

Abstract
Background/Aims
The present study identified genetic predictors of weight change during behavioral weight loss treatment.
Methods
Participants were 3,899 overweight/obese individuals with type 2 diabetes from Look AHEAD, a randomized controlled trial to determine the effects of intensive lifestyle intervention (ILI), including weight loss and physical activity, relative to diabetes support and education, on cardiovascular outcomes. Analyses focused on associations of single nucleotide polymorphisms (SNPs) on the Illumina CARe iSelect (IBC) chip (minor allele frequency >5%; n = 31,959) with weight change at year 1 and year 4, and weight regain at year 4, among individuals who lost ≥ 3% at year 1.
Results
Two novel regions of significant chip-wide association with year-1 weight loss in ILI were identified (p < 2.96E-06). ABCB11 rs484066 was associated with 1.16 kg higher weight per minor allele at year 1, whereas TNFRSF11A, or RANK, rs17069904 was associated with 1.70 kg lower weight per allele at year 1.
Conclusions
This study, the largest to date on genetic predictors of weight loss and regain, indicates that SNPs within ABCB11, related to bile salt transfer, and TNFRSF11A, implicated in adipose tissue physiology, predict the magnitude of weight loss during behavioral intervention. These results provide new insights into potential biological mechanisms and may ultimately inform weight loss treatment.
Keywords: Type 2 diabetes, Obesity, Weight loss, Diet, Genetics
Introduction
Obesity is a major public health problem [1]. Fully 64% of the US population is estimated to be overweight or obese (body mass index ≥ 25) [2] and is at increased risk for weight-related comorbidities, including coronary artery disease, diabetes and certain cancers. Behavioral weight loss intervention, focusing on changes in diet and physical activity, has emerged as a key strategy in combating this rise in obesity and the associated health consequences [3, 4]. These weight loss programs often produce initial weight losses of ≥ 7%, resulting in clinically important health benefits [5, 6]. Nonetheless, partial weight regain is common [6]. A greater understanding of predictors of weight loss and weight maintenance or regain could have important health benefits.
Obesity is also a quintessential phenotype to study the interplay of genetic and environmental factors. Body weight is well known to be heritable [7, 8] and obesity susceptibility loci have been identified through genome-wide association studies (GWAS), although the variance attributable to these loci remains small [9–11]. At the same time, the dramatic rise in obesity rates over the past 30 years suggests environmental influences [12] and evidence of gene × environment interaction can be derived from both twin [13, 14] and molecular genetic studies [15, 16].
Randomized controlled trials (RCTs) comparing behavioral weight loss treatment to a control condition afford a unique opportunity to test gene × environment interaction in the context of gene × treatment arm interaction [17]. Participants are randomly assigned to a treatment that promotes successful weight loss through provision of caloric and physical activity goals and teaching of behavioral strategies, or a control treatment without active weight loss. One type of gene × environment interaction may occur if genetic markers are associated with weight change in a control arm but not a behavioral weight loss treatment arm, suggesting that behavioral weight loss treatment may mitigate genetic effects on the outcome. Alternatively, a gene × environment interaction could occur if genetic markers relate to weight change in the intervention arm but not the control arm, suggesting that genetic factors may influence the ability to lose weight. Both effects are of potential public health importance as they may identify genetic predictors of naturalistic weight gain over time and/or resistance to weight loss. These effects further have potential clinical application as they are examined in the context of a well-established treatment paradigm. It is important to note, nonetheless, that although RCTs with longitudinal follow-up afford many advantages in research design and treatment implications, implementation of such a trial is costly and complex and sample sizes are necessarily smaller than those common in epidemiologic studies of gene × environment interaction.
The goal of the present study is to conduct a chip-wide association study of nearly 32,000 single nucleotide polymorphisms (SNPs) available from the Illumina CARe iSelect (IBC) chip [18] with weight loss at year 1 and year 4 in response to behavioral weight loss intervention and weight change at year 4 among those who lost ≥ 3% weight at year 1. The Look AHEAD study, the largest RCT comparing behavioral weight loss to a control condition with an effective intervention and excellent longitudinal follow-up, provides an excellent opportunity to conduct such analyses.
Materials and Methods
Study Cohort
The Look AHEAD study enrolled 5,145 ethnically diverse overweight and obese subjects with type 2 diabetes and aged 45–76 years. Of these, 1,108 were excluded for lack of consent for genetics studies, lack of institutional review board approval for this ancillary study (including the Southwest American Indian sites), 10 for withdrawn consent for genotyping, and 60 for inadequate DNA samples. This left 4,037 individuals, of which 3,899 contributed genetic data that passed genotyping quality control procedures. These subjects form the basis for the present analyses.
The design and methods of the Look AHEAD trial have been reported elsewhere [19], as have the baseline characteristics of the randomized cohort [20]. Briefly, at baseline, participants were randomized to either an intensive lifestyle intervention (ILI) or a diabetes support and education (DSE) arm. Both the ILI and DSE groups were provided one session of education on diabetes and cardiovascular risk factors. In addition, ILI participants received an intensive lifestyle program, combining diet modification and increased physical activity, designed to produce an average of 7% weight loss and maintain this weight loss. The ILI included 1 individual and 3 group meetings per month for 6 months followed by 1 individual and 2 group meetings per month through 1 year. From years 2 to 4, ILI participants were seen individually at least once a month, contacted another time each month by telephone or e-mail, and offered a variety of ancillary classes. ILI sessions focused on behavioral weight loss strategies, such as self-monitoring, goal setting and stimulus control, to achieve and maintain weight loss. The DSE group received the option of attending 3 sessions per year on nutrition, physical activity and social support with no explicit weight loss goals. In the full trial [6, 21], maximal difference in average weight loss across intervention arm occurred at 1 year follow-up (8.6% in ILI vs. 0.7% in DSE, p < 0.001), with an average weight loss of 4.7% in ILI and 1.1% in DSE at year 4 follow-up. The Look AHEAD trial, including genetic analyses, was approved by local institutional review boards.
Anthropometric Measures
Weight was measured to the nearest 0.1 kg in duplicate at baseline, and year 1 and year 4 follow-ups, using a digital scale. Height was measured in centimeters at baseline using a standard wallmounted stadiometer. Participants wore light clothing or a hospital gown and removed their shoes.
Weight regain was defined as weight change from year 1 to 4 among individuals initially losing at least some weight (≥ 3%) at year 1 following methods used in the Diabetes Prevention Program [22]. As can be seen in online supplementary table 1 (for all online suppl. material, see www.karger.com/doi/10.1159/000353181), among those who lost ≥ 3% weight at year 1, women regained 3.7 ± 8.2 kg and men regained 4.8 ± 7.8 kg from year 1 to 4 on average. It is important to note, however, that only 72.5% of women and 78.5% of men in this subgroup regained weight, as defined by a weight at year 4 greater than their weight at year 1, while the remaining individuals either maintained or continued to lose weight.
Genotyping
The genomic DNA extraction was based on the use of Flexi-Gene DNA Kit (Qiagen Inc., Valencia, Calif., USA) as described by the manufacturer, and DNA quantitation was performed using the PicoGreen dsDNA Quantitation Reagent (Invitrogen, Inc., Carlsbad, Calif., USA). Genotyping was carried out at the Children’s Hospital of Philadelphia using the IBC chip, a gene-centric 50,000 SNP array designed to assess relevant loci across a range of cardiovascular, metabolic and inflammatory syndromes [18]. SNPs were clustered into genotypes using the Illumina Beadstudio software and subjected to quality control filters. Individual samples were excluded for individual call rates <95%, gender mismatch, and duplicate discordance. SNPs were removed for call rates <95%. To facilitate model convergence and minimize the penalty from multiple comparisons due to the many low-frequency SNPs included in the design, we filtered out markers with minor allele frequency (MAF) <0.05. This left 31,692 autosomal SNPs on the IBC chip with MAF >5% whose mean genotyping success rate was 99.8%.
Observed genotype frequencies were compared with those expected under Hardy-Weinberg equilibrium (HWE) using stratified χ2 tests within the two largest racial/ethnic groups (non-Hispanic White and African-American). As the sample is selected for overweight and diabetes, we did not exclude SNPs based on deviation on a chip-wide basis from HWE, but reviewed individual SNP associations to ensure SNPs showing significant associations did not deviate from HWE.
Statistical Analysis
After pruning of SNPs in linkage disequilibrium (LD; r2 > 0.3), the Eigenstrat algorithm [23], as implemented in Golden Helix version 7.1 (Bozeman, Mont., USA), was used to compute principal components for use as covariates to control for ancestry in the regression analyses. Principal component analysis results indicated that the majority of the variance among the multi-racial Look AHEAD cohort was accounted for by the first two principal components, which agreed with self-reported race/ethnicity in distinguishing Caucasians from African-Americans, and Hispanics from these other 2 groups [24]. Preliminary analyses indicated that additional principal components that would have better separated the Hispanic and Asian groups and helped identify the few Native American study participants did not contribute to the weight change models.
Consistent with the parent trial, we focused on weight (not body mass index) change (in kg) as the primary outcome. Therefore, we conducted longitudinal regression analysis of baseline, year-1 and year-4 weight measurements, modeled jointly as a trivariate normal outcome with an unstructured covariance matrix. Three-way interaction models of individual SNP markers with measurement time (year 1 vs. baseline, year 4 vs. baseline) and study arm (ILI vs. DSE) were estimated in Splus 8.2 [25] using restricted maximum likelihood. An additive genetic model was used for all SNP markers, with genotype coded by the number of minor alleles (0/1/2 copies). Minor alleles and allele frequencies were determined from the entire sample of genotyped participants, i.e. race/ethnicity-specific allele frequencies were not used.
