrp22p-K512A with or without rPrp16p-K379A. Reactions were run on glyecerol gradients as above and peak fractions for the lariat intermediate or excised intron were used for immunoprecipitation of spliceosomes associated with rPrp22p. The excised intron migrated with the lariat intermediate and peaked in fractions 16-20; fractions 17 or 18 were used for immunoprecipitation experiments.
Affinity pull-downs
For affinity purification of spliceosomes associated with biotinylated Prp19p, splicing reactions were diluted 1-2 fold in buffer D and incubated for 1-3 hrs at 4 °C with 0.1-0.2 volumes of streptavidin-agarose slurry (Thermo Scientific) pre-washed twice with 25-50 volumes of IPP150 (10 mM Tris-HCl pH 8; 150 mM NaCl; 0.1% NP-40 substitute (Fluka)). Use of streptavidin-agarose for immuniprecipitation of biotinylated Prp19 was verified previously4. Following incubation, beads were washed at 4 °C twice with 50 volumes of IPP150.
For immunoprecipitation of Prp16p-associated spliceosomes, peak glycerol gradient fractions were diluted 1:1 in buffer DK150 (20 mM Hepes, pH 7.9; 150 mM KCl; 60 mM potassium phosphate, pH 7.0) and incubated for 1 hour at 4°C with 0.1-0.2 volumes of protein A-sepharose slurry conjugated to affinity-purified anti-Prp16p antibodies (4-6 mg/mL of bead slurry; gift from C. Guthrie, ref. 38). Following immunoprecipitation, beads were washed at 4 °C twice with 50 volumes of buffer DK150 supplemented with 0.01% NP-40 substitute.
For immunoprecipitation of Prp22p-associated spliceosomes, peak glycerol gradient fractions were diluted 1:1 in buffer DK150 and incubated for 1 hour at 4 °C with 0.1-0.2 volumes of protein A-sepharose slurry conjugated to affinity-purified anti-Prp22p antibodies (1.5 mg/mL of bead slurry; gift from B. Schwer, ref. 40). Following immunoprecipitation, beads were washed at 4 °C twice with 50 volumes of buffer NET-2 (50 mM Tris-HCl, pH 7.4; 150 mM NaCl; 0.05% NP-40 substitute).
Cwc2 depletion and complementation
Splicing extracts prepared from yJPS1489 (Cwc2-TAP) were depleted of Cwc2p essentially as described2.
For expression of rCwc2p, the CWC2 sequence was amplified from a plasmid containing the CWC2 ORF (bJPS2509, Open Biosystems) and cloned into pET15b (bJPS581) using the NdeI and BamHI cloning sites to give bJPS2621. The cloned plasmid was verified by sequencing. Expression of 6His-rCwc2p was performed in E. coli Rosetta2 DE3pLysS cells. Cells were grown at 37°C to OD600 ~ 0.8 and expression was induced with 0.5 mM IPTG at 30 °C for 3 hours. Cells were lysed using a French press and 6His-rCwc2p was purified by Ni2+-NTA affinity chromatography, but binding and washing were performed by gravity flow. Following elution from the Ni2+-NTA resin, the protein was further purified by glycerol gradient centrifugation to more than 95% purity (as estimated by Coomassie Blue staining) and dialyzed against buffer D. For complementation of Cwc2p-depleted extracts, rCwc2p was added back at a final concentration of 2-4 μM.
Original images of gels and autoradiographs used in this study can be found in Supplementary Figure 8.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank S.-C. Cheng (Academia Sinica) for strains; C. Guthrie (UCSF) and B. Schwer (Weill Cornell Medical College) for antibodies; D. Semlow for recombinant proteins and Cy3-labeled UBC4 pre-mRNA; and members of the Staley and Piccirilli labs for critical discussions and comments on the manuscript. M.A.M. was supported by US National Institutes of Health training grant (T32 GM007183); this work was funded by a grant from the National Institutes of Health (R01GM088656) to J.P.S. and J.A.P.
Footnotes
Supplementary Information is available in the online version of the paper.
Author Contributions S.M.F., M.A.M., J.A.P. and J.P.S. designed the study; M.A.M. performed the in vivo genetics; S.M.F. performed the in vitro crosslinking and biochemistry experiments as well as experiments in Supplementary Fig. 7, which were initiated by M.A.M.; S.M.F. and J.P.S wrote the paper with input from M.A.M. and J.A.P.
