

Abstract
We tested whether cardiac myosin binding protein-C (cMyBP-C) affects myosin cross-bridge kinetics in the two cardiac myosin heavy chain (MyHC) isoforms. Mice lacking cMyBP-C (t/t) and transgenic controls (WTt/t) were fed L-thyroxine (T4) to induce 90/10% expression of α/β-MyHC. Non-transgenic (NTG) and t/t mice were fed 6-n-propyl-2-thiouracil (PTU) to induce 100% expression of β-MyHC. Ca2+-activated, chemically-skinned myocardium underwent length perturbation analysis with varying [MgATP] to estimate the MgADP release rate (k−ADP) and MgATP binding rate (k+ATP). Values for k−ADP were not significantly different between t/tT4 (102.2±7.0 s−1) and WTt/tT4 (91.3±8.9 s−1), but k+ATP was lower in t/tT4 (165.9±12.5 mM−1 s−1) compared to WTt/tT4 (298.6±15.7 mM−1 s−1, P<0.01). In myocardium expressing β-MyHC, values for k−ADP were higher in t/tPTU (24.8±1.0 s−1) compared to NTGPTU (15.6±1.3 s−1, P<0.01), and k+ATP was not different. At saturating [MgATP], myosin detachment rate approximates k−ADP, and detachment rate decreased as sarcomere length (SL) was increased in both t/tT4 and WTt/tT4 with similar sensitivities to SL. In myocardium expressing β-MyHC, detachment rate decreased more as SL increased in t/tPTU (21.5±1.3 s−1 at 2.2 μm and 13.3±0.9 s−1 at 3.3 μm) compared to NTGPTU (15.8±0.3 s−1 at 2.2 μm and 10.9±0.3 s−1 at 3.3 μm) as detected by repeated-measures ANOVA (P<0.01). These findings suggest that cMyBP-C reduces MgADP release rate for β-MyHC, but not for α-MyHC, even as the number of cMyBP-C that overlap with the thin filament is reduced to zero. Therefore, cMyBP-C appears to affect β-MyHC kinetics independent of its interaction with the thin filament.
Keywords: cross-bridge, time-on, transgenic
Introduction
Cardiac myosin binding protein-C (cMyBP-C) augments thick filament structural integrity (Zoghbi et al., 2008) and stiffness (Nyland et al., 2009) and can bind actin, myosin and titin (Bhuiyan et al., 2012; Freiburg and Gautel, 1996). These structural characteristics of cMyBP-C play a role in stiffening the contracting myocardium, facilitating cardiac relaxation, prolonging myosin cross-bridge lifetime ton and slowing unloaded shortening velocity, as demonstrated at the whole heart (Carrier et al., 2004; Harris et al., 2002; McConnell et al., 1999), myocardial (Korte et al., 2003; Palmer et al., 2011; Stelzer et al., 2006) and molecular levels (Previs et al., 2012). It is not yet known if cMyBP-C influences specific nucleotide-dependent transition rates in the myosin cross-bridge cycle. Given previous findings that myosin ATPase, loaded and unloaded shortening velocity, myosin cross-bridge detachment rate and MgADP release rate differ by roughly four-fold between cardiac α- and β-MyHC isoforms in the context of an intact myofilament lattice (Korte et al., 2005; Rundell et al., 2005; Wang et al., 2013), rather than the two-fold difference observed using isolated cardiac myosin (Palmiter et al., 1999; Pereira et al., 2001), we hypothesized that the structural protein cMyBP-C would influence cross-bridge kinetics in a myosin isoform-dependent manner.
The present study examines myosin cross-bridge rates of MgADP release and MgATP binding in left ventricular (LV) myocardial strips isolated from mice lacking cMyBP-C vs. controls, which have been matched for MyHC isoform using thyroid hormone manipulation. We also examined the sensitivity of myosin cross-bridge detachment rate to sarcomere length in the two MyHC isoforms. Our results suggest that the presence of cMyBP-C in the myofilament lattice reduces the MgADP release rate in the β-MyHC isoform but not in the α-MyHC and that cMyBP-C has this effect on the β-MyHC isoform as the sarcomere lengthens beyond where cMyBP-C can interact with the thin filament (Pfuhl and Gautel, 2012). The possible structural bases of these effects are discussed.