Therefore, four distinct types of SNP effects were estimated, all of which can be interpreted as the effect of one additional copy of the corresponding minor allele on (a) baseline weight within DSE (SNP main effect); (b) ILI-DSE differences in baseline weight (SNP × study arm interaction); (c) weight change within DSE (SNP × time interaction), and (d) ILI-DSE differences in weight change (SNP × time × study arm interaction). Effects specific to ILI or averaged over the ILI and DSE arms were subsequently obtained by changing the referent group for study arm and re-estimating the model. Of note, in a randomized trial one would expect no ILIDSE differences in SNP effects on baseline weight levels, so set b of model parameters serves solely as a randomization check.
Longitudinal weight outcomes were additionally adjusted for study site, age, gender, and the first two ancestry informative marker principal components [24, 26]. Other than study site, all of these covariates were fully interacted with time, study arm, and time by study arm interaction, so as to allow for these covariate effects to vary across study arm and/or time point, in a manner similar to the SNP effects described above.
For chip-wide analyses, we calculated the effective number of uncorrelated markers among the 31,692 autosomal SNPs under investigation using the Li and Ji approach [27] and found it to equal just 17,254 after LD correction. A chip-wide significance threshold of p = 2.97E-06 would, therefore, be needed to control the more stringent familywise error rate criterion at the 5% level based on Sidak’s multiplicity adjustment [28].
We also used a false discovery rate (FDR) approach to guide our reporting of suggestive (FDR <20%) associations, operationalized via a rank ordering of the genetic markers according to their q values. FDR controls the expected proportion of incorrectly rejected null hypotheses among those deemed significant, rather than across the entire set of hypotheses being tested; this increases power relative to more stringent familywise error rate control at the cost of more type I errors. Calculated using the q value package of Dabney and Storey [29], q values are marker-specific quantities that represent the minimum FDR at which the corresponding hypothesis test could be declared significant [30]. q values preserve the same rank ordering of SNPs as that produced by p value calculations, but recalibrate them by the probability that they represent a false discovery. They were calculated separately for each coefficient of interest (ILI change, DSE change, pooled ILI and DSE and differential change).
We also examined the extent to which the genetic markers predicted weight regain at year 4 among those who lost ≥ 3% of their initial weight at year 1. This method was previously used to characterize weight regain in the Diabetes Prevention Program [22]. The primary outcome for these analyses was SNP × time (year 1 vs. year 4) interaction with the same covariates as above in addition to baseline weight. Sample size for this subanalysis was n = 2,022 (1,545 in ILI and 477 in DSE). All analyses were performed at Brown University.
Results
Descriptive Statistics
Participant characteristics of the subcohort of Look AHEAD used in these analyses are shown in table 1. Individuals were evenly distributed between the ILI and DSE intervention arms and had comparable age and gender as in the entire cohort (data not shown). The number of American Indian participants included in this study is less than that of the parent Look AHEAD trial due to differences in informed consent for genetic ancillary studies. No baseline differences in demographic or clinical characteristics across ILI and DSE were observed. Further, no between-arm differences in baseline means of the outcomes of interest were detected across genotypic groups for any of the markers under consideration (p > 2.96E-06). Similar to the larger Look AHEAD trial [21], individuals assigned to ILI lost significantly more weight at year 1 and 4 than those assigned to DSE.
Table 1.
Population characteristics in the Look AHEAD genetic subcohort
| Characteristic | Total (n = 3,899) | DSE (n = 1,964) | ILI (n = 1,935) |
|---|---|---|---|
| Women, n (%) | 2,192 (56.2) | 1,096 (55.8) | 1,096 (56.6) |
| Ethnicity, n (%) | |||
| African-American | 618 (15.8) | 305 (15.5) | 313 (16.2) |
| American Indiana | 20 (0.5) | 9 (0.5) | 11 (0.6) |
| Asian/Pacific Islander | 41 (1.1) | 19 (1.0) | 22 (1.1) |
| Hispanic/Latino | 307 (7.9) | 159 (8.1) | 148 (7.7) |
| Non-Hispanic White | 2,835 (72.7) | 1,430 (72.8) | 1,405 (72.6) |
| Other (multiple) | 78 (2.0) | 42 (2.1) | 36 (1.9) |
| Age, years | 59.1 ± 6.8 | 59.2 ± 6.8 | 59.0 ± 6.9 |
| BMI | |||
| Women | 36.8 ± 6.2 | 36.9 ± 6.1 | 36.7 ± 6.3 |
| Men | 35.3 ± 5.5 | 35.1 ± 5.2 | 35.5 ± 5.8 |
| Waist circumference, cm | |||
| Women | 111.4 ± 13.7 | 111.5 ± 13.6 | 111.3 ± 13.8 |
| Men | 118.8 ± 13.4 | 118.5 ± 12.9 | 119.2 ± 13.9 |
| Weight at baseline, kg | |||
| Women | 96.7 ± 17.5 | 96.6 ± 17.4 | 96.8 ± 17.7 |
| Men | 109.6 ± 18.5 | 109.4 ± 17.8 | 109.8 ± 19.2 |
| Weight at year 1, kg | |||
| Women | 92.1 ± 17.8 | 95.6 ± 17.5 | 88.7 ± 17.3 |
| Men | 104.1 ± 18.9 | 108.7 ± 17.9 | 99.4 ± 18.8 |
| Weight at year 4, kg | |||
| Women | 93.3 ± 17.8 | 94.4 ± 17.7 | 92.3 ± 17.9 |
| Men | 106.1 ± 19.1 | 108.4 ± 18.2 | 103.7 ± 19.7 |
| Weight change baseline – year 1, kg | |||
| Women | −4.6 ± 7.1 | −0.9 ± 5.1 | −8.1 ± 7.1 |
| Men | −5.6 ± 8.4 | −0.9 ± 5.2 | −10.5 ± 8.3 |
| Weight change baseline – year 4, kg | |||
| Women | −3.3 ± 9.0 | −2.2 ± 9.4 | −4.5 ± 8.5 |
| Men | −3.3 ± 8.8 | −0.9 ± 7.8 | −5.8 ± 9.0 |
Values are means ± SD unless otherwise indicated.
The number of American Indian participants included in this ancillary study is less than that of the parent Look AHEAD trial because not all centers participated.
Demographics and weight change patterns among individuals who lost ≥ 3% of their weight at year 1 are depicted in online supplementary table 1. In this subset, individuals in ILI lost more weight at year 1 and regained more weight from year 1 to year 4. Overall, among those who lost ≥ 3% at year 1, 59% regained weight in DSE and 80% in ILI.
Genetic Associations with Weight Loss at Year 1
Genetic associations of the full set of SNP markers with year-1 weight change in ILI and DSE are depicted in figure 1. The association of two loci with year-1 weight change in the ILI group exceeded chip-wide significance after correcting for chip-wide multiple comparisons (p < 2.96E-06). One intronic locus represented by two SNPs in high LD, rs484066 and rs569805, within ABCB11, showed the strongest association with year-1 weight loss (online suppl. table 2). These SNPs were associated with a 1.16 and 1.24 kg higher weight per minor allele at year 1, respectively, suggesting that the minor allele was associated with resistance to weight loss. A third SNP, rs17069904, within TNFRSF11A, or RANK, also achieved chip-wide significance. This SNP was associated with a 1.70 kg lower weight per allele at year 1, suggesting that the minor allele was associated with greater weight loss. These SNPs had no significant effects in DSE (p > 0.51). The resulting SNP × treatment arm interactions for the lead SNP at each locus were ABCB11 rs484066 interaction, p = 3.98E-05 (fig. 2), and TNFRSF11A rs17069904 interaction, p = 1.57E-04 (fig. 3).
Fig. 1.

IBC chip-wide Manhattan plot for year-1 weight change in ILI (a) and DSE (b). Includes 31,692 autosomal SNPs with MAF >5% (n = 3,889).
Fig. 2.
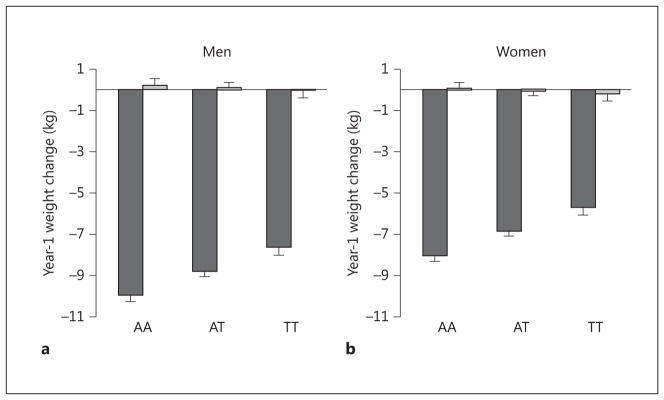
Interaction plot for ABCB11 rs484066 in men (a) and women (b).
Fig. 3.

Interaction plot for TNFRSF11A rs17069904 in men (a) and women (b).
An additional 131 SNPs showed a suggestive association with year-1 weight change (online suppl. table 2; FDR q < 0.20). Eight additional SNPs were in the ABCB11 region and an additional 3 SNPs in the TNFRSF11A region. Regional plots depicting the association of the ABCB11 and TNFRSF11A regions with year-1 weight change are presented in online supplementary figures 1 and 2, respectively.
No additional SNPs showed suggestive evidence for association with year-1 weight change in DSE (FDR q > 0.99), year-1 weight loss as averaged across treatment arms (FDR q > 0.99) or SNP × treatment arm interaction (FDR q > 0.20).
Genetic Associations with Weight Loss from Baseline to Year 4
SNPs with a suggestive association (FDR q < 0.20) with year-4 weight are presented in table 2. No SNPs showed suggestive association with weight change at year 4 in ILI (FDR q > 0.20) or SNP × treatment arm interaction (FDR q > 0.20). It is of note, however, that one of the SNPs identified for year-1 weight change, rs17069904, within TNFRSF11A, continued to show evidence of association with change in ILI at year 4 (p = 0.0002) and SNP × treatment arm interaction (p = 0.0002).
Table 2.