REFERENCES
- 1.Wahl MC, Will CL, Lührmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 2.Rasche N, et al. Cwc2 and its human homologue RBM22 promote an active conformation of the spliceosome catalytic centre. EMBO J. 2012;31:1591–1604. doi: 10.1038/emboj.2011.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galej WP, Oubridge C, Newman AJ, Nagai K. Crystal structure of Prp8 reveals active site cavity of the spliceosome. Nature. 2013;493:638–643. doi: 10.1038/nature11843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fica SM, et al. RNA catalyses nuclear pre-mRNA splicing. Nature. 2013;503:229–234. doi: 10.1038/nature12734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cech TR. The generality of self-splicing RNA: relationship to nuclear mRNA splicing. Cell. 1986;44:207–210. doi: 10.1016/0092-8674(86)90751-8. [DOI] [PubMed] [Google Scholar]
- 6.Steitz TA, Steitz JA. A general two-metal-ion mechanism for catalytic RNA. Proc Natl Acad Sci USA. 1993;90:6498–6502. doi: 10.1073/pnas.90.14.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Toor N, Keating KS, Taylor SD, Pyle AM. Crystal structure of a self-spliced group II intron. Science. 2008;320:77–82. doi: 10.1126/science.1153803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marcia M, Pyle AM. Visualizing group II intron catalysis through the stages of splicing. Cell. 2012;151:497–507. doi: 10.1016/j.cell.2012.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sontheimer EJ, Gordon P, Piccirilli J. Metal ion catalysis during group II intron self-splicing: parallels with the spliceosome. Genes Dev. 1999;13:1729–1741. doi: 10.1101/gad.13.13.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gordon PM, Fong R, Piccirilli JA. A second divalent metal ion in the group II intron reaction center. Chem Biol. 2007;14:607–612. doi: 10.1016/j.chembiol.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 11.Keating KS, Toor N, Perlman PS, Pyle AM. A structural analysis of the group II intron active site and implications for the spliceosome. RNA. 2010;16:1–9. doi: 10.1261/rna.1791310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mikheeva S, Murray HL, Zhou H, Turczyk BM, Jarrell KA. Deletion of a conserved dinucleotide inhibits the second step of group II intron splicing. RNA. 2000;6:1509–1515. doi: 10.1017/s1355838200000972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Lencastre A, Pyle AM. Three essential and conserved regions of the group II intron are proximal to the 5′-splice site. RNA. 2008;14:11–24. doi: 10.1261/rna.774008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacquier A, Michel F. Base-pairing interactions involving the 5′ and 3′-terminal nucleotides of group II self-splicing introns. J Mol Biol. 1990;213:437–447. doi: 10.1016/S0022-2836(05)80206-2. [DOI] [PubMed] [Google Scholar]
- 15.Madhani HD, Guthrie C. A novel base-pairing interaction between U2 and U6 snRNAs suggests a mechanism for the catalytic activation of the spliceosome. Cell. 1992;71:803–817. doi: 10.1016/0092-8674(92)90556-r. [DOI] [PubMed] [Google Scholar]
- 16.Sun JS, Manley JL. A novel U2-U6 snRNA structure is necessary for mammalian mRNA splicing. Genes Dev. 1995;9:843–854. doi: 10.1101/gad.9.7.843. [DOI] [PubMed] [Google Scholar]
- 17.Mefford MA, Staley JP. Evidence that U2/U6 helix I promotes both catalytic steps of pre-mRNA splicing and rearranges in between these steps. RNA. 2009;15:1386–1397. doi: 10.1261/rna.1582609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fabrizio PP, Abelson JJ. Two domains of yeast U6 small nuclear RNA required for both steps of nuclear precursor messenger RNA splicing. Science. 1990;250:404–409. doi: 10.1126/science.2145630. [DOI] [PubMed] [Google Scholar]
- 19.Hilliker AK, Staley JP. Multiple functions for the invariant AGC triad of U6 snRNA. RNA. 2004;10:921–928. doi: 10.1261/rna.7310704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chanfreau G, Jacquier A. Catalytic site components common to both splicing steps of a group II intron. Science. 1994;266:1383–1387. doi: 10.1126/science.7973729. [DOI] [PubMed] [Google Scholar]