Materials and Methods
Animal models
All procedures were reviewed and approved by the Institutional Animal Care and Use Committees of The University of Vermont College of Medicine and The University of Cincinnati Children’s Hospital and complied with the Guide for the Use and Care of Laboratory Animals published by the National Institutes of Health. All mice were of the FVB background. Male, transgenic mice lacking cMyBP-C (t/t) or expressing full length wild-type cMyBP-C in a t/t background (WTt/t) were acquired from University of Cincinnati (Sadayappan et al., 2005). Male, non-transgenic (NTG) mice were acquired from Charles River (Wilmington, MA). WTt/t (n=4) and t/t (n=5) mice were fed 3 mg/kg L-thyroxine (T4) for 10 days, which emulated hyperthyroidism to upregulate α-MyHC expression in the LV for both genotypes (Palmer et al., 2011). NTG (n=5) and t/t (n=5) mice were fed an iodine-deficient 0.15% propylthiouracil (PTU) diet (Harlan Teklad, Indianapolis, IN) for at least 12 weeks, which resulted in hypothyroidism and an upregulation of β-MyHC expressed in the LV (Palmer et al., 2004; Wang et al., 2013). Mice were anaesthetized by inhalation of isoflurane (1.5–3% mg/kg), following which hearts were immediately excised and placed in preoxygenated (95% O2–5% CO2) Krebs solution at room temperature. LVs were placed in 10% paraformaline and morphology was assessed by H&E and Mason’s trichrome stain (American Histolabs, Gaithersburg, MD).
Myosin isoform content
Myosin isoform content in the LV was determined by gel electrophoresis (Reiser and Kline, 1998) using Fluormax-2 Imaging analysis (Bio-Rad, Hercules CA). Total optical densities of bands were corrected for background using ImageJ v1.38 (NIH, USA).
Solutions
Chemicals and reagents were obtained from Sigma Corp. (St. Louis, MO) unless otherwise noted. Krebs solution contained (mmol/L) 137 Na+, 153 Cl−, 5.4 K+, 0.2 Ca2+, 1.3 Mg2+, 10 Glucose, 10 Hepes, 30 BDM, pH 7.4. Solutions for skinned strips were formulated by solving equations describing ionic equilibria (Godt and Lindley, 1982). Relaxing solution: pCa 8.0 (pCa= log10[Ca2+]), 5 EGTA, 5 MgATP, 1 Mg2+, 35 phosphocreatine (PCr), 300 U/mL creatine kinase (CK), ionic strength 200, pH 7.0. Activating solution: same as relaxing with pCa 4.0. Rigor solution: same as relaxing with pCa 4.8 and 0 MgATP. Skinning solution: same as relaxing without CK, with 1% Triton-X100 wt/vol and 50% glycerol wt/vol. Storage solution: same as skinning without Triton, with 10 μg/mL leupeptin. Alkaline phosphatase (AP) solution: same as relaxing solution with 6 U/mL recombinant AP from E-coli (P-4252).