SNPs associated with year-4 weight change (FDR q < 0.20)
| SNP | Chr | Closest gene | Minor allele | MAF, %a | Effect | βb | SE | p value | q valuec |
|---|---|---|---|---|---|---|---|---|---|
| rs13242262 | 7 | IRF5 | T | 40.45 | DSE | 1.414 | 0.303 | 3.00E-06 | 0.051 |
| rs4233788 | 2 | ITGAV | A | 24.74 | DSE | −1.510 | 0.325 | 3.35E-06 | 0.051 |
| rs10229001 | 7 | IRF5 | G | 39.43 | DSE | 1.379 | 0.303 | 5.33E-06 | 0.054 |
| rs4788114 | 16 | LAT | A | 7.50 | DSE | −2.371 | 0.538 | 1.05E-05 | 0.081 |
| rs3911084 | 2 | ITGAV | C | 28.20 | DSE | −1.347 | 0.316 | 2.06E-05 | 0.104 |
| rs17166351 | 7 | IRF5 | A | 49.47 | DSE | −1.252 | 0.295 | 2.24E-05 | 0.104 |
| rs11240089 | 1 | BCL9 | G | 23.17 | DSE | 1.731 | 0.410 | 2.47E-05 | 0.104 |
| rs4244559 | 15 | MEIS2 | G | 33.54 | DSE | 1.253 | 0.298 | 2.72E-05 | 0.104 |
| rs720475 | 7 | ARHGEF5 | A | 23.46 | DSE | −1.403 | 0.340 | 3.71E-05 | 0.126 |
| rs12490899 | 3 | TGFBR2 | A | 5.39 | DSE | −2.735 | 0.667 | 4.15E-05 | 0.127 |
| rs1874328 | 7 | IRF5 | G | 39.45 | DSE | −1.217 | 0.300 | 5.06E-05 | 0.141 |
| rs7808907 | 7 | IRF5 | G | 49.90 | DSE | −1.164 | 0.289 | 5.84E-05 | 0.149 |
| rs6695965 | 1 | BCL9 | G | 23.71 | DSE | 1.616 | 0.412 | 8.84E-05 | 0.197 |
| rs6434188 | 2 | ITGAV | A | 22.48 | DSE | −1.349 | 0.345 | 9.21E-05 | 0.197 |
| rs977483 | 5 | Intergenic | G | 8.80 | DSE | 2.019 | 0.520 | 1.05E-04 | 0.197 |
| rs10505996 | 12 | ITPR2 | G | 8.06 | DSE | −2.137 | 0.551 | 1.07E-04 | 0.197 |
| rs10760707 | 9 | TXNDC4 | T | 15.95 | DSE | 1.597 | 0.413 | 1.10E-04 | 0.197 |
|
| |||||||||
| rs649526 | 15 | PLA2G4F | A | 36.96 | Pooled | −0.936 | 0.216 | 1.43E-05 | 0.186 |
| rs11165294 | 1 | TGFBR3 | A | 11.01 | Pooled | −1.378 | 0.327 | 2.59E-05 | 0.186 |
| rs11063488 | 12 | Intergenic | C | 43.93 | Pooled | 0.901 | 0.214 | 2.68E-05 | 0.186 |
| rs913059 | 1 | TGFBR3 | C | 24.86 | Pooled | 0.998 | 0.239 | 2.92E-05 | 0.186 |
| rs621560 | 15 | PLA2G4F | A | 41.23 | Pooled | −0.874 | 0.210 | 3.24E-05 | 0.186 |
| rs8114057 | 20 | HNF4A | A | 47.13 | Pooled | −0.857 | 0.208 | 3.91E-05 | 0.186 |
| rs11213865 | 11 | POU2AF1 | A | 6.30 | Pooled | −1.694 | 0.413 | 4.09E-05 | 0.186 |
Chr = Chromosome.
MAF in the full sample.
Effect of each copy of the minor allele on baseline to year-4 weight change (in kg) within DSE, and baseline to year-4 weight change (in kg) pooled across ILI and DSE arms.
Ranking based on q values (FDR <20%).
For year-4 weight change in DSE, in contrast, 17 SNPs were identified with suggestive association (FDR q < 0.20). The strongest two loci occurred within IRF5 and ITGAV, represented by 5 and 3 SNPs, respectively. Seven SNPs also showed a suggestive association with weight change as averaged across ILI and DSE (FDR q < 0.20). Two of these SNPs were within PLA2G4F and another 2 within TGFBR3.
Weight Change from Year 1 to 4 among Those Who Lost ≥ 3% at Year 1
SNPs with a suggestive association (FDR q < 0.20) with weight change at year 4 among those who initially lost ≥ 3% of their weight at year 1 are presented in table 3. No suggestive associations were observed within the ILI arm (FDR q > 0.99). For weight change within the DSE arm, 11 SNPs showed suggestive evidence of association (FDR q < 0.20). The closest genes for these SNPs included FOXP1, GRB2, COL1A2, RARB, MMP13, JUN and C8orf49. Eight SNPs showed suggestive evidence of a main effect on weight change across treatment arms (FDR q < 0.20). The closest genes for these SNPs were FOXP1, MMP13, TGFBR3, C8orf49, FDFT1 and ST8SIA4.
Table 3.
SNPs associated with weight change from year 1 to 4 among participants who lost 3% weight by year 1
| SNP | Chr | Closest gene | Minor allele | MAF, %a | Effect | βb | SE | p value | q valuec |
|---|---|---|---|---|---|---|---|---|---|
| rs830653 | 3 | FOXP1 | A | 10.4 | DSE | −3.894 | 0.843 | 3.98E-06 | 0.121 |
| rs9302989 | 17 | GRB2 | G | 26.79 | DSE | 2.964 | 0.680 | 1.33E-05 | 0.172 |
| rs1800222 | 7 | COL1A2 | G | 19.17 | DSE | −3.052 | 0.711 | 1.79E-05 | 0.172 |
| rs2053156 | 17 | GRB2 | C | 27.67 | DSE | 2.762 | 0.659 | 2.82E-05 | 0.172 |
| rs959260 | 17 | GRB2 | G | 27.68 | DSE | 2.762 | 0.659 | 2.82E-05 | 0.172 |
| rs17526019 | 3 | RARB | A | 6.67 | DSE | −4.294 | 1.041 | 3.80E-05 | 0.178 |
| rs10895373 | 11 | MMP13 | A | 15.16 | DSE | −3.008 | 0.733 | 4.10E-05 | 0.178 |
| rs10895372 | 11 | MMP13 | G | 15.41 | DSE | −2.970 | 0.729 | 4.69E-05 | 0.179 |
| rs2760494 | 1 | JUN | G | 16.02 | DSE | −3.078 | 0.767 | 6.14E-05 | 0.189 |
| rs11225490 | 11 | MMP13 | G | 15.65 | DSE | −2.892 | 0.723 | 6.45E-05 | 0.189 |
| rs2740434 | 8 | C8orf49 | A | 31.8 | DSE | −2.321 | 0.582 | 6.82E-05 | 0.189 |
|
| |||||||||
| rs830653 | 3 | FOXP1 | A | 10.4 | Pooled | −2.208 | 0.493 | 7.65E-06 | 0.093 |
| rs10895373 | 11 | MMP13 | A | 15.16 | Pooled | −1.807 | 0.415 | 1.37E-05 | 0.093 |
| rs11225490 | 11 | MMP13 | G | 15.65 | Pooled | −1.780 | 0.409 | 1.40E-05 | 0.093 |
| rs12567680 | 1 | TGFBR3 | A | 32.53 | Pooled | −1.325 | 0.305 | 1.40E-05 | 0.093 |
| rs10895372 | 11 | MMP13 | G | 15.41 | Pooled | −1.790 | 0.413 | 1.48E-05 | 0.093 |
| rs2740434 | 8 | C8orf49 | A | 31.8 | Pooled | −1.391 | 0.329 | 2.41E-05 | 0.113 |
| rs1616534 | 8 | FDFT1 | G | 46.36 | Pooled | −1.274 | 0.304 | 2.76E-05 | 0.113 |
| rs7703643 | 5 | ST8SIA4 | A | 39.57 | Pooled | −1.271 | 0.304 | 2.91E-05 | 0.113 |
Chr = Chromosome.
MAF in the full sample.
Effect of each copy of the minor allele on year-1 to -4 weight change (in kg) within DSE and as pooled across ILI and DSE arms.
Ranking based on q values (FDR <20%).
For SNP × treatment arm interaction (table 4), 5 SNPs showed a suggestive association (FDR q < 0.20). Three of these SNPs occurred within GRB2, 1 within TIMP3 and the last in the region of INSR.
Table 4.
SNPs associated with differential weight change (ILI–DSE) from year 1 to 4 among participants who lost 3% weight at year 1
| SNP | Chr | Closest gene | Minor allele | MAF, %a | Effect | βb | SE | p value | q valuec |
|---|---|---|---|---|---|---|---|---|---|
| rs2053156 | 17 | GRB2 | C | 27.67 | Interaction | −3.506 | 0.751 | 3.14E-06 | 0.034 |
| ILI | −0.744 | 0.361 | 3.92E-02 | ||||||
| DSE | 2.762 | 0.659 | 2.82E-05 | ||||||
|
| |||||||||
| rs959260 | 17 | GRB2 | G | 27.68 | Interaction | −3.504 | 0.751 | 3.21E-06 | 0.034 |
| ILI | −0.742 | 0.361 | 4.00E-02 | ||||||
| DSE | 2.762 | 0.659 | 2.82E-05 | ||||||
|
| |||||||||
| rs713685 | 22 | TIMP3 | A | 8.90 | Interaction | 4.858 | 1.045 | 3.43E-06 | 0.034 |
| ILI | 1.895 | 0.500 | 1.53E-04 | ||||||
| DSE | −2.963 | 0.917 | 1.25E-03 | ||||||
|
| |||||||||
| rs9302989 | 17 | GRB2 | G | 26.79 | Interaction | −3.562 | 0.776 | 4.52E-06 | 0.034 |
| ILI | −0.598 | 0.374 | 1.10E-01 | ||||||
| DSE | 2.964 | 0.680 | 1.33E-05 | ||||||
|
| |||||||||
| rs4804366 | 19 | INSR | G | 12.59 | Interaction | 4.039 | 0.957 | 2.50E-05 | 0.152 |
| ILI | 0.881 | 0.444 | 4.72E-02 | ||||||
| DSE | −3.158 | 0.848 | 1.99E-04 | ||||||
Chr = Chromosome.