- 21.Lesser C, Guthrie C. Mutations in U6 snRNA that alter splice site specificity: implications for the active site. Science. 1993;262:1982–1988. doi: 10.1126/science.8266093. [DOI] [PubMed] [Google Scholar]
- 22.Kandels-Lewis S, Séraphin B. Role of U6 snRNA in 5′ splice site selection. Science. 1993;262:2035–2039. doi: 10.1126/science.8266100. [DOI] [PubMed] [Google Scholar]
- 23.Anokhina M, et al. RNA structure analysis of human spliceosomes reveals a compact 3D arrangement of snRNAs at the catalytic core. EMBO J. 2013;32:2804–2818. doi: 10.1038/emboj.2013.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sashital DG, Cornilescu G, McManus CJ, Brow DA, Butcher SE. U2-U6 RNA folding reveals a group II intron-like domain and a four-helix junction. Nat Struct Mol Biol. 2004;11:1237–1242. doi: 10.1038/nsmb863. [DOI] [PubMed] [Google Scholar]
- 25.Brow DA, Guthrie C. Spliceosomal RNA U6 is remarkably conserved from yeast to mammals. Nature. 1988;334:213–218. doi: 10.1038/334213a0. [DOI] [PubMed] [Google Scholar]
- 26.Qiao F, Cech TR. Triple-helix structure in telomerase RNA contributes to catalysis. Nat Struct Mol Biol. 2008;15:634–640. doi: 10.1038/nsmb.1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitton-Fry RM, DeGregorio SJ, Wang J, Steitz TA, Steitz JA. Poly(A) tail recognition by a viral RNA element through assembly of a triple helix. Science. 2010;330:1244–1247. doi: 10.1126/science.1195858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cash DD, et al. Pyrimidine motif triple helix in the Kluyveromyces lactis telomerase RNA pseudoknot is essential for function in vivo. Proc. Nat. Acad. Sci. USA. 2013;110:10970–10975. doi: 10.1073/pnas.1309590110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leontis NB, Stombaugh J, Westhof E. The non-Watson-Crick base pairs and their associated isostericity matrices. Nucleic Acids Res. 2002;30:3497–3531. doi: 10.1093/nar/gkf481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sontheimer EJ. Site-specific RNA crosslinking with 4-thiouridine. Mol Biol Rep. 1994;20:35–44. doi: 10.1007/BF00999853. [DOI] [PubMed] [Google Scholar]
- 31.Ryan DE, et al. New tertiary constraints between the RNA components of active yeast spliceosomes: a photo-crosslinking study. RNA. 2004;10:1251–1265. doi: 10.1261/rna.7060404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Volbeda A, Lahm A, Sakiyama F, Suck D. Crystal structure of Penicillium citrinum P1 nuclease at 2.8 A resolution. EMBO J. 1991;10:1607–1618. doi: 10.1002/j.1460-2075.1991.tb07683.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yaniv M, Favre A, Barrell BG. Structure of transfer RNA: evidence for interaction between two non-adjacent nucleotide residues in tRNAVal1 from Escherichia coli. Nature. 1969;223:1331–1333. doi: 10.1038/2231331a0. [DOI] [PubMed] [Google Scholar]
- 34.Grishaev A, Ying J, Canny MD, Pardi A, Bax A. Solution structure of tRNAVal from refinement of homology model against residual dipolar coupling and SAXS data. J. Biomol. NMR. 2008;42:99–109. doi: 10.1007/s10858-008-9267-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Favre A, Saintomé C, Fourrey JL, Clivio P, Laugâa P. Thionucleobases as intrinsic photoaffinity probes of nucleic acid structure and nucleic acid-protein interactions. J. Photochem. Photobiol. B, Biol. 1998;42:109–124. doi: 10.1016/s1011-1344(97)00116-4. [DOI] [PubMed] [Google Scholar]
- 36.Chan S-P, Kao D-I, Tsai W-Y, Cheng S-C. The Prp19p-associated complex in spliceosome activation. Science. 2003;302:279–282. doi: 10.1126/science.1086602. [DOI] [PubMed] [Google Scholar]
- 37.Ohrt T, et al. Molecular dissection of step 2 catalysis of yeast pre-mRNA splicing investigated in a purified system. RNA. 2013;19:902–915. doi: 10.1261/rna.039024.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schwer B, Guthrie C. PRP16 is an RNA-dependent ATPase that interacts transiently with the spliceosome. Nature. 1991;349:494–499. doi: 10.1038/349494a0. [DOI] [PubMed] [Google Scholar]