Skinned myoc ardial strips
Skinned papillary muscle strips were prepared and studied as previously described (Wang et al., 2013). Immediately before mechanical analysis, all strips were incubated for 10 min in AP solution at room temperature. The combination of extended BDM incubation and AP pretreatment of experimental and control groups provides a useful approach to better match the phosphorylation status of sarcomeric proteins by reducing any differential phosphorylation profiles at troponin-I serines-21/22 and myosin regulatory light chain serine-19, as demonstrated by phospho-specific antibodies (Wang et al., 2013). Strips were mounted between a piezoelectric motor (P841.60, Physik Instrumente, Auburn, MA) and a strain gauge (AE801, Kronex, Walnut Creek, CA), lowered into a 30μL droplet of relaxing solution maintained at 17 °C, and stretched to 2.2 μm sarcomere length (SL) as measured by digital Fourier Transform (IonOptix Corp, Milton, MA). Strips were calcium activated to pCa 4.8, following which either rigor solution was exchanged for activating solution to lower [MgATP] from 5 to <0.01 mM or SL was increased from 2.2 to 3.3 μm. In the latter case, SL was visualized up to 2.8 μm and then stretched by 15% to reach 3.3 μm, which could not be reliably visualized. At least two strips per heart underwent either the rigor titration or SL extension protocol, and data were averaged for each heart.
Mechanical system analysis
Sinusoidal length perturbations of amplitude 0.125% strip length were applied at 0.125–100 Hz (Wang et al., 2013). Elastic and viscous moduli as a function of angular frequency ω, E(ω) and V(ω), were used to define the complex modulus, Y(ω)=E(ω) + iV(ω) where i = √−1. Fitting Eq. 1 to the entire frequency range of moduli values provided estimates of six model parameters (A, k, B, 2πb, C, 2πc).
| (Equation 1) |
The A-term in Eq. 1 reflects the visco-elastic mechanical response of passive elements in the muscle. In our interpretation, parameter A represents the combined mechanical stiffness of the parallel elastic elements, the myofilaments and the number of strongly bound cross-bridges, and k describes the degree of the viscoelasticity in the response, where k→0 is a purely elastic response and k→1 is a purely viscous response. The B- and C-terms of Eq. 1 reflect enzymatically driven myosin cross-bridge formation in activated muscle. Parameters B and C reflect the number of cross-bridges formed × their mean stiffness (or total myosin cross-bridges available × duty ratio × mean stiffness), and the rate parameters 2πb and 2πc reflect cross-bridge kinetics sensitive to biochemical perturbations known to affect enzymatic activity, such as [MgATP], [MgADP], or [Pi]. The B-term underlies processes of cross-bridge attachment or force generation, such that 2πb describes rate of force generation and has been proposed to be due to cross-bridge recruitment (Campbell et al., 2004) or the characteristic rates of myosin isomerization including Pi-dependent cross-bridge detachment (Kawai and Halvorson, 1991; Zhao and Kawai, 1993). The C-term underlies processes of cross-bridge detachment or force decay, such that 2πc describes to the rate of cross-bridge detachment, which is inversely related to the average myosin attachment time, ton=(2πc)−1 (Wang et al., 2013).
Myosin enzyme kinetics
Assuming that the myosin ton consists almost entirely of the time periods of myosin ADP and rigor states, the myosin detachment rate, 2πc, is therefore related to MgATP-dependent parameters of myosin detachment as explained in detail in Tyska and Warshaw and implemented in our previous publication (Tyska and Warshaw, 2002; Wang et al., 2013):
| (Equation 2) |
where k−ADP = rate of MgADP release and the asymptotic, maximal myosin detachment rate (s−1), k+ATP = rate of MgATP binding per MgATP concentration (M−1 s−1), and the ratio (k−ADP/k+ATP) = concentration of MgATP producing half the maximal myosin detachment rate ([MgATP]50). One or two strips per heart underwent these measured of MgADP and MgATP kinetics.
Statistical analysis
Multiple measurements from the same heart were averaged to provide a single measure for that heart. All values are mean±SE, where n represents the number of hearts. Moduli data were fit to Eq. 1 using IDL (v. 7, Exelis Visual Information Solutions, Inc.; Boulder, CO) as previously described (Wang et al., 2013). The differential sensitivity of myosin detachment rate to varying sarcomere length was analyzed using repeated-measures ANOVA with sarcomere length as within-subject variable and genotype as between-subject variable. Statistical analyses were performed using SPSS (v.20.0, IBM SPSS Statistics, Chicago, IL).