MAF in the full sample.
Effects of each copy of the minor allele on within-arm weight change for ILI and DSE as well as differential weight change (ILI–DSE) from year 1 to 4 (in kg).
Ranking based on q values (FDR <20%).
Discussion
This paper presents the largest study and the first chipwide analysis of weight loss in response to behavioral treatment and weight regain after successful weight loss. Our results identify novel regions of significant chipwide association with magnitude of weight loss in response to behavioral treatment, including ABCB11 and TNFRSF11A (p < 2.96E-06), as well as a number of suggestive associations (FDR q < 0.20) for weight loss at year 1, weight change from baseline to year 4, and weight regain at year 4 among those who lost ≥ 3% at year 1. These results suggest new potential mechanisms contributing to weight loss and regain and have the potential to inform behavioral weight loss treatment.
Prior genetic studies of weight loss had focused on variation within PPARG or obesity risk SNPs from GWAS. The Ala12Ala genotype in PPARG (rs1801282) has been associated with greater weight loss in response to lifestyle intervention both in the US Diabetes Prevention Program (DPP) [31] and the Finnish Diabetes Prevention Study (DPS) [32]. The association of FTO with weight loss is less clear. Obesity risk alleles within FTO predicted a greater increase in subcutaneous adipose tissue in the placebo group in the DPP [33] but greater free fatty mass in response to a low-protein diet and less free fatty mass in response to a high-fat diet in the POUNDS LOST trial [34]. In the DPP, the obesity risk allele at rs6265 in BDNF was also associated with greater weight regain over 2 years among those who had initially lost ≥ 3% at 6 months [22]. In a prior Look AHEAD report [24], obesity risk SNPs derived from GWAS were not significantly associated with magnitude of initial weight loss. However, FTO risk alleles predicted weight regain in the DSE group, but not within the ILI group, resulting in SNP × treatment arm interaction. The obesity risk allele at BDNF rs6265 was also associated with marginally greater weight regain across treatment arms, consistent with the findings of the DPP.
In the present paper, we sought to broaden the prior literature by examining whether any additional SNPs represented on the IBC chip predict the extent of weight loss or weight regain during behavioral intervention. The IBC chip assays loci across a range of cardiovascular, metabolic and inflammatory syndromes [18], the majority of which had not previously been implicated in obesity or weight change. Thus, we could determine whether any of these regions might be relevant to weight-related phenotypes.
Genetic Predictors of Year-1 Weight Loss
For weight change at year 1, all SNP associations reaching chip-wide statistical significance or suggestive significance based on FDR occurred within ILI. This indicates that, at year 1, all of the SNP associations appear to influence ability to lose weight in response to intervention, whereas little to no effect of the SNPs was observed for naturalistic weight change in DSE over the year.
The strongest association with year-1 weight change in ILI was an intronic locus represented by 2 SNPs in high LD, rs484066 and rs569805, in ABCB11, or ATP-binding cassette, subfamily B, member 11, also called bile salt export pump (BSEP). ABCB11 is the primary mediator of bile salt secretion across the canalicular membrane and plays a critical role in absorption of dietary fat from the gut and counter transport of hepatic cholesterol from the liver to the intestine for elimination [35]. Mutations of ABCB11 are known to cause progressive intrahepatic cholestasis [36, 37] and benign recurrent intrahepatic cholestasis [38, 39], and GWAS identify the ABCB11 region as a predictor of alkaline phosphatase, a marker of biliary obstruction [40].
Perturbations of bile salt transport appear to alter body weight and serum cholesterol levels. ABCB11 knockout mice have smaller body size than wild-type litter mates [41], while overexpression of ABCB11 in mice leads to greater fat absorption from the intestine, more rapid weight gain and a reduction in energy expenditure in response to a high-fat diet but not on a control diet [42]. ABCB11 variation has also been implicated in obesity in candidate gene studies [43], and, in GWAS, in fasting high-density lipoprotein and glucose [44]. It is of note that this locus is also proximal to glucose-6-phosphatase, catalytic, 2 (G6PC2), associated with fasting glucose in GWAS [45].
The TNFRSF11A, or as more commonly known RANK, polymorphism rs17069904 was also associated with weight loss at year 1 in response to ILI and nominally associated with year-4 weight change, with nominal SNP × treatment interactions at each time point. RANK, along with the RANK ligand, are members of the TNF family of genes that, with osteoprotegerin (OPG), form a signaling network that regulates bone mineral density [46]. Intriguingly, RANK ligand and OPG are expressed in adipose tissue [47], and OPG levels have been shown to be reduced by a weight loss intervention [48]. Links between change in weight and bone mass density are well described and further highlight reciprocal pathways of influence in adipose and bone [46]. RANK has been associated with percentage fat mass in animal linkage studies [49] and body mass index in humans [50]. Our results provide support for a novel role for the RANK - RANK ligand-OPG pathway in adipose tissue response to a lifestyle intervention in addition to their well-established role in bone.
Of the suggestive associations with year-1 weight loss, at least 2 were of particular note. The strongest suggestive association occurred within AANAT (rs12452844, p = 3.12E-06). This gene codes for arylalkylamine N-acetyltransferase, a critical enzyme in melatonin production and regulation of circadian rhythm, and has previously been associated with delayed sleep onset syndrome [51]. As epidemiologic studies document associations between short sleep and body mass index [52], this provides sug gestive evidence that variation in AANAT may contribute to that association. A suggestive association was also seen for TCD1B1. TBC1D1 is an insulin-sensitive regulator of GLUT4 function in skeletal muscle, and variation in TBC1D1 may alter glucose uptake during exercise [53]. A non-synonymous polymorphism in the TBC1D1 gene has also been associated with severe familial obesity in at least two independent studies [54, 55].
Genetic Predictors of Year-4 Weight Change and Weight Regain
For weight change between baseline and year 4, a different pattern of results emerged. The effects on weight change occurred primarily within the DSE group, with no weight loss intervention, or when pooled across the ILI and DSE. This suggests that SNPs were often associated with naturalistic change in weight over time in DSE or that SNPs were associated with naturalistic weight change in DSE and resistance to weight loss or weight regain in ILI. It is of note, however, that TNFRSF11A rs17069904 continued to show evidence of association with weight change in ILI at year 4 (p = 0.0002) and SNP × treatment arm interaction (p = 0.0002).
For weight change from year 1 to 4 among those who had lost ≥ 3% at year 1, SNP associations were also primarily seen in the subset of individuals randomized to DSE who had lost at least some weight at year 1. In several instances, a weaker but consistent effect in ILI resulted in a SNP effect on weight change as averaged across treatment arms. In a few instances, a weaker effect in the opposite direction occurred resulting in a suggestive interaction (FDR q < 0.20).
The causes of weight changes during the first 4 years after randomization are likely to be different between the ILI and DSE groups at different time points. The mean weight loss trajectory in the ILI group was typical of most behavioral weight loss programs, i.e. maximal weight loss at 1 year with a tendency for partial regain in the next 3 years. The initial weight loss was presumably due to intensive behavioral efforts with the regain representing a combination of inability to sustain the behavioral changes and potentially physiologic adaptations to weight loss. By contrast, the gradual, relatively steady, weight loss in the DSE group was presumably due primarily to joint effects of aging and increasing duration of diabetes which, at least in some observational studies, are associated with weight loss [56]. Therefore, one might expect the predictors (including genetic) of weight change to differ between the two treatment groups and at different time points of follow-up.
It is nonetheless notable that several of the SNP associations with year-4 weight change, either from baseline to year 4, or year 1 to 4, among those who initially lost ≥ 3%, occur in pathways integrally involved in structural remodeling of adipose tissue and fibrosis accumulation with increasing fat mass [57]. Proteolytic systems, such as the matrix metalloproteinase (MMP) system, contribute to tissue remodeling by degradation of the extracellular matrix and membrane components or by activation of latent growth factors [58]. MMP 13 (collagenase 3; MMP13) is expressed in adipose tissue with increases in expression after high-fat feeding [59]. Tissue inhibitor of metalloproteinases-3 (TIMP3) is 1 of 4 tissue inhibitors of metalloproteinases that have been characterized and are able to inhibit the activities of MMPs [60, 61]. MMPs release transforming growth factor-β (TGF-β), which alters cell migration and further regulates extracellular matrix and promotes fibrosis [62]. TGFB3 is expressed in adipose tissue and downregulated in response to caloric restriction [63]. ITGAV encodes integrin α chain V. Integrins also interact with extracellular matrix ligands and may play a role in adipocyte apoptosis [57]. PLA2G4F is a member of the phospholipase family that hydrolyzes phospholipids into arachidonic acid and may play a role in fat deposition and the storage of lipids in adipose tissue [64]. The MMP and PLA2 pathways have been shown to alter the degree of weight gain in response to high-fat diets in animal models [65–67].
Other SNP associations with weight change between baseline and year 4 were related to immune function. IRF5 is expressed in adipose tissue and mediates proinflammatory cytokine release. A high-fat diet has been shown to upregulate IRF5. FOXP1 has been associated with cytokine production of macrophages [68]. Finally, GRB2 and INSR increase PI3K signaling to regulate glucose uptake. Thus, a number of the SNP associations with year-4 weight change or regain occur within plausible biological pathways of direct relevance to adipose tissue and inflammation.