- 39.James S-A, Turner W, Schwer B. How Slu7 and Prp18 cooperate in the second step of yeast pre-mRNA splicing. RNA. 2002;8:1068–1077. doi: 10.1017/s1355838202022033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwer B, Gross CH. Prp22, a DExH-box RNA helicase, plays two distinct roles in yeast pre-mRNA splicing. EMBO J. 1998;17:2086–2094. doi: 10.1093/emboj/17.7.2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chan S-P, Cheng S-C. The Prp19-associated complex is required for specifying interactions of U5 and U6 with pre-mRNA during spliceosome activation. J Biol Chem. 2005;280:31190–31199. doi: 10.1074/jbc.M505060200. [DOI] [PubMed] [Google Scholar]
- 42.Semlow DR, Staley JP. Staying on message: ensuring fidelity in pre-mRNA splicing. Trends in Biochemical Sciences. 2012;37:263–273. doi: 10.1016/j.tibs.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wlodaver AM, Staley JP. The DExD/H-box ATPase Prp2p destabilizes and proofreads the catalytic RNA core of the spliceosome. RNA. 2014;20:1–13. doi: 10.1261/rna.042598.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Konarska MM, Vilardell J, Query CC. Repositioning of the reaction intermediate within the catalytic center of the spliceosome. Mol Cell. 2006;21:543–553. doi: 10.1016/j.molcel.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 45.Hilliker AK, Mefford MA, Staley JP. U2 toggles iteratively between the stem IIa and stem IIc conformations to promote pre-mRNA splicing. Genes Dev. 2007;21:821–834. doi: 10.1101/gad.1536107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Branch AD, Levine BJ, Polaskova JA. An RNA tertiary structure of the hepatitis delta agent contains UV-sensitive bases U-712 and U-865 and can form in a bimolecular complex. Nucleic Acids Res. 1995;23:491–499. doi: 10.1093/nar/23.3.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pecoraro VL, Hermes JD, Cleland WW. Stability constants of Mg2+ and Cd2+ complexes of adenine nucleotides and thionucleotides and rate constants for formation and dissociation of MgATP and MgADP. Biochemistry. 1984;23:5262–5271. doi: 10.1021/bi00317a026. [DOI] [PubMed] [Google Scholar]
- 48.Shuster EO, Guthrie C. Two conserved domains of yeast U2 snRNA are separated by 945 nonessential nucleotides. Cell. 1988;55:41–48. doi: 10.1016/0092-8674(88)90007-4. [DOI] [PubMed] [Google Scholar]
- 49.Fabrizio P, McPheeters DS, Abelson J. In vitro assembly of yeast U6 snRNP: a functional assay. Genes Dev. 1989;3:2137–2150. doi: 10.1101/gad.3.12b.2137. [DOI] [PubMed] [Google Scholar]
- 50.Ghaemmaghami S, et al. Global analysis of protein expression in yeast. Nat Cell Biol. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 51.Chiu Y-F, et al. Cwc25 is a novel splicing factor required after Prp2 and Yju2 to facilitate the first catalytic reaction. Mol Cell Biol. 2009;29:5671–5678. doi: 10.1128/MCB.00773-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sikorski RS, Boeke JD. In vitro mutagenesis and plasmid shuffling: from cloned gene to mutant yeast. Meth Enzymol. 1991;194:302–318. doi: 10.1016/0076-6879(91)94023-6. [DOI] [PubMed] [Google Scholar]
- 53.Abelson J, et al. Conformational dynamics of single pre-mRNA molecules during in vitro splicing. Nat Struct Mol Biol. 2010;17:504–512. doi: 10.1038/nsmb.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hotz H, Schwer B. Characterization of dominant-negative mutants of the DEAH-box splicing factors Prp22 and Prp16. Journal of Biological Chemistry. 2002;18:15452–15458. doi: 10.1074/jbc.M112473200. [DOI] [PubMed] [Google Scholar]
- 55.Silverman SK. In vitro selection, characterization, and application of deoxyribozymes that cleave RNA. Nucleic Acids Res. 2005;33:6151–6163. doi: 10.1093/nar/gki930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tarn WY, Lee KR, Cheng SC. Yeast precursor mRNA processing protein PRP19 associates with the spliceosome concomitant with or just after dissociation of U4 small nuclear RNA. Proc Natl Acad Sci USA. 1993;90:10821–10825. doi: 10.1073/pnas.90.22.10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cheng SC, Newman AN, Lin RJ, McFarland GD, Abelson JN. Preparation and fractionation of yeast splicing extract. Meth Enzymol. 1990;181:89–96. doi: 10.1016/0076-6879(90)81114-a. [DOI] [PubMed] [Google Scholar]
- 58.Tarn WY, et al. Functional association of essential splicing factor(s) with PRP19 in a protein complex. EMBO J. 1994;13:2421–2431. doi: 10.1002/j.1460-2075.1994.tb06527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.