Results
Cardiac morphology and myosin isoform profiles
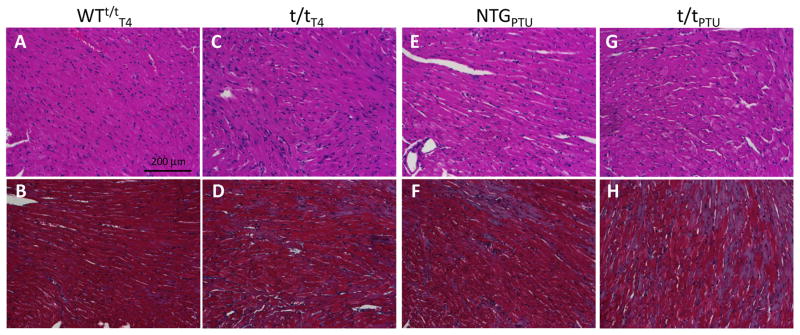
Qualitative visual assessment of LV morphology suggests the effects of a 10 day T4 diet did not change LV structure in the WTt/tT4 compared to a normal mouse LV evidenced by the relatively well organized nuclei (stained blue by H&E, Fig. 1A) and myocytes (stained red by H&E, Fig. 1A) with no fibrosis (stained blue by trichrome, Fig. 1B). There was also significant myocyte disarray and fibrosis in the t/tT4 compared to WTt/tT4 mice (Fig. 1A–D). These findings are consistent with those reported for cMyBP-C knockout mice (Carrier et al., 2004; Harris et al., 2002; McConnell et al., 1999) and WTt/t (Sadayappan et al., 2005).
Figure 1. Cardiac morphology assessed by H&E and Mason’s trichrome staining.

The WTt/tT4 shows normal myocyte disarray and fibrosis (A and B), comparable to that observed for a normal mouse, while the t/tT4 myocardium displayed disarray and fibrosis (C and D) relative to the WTt/tT4. These observations in the T4-fed mice were similar to those expected without any special diet. After 12 weeks of hypothyroidism due to PTU diet, however, both NTGPTU (E and F) and t/tPTU (G and H) show significant fibrosis as indicated by blue color in the trichrome stain (F and H).
The NTGPTU hearts showed limited myocyte disarray (Fig. 1E), whereas the t/tPTU hearts display a moderate level of myocyte disarray due to the loss of cMyBP-C (Fig. 1G). Both NTGPTU and t/tPTU also demonstrate cardiac fibrosis, as indicated by blue stain in Fig. 1F & 1H, observed after 12 weeks of PTU diet and subsequent hypothyroidism. These findings suggest that secondary responses to the lack of cMyBP-C led to some differences in myocyte disarray.
Consistent with our previous observations (Palmer et al., 2011), regimens of T4 diet successfully resulted in complimentary LV α-MyHC content in the WTt/tT4 (87 ± 1 %) and t/tT4 (89 ± 1 %) populations (Fig. 2A). The PTU diet resulted in 100% expression of β-MyHC in both the NTGPTU and t/tPTU populations (Fig. 2B).
Figure 2. Complex modulus of skinned myocardial strips from mice expressing α- vs. β-MyHC.

Gel electrophoresis of left ventricle tissue homogenate stained with Coomassie-Blue demonstrate (A) a ~90/10% α/β-MyHC expression ratio for T4-fed transgenic control mice (WTt/tT4) and mice lacking cMyBP-C (t/tT4), and (B) ~100% β-MyHC expression in PTU-fed non-transgenic mice (NTGPTU) and t/t lacking cMyBP-C (t/tPTU). Bands for T4-fed mice are from the same gel, but not contiguous lanes, whereas bands for PTU-fed mice are from contiguous lanes of the same gel. Frequency characteristics of the elastic (C and D) and viscous (E and F) moduli of myocardium at maximum calcium activation and 5 mM MgATP are similar between groups bearing α-MyHC, but noticeably different between groups bearing β-MyHC.