Strengths and Limitations
This study has several strengths. It is the largest RCT of behavioral weight loss, with nearly 3,900 individuals with genetic consent randomly assigned to either an effective ILI focusing on weight loss and physical activity promotion or a minimal contact control group. This design presents several distinct advantages to studying gene × environment interaction relative to epidemiologic studies, including random assignment on a 1: 1 basis to an environmental exposure with a large effect size. Further, we accurately measure degree of intentional weight loss and extent of weight regain with longitudinal measures and excellent retention rates. These phenotypes are of key public health importance given the well-recognized obesity epidemic and success of behavioral weight loss in reducing cardiovascular and diabetes risk. Yet, they are inherently difficult to assess in epidemiologic studies due to difficulties with self-report and the potential for confounding by unintentional weight loss due to illness. The potential for more direct clinical application is also augmented using this design to study gene × environment interaction as the genetic effects are established in the context of a well-established and effective treatment protocol.
Importantly, we also present the first chip-wide analysis of intentional weight loss and regain and identify novel loci related to weight change phenotypes. Our genotyping platform was comprised of SNPs within over 2,100 genes related to cardiovascular disease and its risk factors. Thus, these SNPs may not be newly discovered as related to cardiometabolic phenotypes, but none had been identified previously in relation to weight change. This supports the possibility that careful genetic study of phenotypes in different environments or in response to different treatments may yield new insights into genetic architecture. It is further plausible that the inclusion of a greater number of SNPs selected with a more agnostic approach, such as GWAS or next-generation sequencing, may discover additional loci related to weight loss and regain. Finally, replication of these loci in an independent sample would further strengthen the interpretation of these results. We presented suggestive associations in addition to those reaching chip-wide significance to support these efforts. Nonetheless, it is important to note that this cohort was selected for type 2 diabetes and overweight, which may complicate replication efforts.
Conclusions
Overall, this largest chip-wide study of genetic predictors of weight loss and weight regain identifies SNPs within ABCB11 and TNFRSF11A as predictors of the magnitude of weight loss during a behavioral weight loss intervention. These genetic associations highlight the potential role of bile salt transport and the TNF superfamily as novel mechanisms contributing success in weight loss with behavioral efforts. Furthermore, these results indicate that discovery efforts for genetic predictors of novel phenotypes, such as weight loss and weight regain, may yield new insights into the genetics of obesity and treatment response.
Acknowledgments
We acknowledge the Look AHEAD sites that participated in this ancillary study.
Funding and Support
This study was supported by the Department of Health and Human Services through the following cooperative agreements from the National Institutes of Health: DK57136, DK57149, DK56990, DK57177, DK57171, DK57151, DK57182, DK57131, DK57002, DK57078, DK57154, DK57178, DK57219, DK57008, DK57135 and DK56992. The following federal agencies have contributed support: National Institute of Diabetes and Digestive and Kidney Diseases; National Heart, Lung, and Blood Institute; National Institute of Nursing Research; National Center on Minority Health and Health Disparities; NIH Office of Research on Women’s Health, and the Centers for Disease Control and Prevention. This research was supported in part by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases. The Indian Health Service (IHS) provided personnel, medical oversight, and use of facilities. The opinions expressed in this paper are those of the authors and do not necessarily reflect the views of the IHS or other funding sources.
Additional support was received from The Johns Hopkins Medical Institutions Bayview General Clinical Research Center (M01RR02719); the Massachusetts General Hospital Mallinckrodt General Clinical Research Center and the Massachusetts Institute of Technology General Clinical Research Center (M01RR01066); the University of Colorado Health Sciences Center General Clinical Research Center (M01RR00051) and Clinical Nutrition Research Unit (P30 DK48520); the University of Tennessee at Memphis General Clinical Research Center (M01RR0021140); the University of Pittsburgh General Clinical Research Center (GCRC) (M01RR000056); the Clinical Translational Research Center (CTRC) funded by the Clinical and Translational Science Award (UL1 RR 024153) and NIH grant (DK 046204); the VA Puget Sound Health Care System Medical Research Service, Department of Veterans Affairs, and the Frederic C. Bartter General Clinical Research Center (M01RR01346).
The following organizations have committed to make major contributions to Look AHEAD: FedEx Corporation; Health Management Resources; LifeScan, Inc., a Johnson & Johnson Company; OPTIFAST® of Nestlé HealthCare Nutrition, Inc.; Hoffmann-La Roche Inc.; Abbott Nutrition; and Slim-Fast Brand of Unilever North America.
Some of the information contained herein was derived from data provided by the Bureau of Vital Statistics, New York City Department of Health and Mental Hygiene for providing vital statistics data.
Clinical Sites
The Johns Hopkins Medical Institutions
Frederick L. Brancati, MD, MHS1; Lee Swartz2; Lawrence Cheskin, MD3; Jeanne M. Clark, MD, MPH3; Kerry Stewart, EdD3; Richard Rubin, PhD3; Jean Arceci, RN4; Suzanne Ball; Jeanne Charleston, RN; Danielle Diggins; Mia Johnson; Joyce Lambert; Kathy Michalski, RD; Dawn Jiggetts; Chanchai Sapun.
Pennington Biomedical Research Center
George A. Bray, MD1; Kristi Rau2; Allison Strate, RN2; Frank L. Greenway, MD3; Donna H. Ryan, MD3; Donald Williamson, PhD3; Brandi Armand, LPN; Jennifer Arceneaux; Amy Bachand, MA; Michelle Begnaud, LDN, RD, CDE; Betsy Berhard; Elizabeth Caderette; Barbara Cerniauskas, LDN, RD, CDE; David Creel, MA; Diane Crow; Crystal Duncan; Helen Guay, LDN, LPC, RD; Carolyn Johnson, Lisa Jones; Nancy Kora; Kelly LaFleur; Kim Landry; Missy Lingle; Jennifer Perault; Cindy Puckett; Mandy Shipp, RD; Marisa Smith; Elizabeth Tucker.
The University of Alabama at Birmingham
Cora E. Lewis, MD, MSPH1; Sheikilya Thomas, MPH2; Monika Safford, MD3; Vicki DiLillo, PhD; Charlotte Bragg, MS, RD, LD; Amy Dobelstein; Stacey Gilbert, MPH; Stephen Glasser, MD3; Sara Hannum, MA; Anne Hubbell, MS; Jennifer Jones, MA; DeLavallade Lee; Ruth Luketic, MA, MBA, MPH; L. Christie Oden; Janet Raines, MS; Cathy Roche, RN, BSN; Janet Truman; Nita Webb, MA; Casey Azuero, MPH; Jane King, MLT; Andre Morgan.
Harvard Center
Massachusetts General Hospital: David M. Nathan, MD1; Enrico Cagliero, MD3; Kathryn Hayward, MD3; Heather Turgeon, RN, BS, CDE2; Linda Delahanty, MS, RD3; Ellen Anderson, MS, RD3; Laurie Bissett, MS, RD; Valerie Goldman, MS, RD; Virginia Harlan, MSW; Theresa Michel, DPT, DSc, CCS; Mary Larkin, RN; Christine Stevens, RN; Kylee Miller, BA; Jimmy Chen, BA; Karen Blumenthal, BA; Gail Winning, BA; Rita Tsay, RD; Helen Cyr, RD; Maria Pinto.
Joslin Diabetes Center: Edward S. Horton, MD1; Sharon D. Jackson, MS, RD, CDE2; Osama Hamdy, MD, PhD3; A. Enrique Caballero, MD3; Sarah Bain, BS; Elizabeth Bovaird, BSN, RN; Barbara Fargnoli, MS, RD; Jeanne Spellman, BS, RD; Ann Goebel-Fabbri, PhD; Lori Lambert, MS, RD; Sarah Ledbury, MEd, RD; Maureen Malloy, BS; Kerry Ovalle, MS, RCEP, CDE.
Beth Israel Deaconess Medical Center: George Blackburn, MD, PhD1; Christos Mantzoros, MD, DSc3; Ann McNamara, RN; Kristina Spellman, RD.
University of Colorado Health Sciences Center
James O. Hill, PhD1; Marsha Miller, MS, RD2; Brent Van Dorsten, PhD3; Judith Regensteiner, PhD3; Ligia Coelho, BS; Paulette Cohrs, RN, BSN; Susan Green; April Hamilton, BS, CCRC; Jere Hamilton, BA; Eugene Leshchinskiy; Lindsey Munkwitz, BS; Loretta Rome, TRS; Terra Worley, BA; Kirstie Craul, RD, CDE; Sheila Smith, BS.
Baylor College of Medicine
John P. Foreyt, PhD1; Rebecca S. Reeves, DrPH, RD2; Henry Pownall, PhD3; Ashok Balasubramanyam, MBBS3; Peter Jones, MD3; Michele Burrington, RD, RN; Chu-Huang Chen, MD, PhD3; Allyson Clark Gardner, MS, RD; Molly Gee, MEd, RD; Sharon Griggs; Michelle Hamilton; Veronica Holley; Jayne Joseph, RD; Julieta Palencia, RN; Jennifer Schmidt; Carolyn White.
The University of Tennessee Health Science Center
University of Tennessee East: Karen C. Johnson, MD, MPH1; Carolyn Gresham, RN2; Stephanie Connelly, MD, MPH3; Amy Brewer, RD, MS; Mace Coday, PhD; Lisa Jones, RN; Lynne Lichtermann, RN, BSN; Shirley Vosburg, RD, MPH; J. Lee Taylor, MEd, MBA.
University of Tennessee Downtown: Abbas E. Kitabchi, PhD, MD1; Ebenezer Nyenwe, MD3; Helen Lambeth, RN, BSN2; Amy Brewer, MS, RD, LDN; Debra Clark, LPN; Andrea Crisler, MT; Debra Force, MS, RD, LDN; Donna Green, RN; Robert Kores, PhD.
University of Minnesota
Robert W. Jeffery, PhD1; Carolyn Thorson, CCRP2; John P. Bantle, MD3; J. Bruce Redmon, MD3; Richard S. Crow, MD3; Scott Crow, MD3; Susan K. Raatz, PhD, RD3; Kerrin Brelje, MPH, RD; Carolyne Campbell; Jeanne Carls, MEd; Tara Carmean-Mihm, BA; Julia Devonish, MS; Emily Finch, MA; Anna Fox, MA; Elizabeth Hoelscher, MPH, RD, CHES; La Donna James; Vicki A. Maddy, BS, RD; Therese Ockenden, RN; Birgitta I. Rice, MS, RPh, CHES; Tricia Skarphol, BS; Ann D. Tucker, BA; Mary Susan Voeller, BA; Cara Walcheck, BS, RD.
St. Luke’s Roosevelt Hospital Center
Xavier Pi-Sunyer, MD1; Jennifer Patricio, MS2; Stanley Heshka, PhD3; Carmen Pal, MD3; Lynn Allen, MD; Lolline Chong, BS, RD; Marci Gluck, PhD; Diane Hirsch, RNC, MS, CDE; Mary Anne Holowaty, MS, CN; Michelle Horowitz, MS, RD; Nancy Rau, MS, RD, CDE; Dori Brill Steinberg, BS.
University of Pennsylvania
Thomas A. Wadden, PhD1; Barbara J. Maschak-Carey, MSN, CDE2; Robert I. Berkowitz, MD3; Seth Braunstein, MD, PhD3; Gary Foster, PhD3; Henry Glick, PhD3; Shiriki Kumanyika, PhD, RD, MPH3; Stanley S. Schwartz, MD3; Michael Allen, RN; Yuliis Bell; Johanna Brock; Susan Brozena, MD; Ray Carvajal, MA; Helen Chomentowski; Canice Crerand, PhD; Renee Davenport; Andrea Diamond, MS, RD; Anthony Fabricatore, PhD; Lee Goldberg, MD; Louise Hesson, MSN, CRNP; Thomas Hudak, MS; Nayyar Iqbal, MD; LaShanda Jones-Corneille, PhD; Andrew Kao, MD; Robert Kuehnel, PhD; Patricia Lipschutz, MSN; Monica Mullen, RD, MPH.
University of Pittsburgh
John M. Jakicic, PhD1; David E. Kelley, MD1; Jacqueline Wesche-Thobaben, RN, BSN, CDE2; Lewis H. Kuller, MD, DrPH3; Andrea Kriska, PhD3; Amy D. Otto, PhD, RD, LDN3; Lin Ewing, PhD, RN3; Mary Korytkowski, MD3; Daniel Edmundowicz, MD3; Monica E. Yamamoto, DrPH, RD, FADA3; Rebecca Danchenko, BS; Barbara Elnyczky; David O. Garcia, MS; George A. Grove, MS; Patricia H. Harper, MS, RD, LDN; Susan Harrier, BS; Nicole L. Helbling, MS, RN; Diane Ives, MPH; Juliet Mancino, MS, RD, CDE, LDN; Anne Mathews, PhD, RD, LDN; Tracey Y. Murray, BS; Joan R. Ritchea; Susan Urda, BS, CTR; Donna L. Wolf, PhD.
The Miriam Hospital/Brown Medical School
Rena R. Wing, PhD1; Renee Bright, MS2; Vincent Pera, MD3; John Jakicic, PhD3; Deborah Tate, PhD3; Amy Gorin, PhD3; Kara Gallagher, PhD3; Amy Bach, PhD; Barbara Bancroft, RN, MS; Anna Bertorelli, MBA, RD; Richard Carey, BS; Tatum Charron, BS; Heather Chenot, MS; Kimberley Chula-Maguire, MS; Pamela Coward, MS, RD; Lisa Cronkite, BS; Julie Currin, MD; Maureen Daly, RN; Caitlin Egan, MS; Erica Ferguson, BS, RD; Linda Foss, MPH; Jennifer Gauvin, BS; Don Kieffer, PhD; Lauren Lessard, BS; Deborah Maier, MS; J.P. Massaro, BS; Tammy Monk, MS; Rob Nicholson, PhD; Erin Patterson, BS; Suzanne Phelan, PhD; Hollie Raynor, PhD, RD; Douglas Raynor, PhD; Natalie Robinson, MS, RD; Deborah Robles; Jane Tavares, BS.
The University of Texas Health Science Center at San Antonio
Steven M. Haffner, MD1; Helen P. Hazuda, PhD1; Maria G. Montez, RN, MSHP, CDE2; Carlos Lorenzo, MD3; Charles F. Coleman, MS, RD; Domingo Granado, RN; Kathy Hathaway, MS, RD; Juan Carlos Isaac, RC, BSN; Nora Ramirez, RN, BSN; Ronda Saenz, MS, RD.
VA Puget Sound Health Care System/University of Washington
Steven Kahn MB, ChB1; Brenda Montgomery, RN, MS, CDE2; Robert Knopp, MD3; Edward Lipkin, MD3; Dace Trence, MD3; Terry Barrett, BS; Joli Bartell, BA; Diane Greenberg, PhD; Anne Murillo, BS; Betty Ann Richmond, MEd; Jolanta Socha, BS; April Thomas, MPH, RD; Alan Wesley, BA.
Southwestern American Indian Center, Phoenix, Arizona and Shiprock, New Mexico
William C. Knowler, MD, DrPH1; Paula Bolin, RN, MC2; Tina Killean, BS2; Cathy Manus, LPN3; Jonathan Krakoff, MD3; Jeffrey M. Curtis, MD, MPH3; Justin Glass, MD3; Sara Michaels, MD3; Peter H. Bennett, MB, FRCP3; Tina Morgan3; Shandiin Begay, MPH; Paul Bloomquist, MD; Teddy Costa, BS; Bernadita Fallis, RN, RHIT, CCS; Jeanette Hermes, MS, RD; Diane F. Hollowbreast; Ruby Johnson; Maria Meacham, BSN, RN, CDE; Julie Nelson, RD; Carol Percy, RN; Patricia Poorthunder; Sandra Sangster; Nancy Scurlock, MSN, ANP-C, CDE; Leigh A. Shovestull, RD, CDE; Janelia Smiley; Katie Toledo, MS, LPC; Christina Tomchee, BA; Darryl Tonemah, PhD.
University of Southern California
Anne Peters, MD1; Valerie Ruelas, MSW, LCSW2; Siran Ghazarian Sengardi, MD2; Kathryn (Mandy) Graves Hillstrom, EdD, RD, CDE; Kati Konersman, MA, RD, CDE; Sara Serafin-Dokhan.
Coordinating Center
Wake Forest University
Mark A. Espeland, PhD1; Judy L. Bahnson, BA, CCRP3; Lynne E. Wagenknecht, DrPH3; David Reboussin, PhD3; W. Jack Rejeski, PhD3; Alain G. Bertoni, MD, MPH3; Wei Lang, PhD3; Michael S. Lawlor, PhD3; David Lefkowitz, MD3; Gary D. Miller, PhD3; Patrick S. Reynolds, MD3; Paul M. Ribisl, PhD3; Mara Vitolins, DrPH3; Haiying Chen, PhD3; Delia S. West, PhD3; Lawrence M. Friedman, MD3; Brenda L. Craven, MS, CCRP2; Kathy M. Dotson, BA2; Amelia Hodges, BS, CCRP2; Carrie C. Williams, MA, CCRP2; Andrea Anderson, MS; Jerry M. Barnes, MA; Mary Barr; Daniel P. Beavers, PhD; Tara Beckner; Cralen Davis, MS; Thania Del Valle-Fagan, MD; Patricia A. Feeney, MS; Candace Goode; Jason Griffin, BS; Lea Harvin, BS; Patricia Hogan, MS; Sarah A. Gaussoin, MS; Mark King, BS; Kathy Lane, BS; Rebecca H. Neiberg, MS; Michael P. Walkup, MS; Karen Wall, AAS; Terri Windham.
Central Resources Centers
DXA Reading Center, University of California at San Francisco
Michael Nevitt, PhD1; Ann Schwartz, PhD2; John Shepherd, PhD3; Michaela Rahorst; Lisa Palermo, MS, MA; Susan Ewing, MS; Cynthia Hayashi; Jason Maeda, MPH.
Central Laboratory, Northwest Lipid Metabolism and Diabetes Research Laboratories
Santica M. Marcovina, PhD, ScD1; Jessica Chmielewski2; Vinod Gaur, PhD4.
ECG Reading Center, EPICARE, Wake Forest University School of Medicine
Elsayed Z. Soliman, MD, MSc, MS1; Ronald J. Prineas, MD, PhD1; Charles Campbell2; Zhu-Ming Zhang, MD3; Teresa Alexander; Lisa Keasler; Susan Hensley; Yabing Li, MD.
Diet Assessment Center, University of South Carolina, Arnold School of Public Health, Center for Research in Nutrition and Health Disparities
Robert Moran, PhD1.
Hall-Foushee Communications, Inc
Richard Foushee, PhD; Nancy J. Hall, MA.
Federal Sponsors
National Institute of Diabetes and Digestive and Kidney Diseases
Mary Evans, PhD; Barbara Harrison, MS; Van S. Hubbard, MD, PhD; Susan Z. Yanovski, MD; Robert Kuczmarski, PhD.
National Heart, Lung, and Blood Institute
Lawton S. Cooper, MD, MPH; Peter Kaufman, PhD, FABMR.
Centers for Disease Control and Prevention
Edward W. Gregg, PhD; David F. Williamson, PhD; Ping Zhang, PhD.
Footnotes
Principal investigator.
Program coordinator.
Co-investigator.
All other Look AHEAD staffs are listed alphabetically by site.
Look AHEAD Genetic Ancillary Study Clinical Registry Number: NCT01270763; Look AHEAD Clinical Registry Number: NCT00017953.
Disclosure Statement
The authors have no conflict of interest to declare.