It should be noted that we used the WTt/t as a control for t/t in the α-MyHC background, because feeding T4 to NTG mice results in 0% β-MyHC and therefore does not match the ~10% β-MyHC isoform content in the t/tT4. Furthermore, we did not use the WTt/tT4 as a control for t/t in the β-MyHC background, because PTU fed to the WTt/t would result in the loss of cMyBP-C from the sarcomere because cMyBP-C is expressed with the α-MyHC promoter in this transgenic mouse (Sadayappan et al., 2005). Further consideration of the control mice are presented in the Limitations subsection of the Discussion.
Myosin enzyme kinetics
In the WTt/tT4 and t/tT4 myocardium expressing predominately α-MyHC, the elastic and viscous moduli at saturating [MgATP] exhibited similar frequency characteristics between the genotypes (Fig. 2C and 2E). In contrast, the respective dips and shoulders of the elastic and viscous moduli-frequency responses were shifted to higher frequencies in the t/tPTU compared to NTGPTU controls expressing β-MyHC (Fig. 2D and 2F), indicating faster cross-bridge kinetics for t/tPTU vs. NTGPTU. Complex moduli were fit to Eq. 1 to estimate model parameters describing viscoelastic muscle mechanics and rate constants associated with cross-bridge kinetics as [MgATP] decreased from 5 to ≥ 0.1 mM. Although not shown here, the moduli-frequency relationship shifted towards lower frequencies as [MgATP] was reduced, similar to that illustrated in the previous report (Wang et al., 2013), which reflects a prolonged myosin cross-bridge rigor state at lower [MgATP].
For all populations, the magnitude A was enhanced and the index k was depressed as [MgATP] was reduced. These findings correspond to a greater number and longer duration strongly bound cross-bridges (Fig. 3A–B) and a more elastic vs. viscous characteristic (Fig. 3C–D) of the muscle, respectively, as [MgATP] decreased. Magnitudes B and C displayed high variation particularly as [MgATP] fell below 0.5 mM (Fig. 3E–F and 3I–J). The rate of force development 2πb rose to a maximum value at saturating [MgATP] and was faster in the α-MyHC compared to β-MyHC as would be expected for these isoforms (Fig. 3G–H). For all populations, cross-bridge detachment rate 2πc rose hyperbolically with increasing [MgATP] (Fig. 4A). These 2πc-[MgATP] relationships were fit to Eq. 2 to estimate MgADP release (k−ADP) and MgATP attachment (k+ATP) rates for α- and β-MyHC in the presence and absence of cMyBP-C (Table 1).
Figure 3. Effect of [MgATP] on parameters of Eq. 1.

For T4 and PTU-fed populations, myocardial viscoelastic stiffness rose as [MgATP] was lowered as reflected by the reduction in magnitude A (A and B) and became increasingly elastic as reflected by the decrease in k (C and D). The rate of force redevelopment 2πb was reduced as [MgATP] was lowered among all populations (G and H), while 2πb was ~2-fold faster for α- vs. β-MyHC. Compared to other model parameters, the magnitudes B (E and F) and C (I and J) were relatively insensitive to [MgATP]. †P<0.01; *P<0.05 between experimental and control groups within a panel.
Figure 4. MgATP-sensitivity of myosin cross-bridge detachment rate 2πc at maximal Ca2+ activation (pCa 4.8).

(A) 2πc was elevated as [MgATP] increased from <0.01 to 5 mM in skinned myocardial strips from thyroxin-fed and PTU-fed mice. Solid (controls) and dashed (t/t) lines show fits to Eq. 2. Normalizing 2πc to the asymptotic MgADP release rate (k−ADP; Table 2) demonstrates relative sensitivities to [MgATP] for each population, effectively highlighting (B) the ~2-fold faster MgATP release rate (k+ATP) from α-MyHC for WT t/tT4 vs. t/tT4 and (C) the ~1.5-fold faster k−ADP from β-MyHC for NTGPTU vs. t/tPTU. Insets within panels B and C show data vs. log[MgATP]. †P<0.01; *P<0.05 between experimental and control groups within a panel.