References
- 1.NHLBI: Clinical guidelines on the identification, evaluation, and treatment of overweight and obesity in adults: the evidence report. Obes Res. 1998;6:51S–210S. [PubMed] [Google Scholar]
- 2.Ogden CL, Carroll MD, Curtin LR, McDowell MA, Tabak CJ, Flegal KM. Prevalence of overweight and obesity in the United States, 1999–2004. JAMA. 2006;295:1549–1555. doi: 10.1001/jama.295.13.1549. [DOI] [PubMed] [Google Scholar]
- 3.Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393–403. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Diabetes Prevention Program Research Group: The Diabetes Prevention Program. Design and methods for a clinical trial in the prevention of type 2 diabetes. Diabetes Care. 1999;22:623–634. doi: 10.2337/diacare.22.4.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, Nathan DM. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393–403. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wing RR. Long-term effects of a lifestyle intervention on weight and cardiovascular risk factors in individuals with type 2 diabetes mellitus: four-year results of the Look AHEAD trial. Arch Intern Med. 2010;170:1566–1575. doi: 10.1001/archinternmed.2010.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stunkard AJ, Foch TT, Hrubec Z. A twin study of human obesity. JAMA. 1986;256:51–54. [PubMed] [Google Scholar]
- 8.Maes HH, Neale MC, Eaves LJ. Genetic and environmental factors in relative body weight and human adiposity. Behav Genet. 1997;27:325–351. doi: 10.1023/a:1025635913927. [DOI] [PubMed] [Google Scholar]
- 9.Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42:937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thorleifsson G, Walters GB, Gudbjartsson DF, Steinthorsdottir V, Sulem P, et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat Genet. 2009;41:18–24. doi: 10.1038/ng.274. [DOI] [PubMed] [Google Scholar]
- 11.Willer CJ, Speliotes EK, Loos RJ, Li S, Lindgren CM, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41:25–34. doi: 10.1038/ng.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McAllister EJ, Dhurandhar NV, Keith SW, Aronne LJ, Barger J, Baskin M, Benca RM, Biggio J, Boggiano MM, Eisenmann JC, Elobeid M, Fontaine KR, Gluckman P, Hanlon EC, Katzmarzyk P, Pietrobelli A, Redden DT, Ruden DM, Wang C, Waterland RA, Wright SM, Allison DB. Ten putative contributors to the obesity epidemic. Crit Rev Food Sci Nutr. 2009;49:868–913. doi: 10.1080/10408390903372599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCaffery JM, Papandonatos GD, Bond DS, Lyons MJ, Wing RR. Gene × environment interaction of vigorous exercise and body mass index among male Vietnam-era twins. Am J Clin Nutr. 2009;89:1011–1018. doi: 10.3945/ajcn.2008.27170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lajunen HR, Kaprio J, Rose RJ, Pulkkinen L, Silventoinen K. Genetic and environmental influences on BMI from late childhood to adolescence are modified by parental education. Obesity (Silver Spring) 2012;20:583–589. doi: 10.1038/oby.2011.304. [DOI] [PubMed] [Google Scholar]
- 15.Qi Q, Chu AY, Kang JH, Jensen MK, Curhan GC, Pasquale LR, Ridker PM, Hunter DJ, Willett WC, Rimm EB, Chasman DI, Hu FB, Qi L. Sugar-sweetened beverages and genetic risk of obesity. N Engl J Med. 2012;367:1387–1396. doi: 10.1056/NEJMoa1203039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qi Q, Li Y, Chomistek AK, Kang JH, Curhan GC, Pasquale LR, Willett WC, Rimm EB, Hu FB, Qi L. Television watching, leisure time physical activity, and the genetic predisposition in relation to body mass index in women and men. Circulation. 2012;126:1821–1827. doi: 10.1161/CIRCULATIONAHA.112.098061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thomas D. Gene–environment-wide association studies: emerging approaches. Nat Rev Genet. 2010;11:259–272. doi: 10.1038/nrg2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keating BJ, Tischfield S, Murray SS, Bhangale T, Price TS, et al. Concept, design and implementation of a cardiovascular genecentric 50 k SNP array for large-scale genomic association studies. PLoS One. 2008;3:e3583. doi: 10.1371/journal.pone.0003583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ryan DH, Espeland MA, Foster GD, Haffner SM, Hubbard VS, Johnson KC, Kahn SE, Knowler WC, Yanovski SZ. Look AHEAD (Action for Health in Diabetes): design and methods for a clinical trial of weight loss for the prevention of cardiovascular disease in type 2 diabetes. Control Clin Trials. 2003;24:610–628. doi: 10.1016/s0197-2456(03)00064-3. [DOI] [PubMed] [Google Scholar]
- 20.Bray G, Gregg E, Haffner S, Pi-Sunyer XF, WagenKnecht LE, Walkup M, Wing R. Baseline characteristics of the randomised cohort from the Look AHEAD (Action for Health in Diabetes) study. Diab Vasc Dis Res. 2006;3:202–215. doi: 10.3132/dvdr.2006.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pi-Sunyer X, Blackburn G, Brancati FL, Bray GA, Bright R, et al. Reduction in weight and cardiovascular disease risk factors in individuals with type 2 diabetes: one-year results of the look AHEAD trial. Diabetes Care. 2007;30:1374–1383. doi: 10.2337/dc07-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delahanty LM, Pan Q, Jablonski KA, Watson KE, McCaffery JM, Shuldiner A, Kahn SE, Knowler WC, Florez JC, Franks PW. Genetic predictors of weight loss and weight regain after intensive lifestyle modification, metformin treatment, or standard care in the Diabetes Prevention Program. Diabetes Care. 2012;35:363–366. doi: 10.2337/dc11-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 24.McCaffery JM, Papandonatos GD, Huggins GS, Peter I, Kahn SE, Knowler WC, Hudnall GE, Lipkin EW, Kitabchi AE, Wagenknecht LE, Wing RR. FTO predicts weight regain in the Look AHEAD clinical trial. Int J Obes (Lond) 2013 doi: 10.1038/ijo.2013.54. E-pub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.TIBCO Software I. TIBCO Spotfire SPLUS 8.2 for Solaris/Linux User’s Guide. Seattle, WA: TIBCO Software, Inc; 2010. [Google Scholar]
- 26.McCaffery JM, Papandonatos GD, Peter I, Huggins GS, Raynor HA, Delahanty LM, Cheskin LJ, Balasubramanyam A, Wagenknecht LE, Wing RR. Obesity susceptibility loci and dietary intake in the Look AHEAD Trial. Am J Clin Nutr. 2012;95:1477–1486. doi: 10.3945/ajcn.111.026955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li J, Ji L. Adjusting multiple testing in multilocus analyses using the eigenvalues of a correlation matrix. Heredity. 2005;95:221–227. doi: 10.1038/sj.hdy.6800717. [DOI] [PubMed] [Google Scholar]
- 28.Sidak Z. Rectangular confidence region for the means of multivariate normal distributions. J Am Stat Assoc. 1967;62:626–633. [Google Scholar]
- 29.Dabney A, Storey JD. Q-value estimation for false discovery rate control. R package version 1.30.0 [Google Scholar]
- 30.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Franks PW, Jablonski KA, Delahanty L, Hanson RL, Kahn SE, Altshuler D, Knowler WC, Florez JC. The Pro12Ala variant at the peroxisome proliferator-activated receptor gamma gene and change in obesity-related traits in the Diabetes Prevention Program. Diabetologia. 2007;50:2451–2460. doi: 10.1007/s00125-007-0826-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lindi VI, Uusitupa MI, Lindstrom J, Louheranta A, Eriksson JG, Valle TT, Hamalainen H, Ilanne-Parikka P, Keinanen-Kiukaanniemi S, Laakso M, Tuomilehto J. Association of the Pro12Ala polymorphism in the PPAR-gamma2 gene with 3-year incidence of type 2 diabetes and body weight change in the Finnish Diabetes Prevention Study. Diabetes. 2002;51:2581–2586. doi: 10.2337/diabetes.51.8.2581. [DOI] [PubMed] [Google Scholar]
- 33.Franks PW, Jablonski KA, Delahanty LM, McAteer JB, Kahn SE, Knowler WC, Florez JC. Assessing gene-treatment interactions at the FTO and INSIG2 loci on obesity-related traits in the Diabetes Prevention Program. Diabetologia. 2008;51:2214–2223. doi: 10.1007/s00125-008-1158-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang X, Qi Q, Zhang C, Smith SR, Hu FB, Sacks FM, Bray GA, Qi L. FTO genotype and 2-year change in body composition and fat distribution in response to weight-loss diets: the POUNDS LOST Trial. Diabetes. 2012;61:3005–3011. doi: 10.2337/db11-1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stieger B. Recent insights into the function and regulation of the bile salt export pump (ABCB11) Curr Opin Lipidol. 2009;20:176–181. doi: 10.1097/MOL.0b013e32832b677c. [DOI] [PubMed] [Google Scholar]
- 36.Jansen PL, Strautnieks SS, Jacquemin E, Hadchouel M, Sokal EM, Hooiveld GJ, Koning JH, De Jager-Krikken A, Kuipers F, Stellaard F, Bijleveld CM, Gouw A, Van Goor H, Thompson RJ, Muller M. Hepatocanalicular bile salt export pump deficiency in patients with progressive familial intrahepatic cholestasis. Gastroenterology. 1999;117:1370–1379. doi: 10.1016/s0016-5085(99)70287-8. [DOI] [PubMed] [Google Scholar]
- 37.Strautnieks SS, Bull LN, Knisely AS, Kocoshis SA, Dahl N, Arnell H, Sokal E, Dahan K, Childs S, Ling V, Tanner MS, Kagalwalla AF, Nemeth A, Pawlowska J, Baker A, Mieli-Vergani G, Freimer NB, Gardiner RM, Thompson RJ. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet. 1998;20:233–238. doi: 10.1038/3034. [DOI] [PubMed] [Google Scholar]