Table 1.
Parameters representing myosin enzyme kinetics were estimated by fitting myosin cross-bridge detachment rate (2πc) vs. MgATP relationships to Eq. 2.
| WTt/tT4 | t/tT4 | NTGPTU | t/tPTU | |
|---|---|---|---|---|
|
|
||||
| k−ADP (s−1) | 91.3±8.9 | 102.2±7.0 | 15.6±1.3 | 24.8±1.0† |
| k+ATP (mM−1s−1) | 298.6±15.7 | 165.9±12.5† | 99.8±20.0 | 77.5±14.5 |
| t−ADP (ms) | 11.3±1.0 | 10.0±0.6 | 65.6±10.6 | 40.6±17.1† |
| MgATP50 (μM) | 333±34 | 689±25† | 188±45 | 361±58* |
P<0.05;
P<0.01 against respective controls.
Values for k−ADP did not differ between WTt/tT4 and t/tT4, while k+ATP was nearly twice as fast in the WTt/tT4 compared to t/tT4 (Table 1). This effect of cMyBP-C on the rate MgATP binding can be observed via higher 2πc values in the WTt/tT4 at sub-saturating [MgATP] (Fig 4A–C). In contrast, values for k−ADP were significantly higher in t/tPTU compared to NTGPTU, and k+ATP did not differ between these genotypes expressing β-MyHC. These findings show that cMyBP-C does not significantly affect MgADP release rate for α-MyHC in the intact myofilament lattice, and that cMyBP-C would not be expected to affect α-MyHC cross-bridge ton under physiological conditions that include saturating [MgATP] and an intact myofilament lattice. In contrast, these findings suggest that cMyBP-C slows MgADP release in the β-MyHC to prolong the myosin cross-bridge ton under physiological conditions.
Myosin cross-bridge detachment rate and sarcomere length
No differences in isometric tension were observed between t/t mice and their respective controls. Passive tension values (mean±SE in kPa, pCa 8.0, 5 mM MgATP) were 5.5±0.1 and 5.6±0.4 for WTt/tT4 and t/tT4, respectively, at 2.2 μm SL and then 27.2±6.7 and 22.9±3.0 for WTt/tT4 and t/tT4, respectively, at 3.3 μm SL. Passive tension in the myocardial strips bearing β-MyHC were 5.3±0.3, and 5.5±0.1 for NTGPTU and t/tPTU, respectively, at 2.2 μm and then 95.9±9.1 and 72.7±17.3 for NTGPTU, and t/tPTU, respectively, at 3.3 um SL.
Developed tension values (pCa 4.8 tension – passive tension) were 24.9±2.2 and 29.6±2.0 for WTt/tT4 and t/tT4, respectively, at 2.2 μm SL and then 19.2±2.4 and 19.1±1.7 for WTt/tT4 and t/tT4, respectively, at 3.3 μm SL. Developed tension in the myocardial strips bearing β-MyHC were 21.6±1.3, and 16.6±2.5 for NTGPTU and t/tPTU, respectively, at 2.2 μm and then 49.9±2.9 and 50.9±10.1 for NTGPTU and t/tPTU, respectively, at 3.3 um SL. These data for isometric tension are indicative of comparable activation of a muscle strip across the range of 2.2–3.3 μm sarcomere length.
It is visually apparent that the elastic and viscous moduli were raised in magnitude and, more importantly, shifted to lower frequencies as sarcomere length was increased from 2.2 μm (Fig. 2) to 3.3 μm (Fig. 5). In myocardium expressing predominately α-MyHC, myosin detachment rate 2πc was not statistically different between t/tT4 and WTt/tT4 (Fig. 6A; P<0.148 for genotype main effect, Table 2), but increasing SL from 2.2–3.3 μm reduced 2πc by ~15% for both genotypes (P<0.001 for SL main effect). Detachment rate was not differentially affected by the absence of cMyBP-C in t/tT4 vs. WTt/tT4 (P=0.307 for SL × genotype interaction), suggesting that cMyBP-C did not influence α-MyHC detachment rate as SL varied.