- 38.Noe J, Kullak-Ublick GA, Jochum W, Stieger B, Kerb R, Haberl M, Mullhaupt B, Meier PJ, Pauli-Magnus C. Impaired expression and function of the bile salt export pump due to three novel ABCB11 mutations in intrahepatic cholestasis. J Hepatol. 2005;43:536–543. doi: 10.1016/j.jhep.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 39.van Mil SW, van der Woerd WL, van der Brugge G, Sturm E, Jansen PL, Bull LN, van den Berg IE, Berger R, Houwen RH, Klomp LW. Benign recurrent intrahepatic cholestasis type 2 is caused by mutations in ABCB11. Gastroenterology. 2004;127:379–384. doi: 10.1053/j.gastro.2004.04.065. [DOI] [PubMed] [Google Scholar]
- 40.Chambers JC, Zhang W, Sehmi J, Li X, Wass MN, et al. Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat Genet. 2011;43:1131–1138. doi: 10.1038/ng.970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang R, Salem M, Yousef IM, Tuchweber B, Lam P, Childs SJ, Helgason CD, Ackerley C, Phillips MJ, Ling V. Targeted inactivation of sister of P-glycoprotein gene (spgp) in mice results in nonprogressive but persistent intrahepatic cholestasis. Proc Natl Acad Sci USA. 2001;98:2011–2016. doi: 10.1073/pnas.031465498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Henkel AS, Kavesh MH, Kriss MS, Dewey AM, Rinella ME, Green RM. Hepatic overexpression of abcb11 promotes hypercholesterolemia and obesity in mice. Gastroenterology. 2011;141:1404–1411. 1411.e1–e2. doi: 10.1053/j.gastro.2011.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andreotti G, Menashe I, Chen J, Chang SC, Rashid A, Gao YT, Han TQ, Sakoda LC, Chanock S, Rosenberg PS, Hsing AW. Genetic determinants of serum lipid levels in Chinese subjects: a population-based study in Shanghai, China. Eur J Epidemiol. 2009;24:763–774. doi: 10.1007/s10654-009-9402-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kraja AT, Vaidya D, Pankow JS, Goodarzi MO, Assimes TL, et al. A bivariate genomewide approach to metabolic syndrome: STAMPEED Consortium. Diabetes. 2011;60:1329–1339. doi: 10.2337/db10-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bouatia-Naji N, Rocheleau G, Van Lommel L, Lemaire K, Schuit F, et al. A polymorphism within the G6PC2 gene is associated with fasting plasma glucose levels. Science. 2008;320:1085–1088. doi: 10.1126/science.1156849. [DOI] [PubMed] [Google Scholar]
- 46.Gomez-Ambrosi J, Rodriguez A, Catalan V, Fruhbeck G. The bone-adipose axis in obesity and weight loss. Obes Surg. 2008;18:1134–1143. doi: 10.1007/s11695-008-9548-1. [DOI] [PubMed] [Google Scholar]
- 47.An JJ, Han DH, Kim DM, Kim SH, Rhee Y, Lee EJ, Lim SK. Expression and regulation of osteoprotegerin in adipose tissue. Yonsei Med J. 2007;48:765–772. doi: 10.3349/ymj.2007.48.5.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Holecki M, Zahorska-Markiewicz B, Janowska J, Nieszporek T, Wojaczynska-Stanek K, Zak-Golab A, Wiecek A. The influence of weight loss on serum osteoprotegerin concentration in obese perimenopausal women. Obesity (Silver Spring) 2007;15:1925–1929. doi: 10.1038/oby.2007.229. [DOI] [PubMed] [Google Scholar]
- 49.Cheverud JM, Ehrich TH, Hrbek T, Kenney JP, Pletscher LS, Semenkovich CF. Quantitative trait loci for obesity- and diabetes-related traits and their dietary responses to high-fat feeding in LGXSM recombinant inbred mouse strains. Diabetes. 2004;53:3328–3336. doi: 10.2337/diabetes.53.12.3328. [DOI] [PubMed] [Google Scholar]
- 50.Zhao LJ, Guo YF, Xiong DH, Xiao P, Recker RR, Deng HW. Is a gene important for bone resorption a candidate for obesity? An association and linkage study on the RANK (receptor activator of nuclear factor-kappaB) gene in a large Caucasian sample. Hum Genet. 2006;120:561–570. doi: 10.1007/s00439-006-0243-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hohjoh H, Takasu M, Shishikura K, Takahashi Y, Honda Y, Tokunaga K. Significant association of the arylalkylamine N-acetyl-transferase (AA-NAT) gene with delayed sleep phase syndrome. Neurogenetics. 2003;4:151–153. doi: 10.1007/s10048-002-0141-9. [DOI] [PubMed] [Google Scholar]
- 52.Cappuccio FP, Taggart FM, Kandala NB, Currie A, Peile E, Stranges S, Miller MA. Meta-analysis of short sleep duration and obesity in children and adults. Sleep. 2008;31:619–626. doi: 10.1093/sleep/31.5.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jessen N, An D, Lihn AS, Nygren J, Hirshman MF, Thorell A, Goodyear LJ. Exercise increases TBC1D1 phosphorylation in human skeletal muscle. Am J Physiol Endocrinol Metab. 2011;301:E164–E171. doi: 10.1152/ajpendo.00042.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meyre D, Farge M, Lecoeur C, Proenca C, Durand E, Allegaert F, Tichet J, Marre M, Balkau B, Weill J, Delplanque J, Froguel P. R125W coding variant in TBC1D1 confers risk for familial obesity and contributes to linkage on chromosome 4p14 in the French population. Hum Mol Genet. 2008;17:1798–1802. doi: 10.1093/hmg/ddn070. [DOI] [PubMed] [Google Scholar]
- 55.Stone S, Abkevich V, Russell DL, Riley R, Timms K, Tran T, Trem D, Frank D, Jammulapati S, Neff CD, Iliev D, Gress R, He G, Frech GC, Adams TD, Skolnick MH, Lanchbury JS, Gutin A, Hunt SC, Shattuck D. TBC1D1 is a candidate for a severe obesity gene and evidence for a gene/gene interaction in obesity predisposition. Hum Mol Genet. 2006;15:2709–2720. doi: 10.1093/hmg/ddl204. [DOI] [PubMed] [Google Scholar]
- 56.Looker HC, Knowler WC, Hanson RL. Changes in BMI and weight before and after the development of type 2 diabetes. Diabetes Care. 2001;24:1917–1922. doi: 10.2337/diacare.24.11.1917. [DOI] [PubMed] [Google Scholar]
- 57.Divoux A, Clement K. Architecture and the extracellular matrix: the still unappreciated components of the adipose tissue. Obes Rev. 2011;12:e494–e503. doi: 10.1111/j.1467-789X.2010.00811.x. [DOI] [PubMed] [Google Scholar]
- 58.Christiaens V, Lijnen HR. Role of the fibrinolytic and matrix metalloproteinase systems in development of adipose tissue. Arch Physiol Biochem. 2006;112:254–259. doi: 10.1080/13813450601093567. [DOI] [PubMed] [Google Scholar]
- 59.de Meijer VE, Sverdlov DY, Le HD, Popov Y, Puder M. Tissue-specific differences in inflammatory infiltrate and matrix metalloproteinase expression in adipose tissue and liver of mice with diet-induced obesity. Hepatol Res. 2012;42:601–610. doi: 10.1111/j.1872-034X.2011.00960.x. [DOI] [PubMed] [Google Scholar]
- 60.Gomez DE, Alonso DF, Yoshiji H, Thorgeirsson UP. Tissue inhibitors of metalloproteinases: structure, regulation and biological functions. Eur J Cell Biol. 1997;74:111–122. [PubMed] [Google Scholar]
- 61.Mohammed FF, Smookler DS, Taylor SE, Fingleton B, Kassiri Z, Sanchez OH, English JL, Matrisian LM, Au B, Yeh WC, Khokha R. Abnormal TNF activity in Timp3−/− mice leads to chronic hepatic inflammation and failure of liver regeneration. Nat Genet. 2004;36:969–977. doi: 10.1038/ng1413. [DOI] [PubMed] [Google Scholar]
- 62.Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol. 2006;7:885–896. doi: 10.1038/nrm2066. [DOI] [PubMed] [Google Scholar]
- 63.Clement K, Viguerie N, Poitou C, Carette C, Pelloux V, Curat CA, Sicard A, Rome S, Benis A, Zucker JD, Vidal H, Laville M, Barsh GS, Basdevant A, Stich V, Cancello R, Langin D. Weight loss regulates inflammation-related genes in white adipose tissue of obese subjects. FASEB J. 2004;18:1657–1669. doi: 10.1096/fj.04-2204com. [DOI] [PubMed] [Google Scholar]
- 64.Ii H, Hatakeyama S, Tsutsumi K, Sato T, Akiba S. Group IVA phospholipase A2 is associated with the storage of lipids in adipose tissue and liver. Prostaglandins Other Lipid Mediat. 2008;86:12–17. doi: 10.1016/j.prostaglandins.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 65.Mancuso DJ, Sims HF, Yang K, Kiebish MA, Su X, Jenkins CM, Guan S, Moon SH, Pietka T, Nassir F, Schappe T, Moore K, Han X, Abumrad NA, Gross RW. Genetic ablation of calcium-independent phospholipase A2gamma prevents obesity and insulin resistance during high fat feeding by mitochondrial uncoupling and increased adipocyte fatty acid oxidation. J Biol Chem. 2010;285:36495–36510. doi: 10.1074/jbc.M110.115766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Song H, Wohltmann M, Bao S, Ladenson JH, Semenkovich CF, Turk J. Mice deficient in group VIB phospholipase A2 (iPLA2gamma) exhibit relative resistance to obesity and metabolic abnormalities induced by a Western diet. Am J Physiol Endocrinol Metab. 2010;298:E1097–E1114. doi: 10.1152/ajpendo.00780.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lijnen HR, Maquoi E, Hansen LB, Van Hoef B, Frederix L, Collen D. Matrix metalloproteinase inhibition impairs adipose tissue development in mice. Arterioscler Thromb Vasc Biol. 2002;22:374–379. doi: 10.1161/hq0302.104522. [DOI] [PubMed] [Google Scholar]
- 68.Shi C, Sakuma M, Mooroka T, Liscoe A, Gao H, Croce KJ, Sharma A, Kaplan D, Greaves DR, Wang Y, Simon DI. Down-regulation of the forkhead transcription factor Foxp1 is required for monocyte differentiation and macrophage function. Blood. 2008;112:4699–4711. doi: 10.1182/blood-2008-01-137018. [DOI] [PMC free article] [PubMed] [Google Scholar]