Figure 5. Complex modulus at 3.3 μm sarcomere length.

Bright field images of papillary muscle illustrate visible sarcomeres at 2.2 μm (A) and 2.8 μm (B) sarcomere length. Further lengthening to 3.3 μm did not routinely permit visualization of sarcomeres. Frequency characteristics of the elastic (C and D) and viscous (E and F) moduli of mouse myocardium were similar between groups bearing α-MyHC, but noticeably different between groups bearing β-MyHC. Specifically, myocardium lacking cMyBP-C in the t/tPTU display higher frequency characteristics compared to NTGPTU.
Figure 6. Effect of sarcomere length on myosin cross-bridge detachment rate 2πc.
(A) Cross-bridge detachment rate decreased and (C) cross-bridge attachment duration (ton) increased for α-MyHC with increasing sarcomere length, independent of the presence (WTt/tT4) or absence (t/tT4) of cMyBP-C. (B) Detachment rate was faster and (D) ton was shorter for β-MyHC in the absence of cMyBP-C at all sarcomere lengths examined, including 3.3 μm where there is no overlap between the thin filament and the thick filament C-zone. The sensitivity of detachment rate reduction with increasing sarcomere length was greater in the absence (t/tPTU) rather than presence (NTGPTU) of cMyBP-C. †P<0.01; *P<0.05 between experimental and control groups.
Table 2.
Results of repeated-measures ANOVA applied to detachment rate 2 c detected at increasing sarcomere lengths (SL).
| WTt/tT4 vs. t/tT4 | NTGPTU vs. t/tPTU | |
|---|---|---|
|
|
||
| SL | <0.001† | <0.001† |
| Genotype | 0.148 | 0.004† |
| SL × genotype | 0.307 | 0.002† |
P<0.01
In myocardium expressing β-MyHC, cross-bridge detachment rate was significantly higher in the t/tPTU compared to NTGPTU over the entire range of SL examined (Fig. 6B; P=0.004 for genotype main effect, Table 2), including 3.3 μm where there is no overlap between the thin filament and the C-zone of thick filaments, where cMyBP-C resides. Increases in SL more greatly reduced 2πc for t/tPTU than for NTGPTU (P=0.002 for SL × genotype interaction). These results suggest that the presence of cMyBP-C reduces myosin detachment rate in β-MyHC both with and without overlap between the thin filament and the C-zone. Furthermore, the relative reduction in β-MyHC detachment rate with increasing SL was amplified in the absence of cMyBP-C.
Discussion
This study presents the effects of cMyBP-C on MgATP-dependent kinetics of the two cardiac myosin isoforms in mouse myocardium. In the α-MyHC k−ADP was not measurably affected by the absence of cMyBP-C, but in β-MyHC k−ADP was enhanced ~35% with the absence of cMyBP-C. Lengthening the sarcomere from 2.2 to 3.3 m reduced the cross-bridge detachment rate in α-MyHC by ~15%, but this effect was not sensitive to the absence of cMyBP-C. Cross-bridge detachment rate in the β-MyHC, in contrast, was reduced ~30% with sarcomere lengthening in the presence of cMyBP-C and ~40% in the absence of cMyBP-C. These findings collectively suggest that under physiological [MgATP] conditions cMyBP-C reduces β-MyHC detachment rate through some structural role that is independent of any possible interaction with the thin filament.
MyHC kinetics measured under isometric conditions
We would contend that the cardiac sarcomere in our assay is effectively isometric and the possible viscous effects of cMyBP-C binding to the thin filament are minimal. The length perturbation analysis technique used in this study relies upon oscillating the ~1100 nm half-sarcomere an amplitude of ~1.3 nm. The N-terminus of cMyBP-C emanating from the surface of the thick filament is on the order of 29–34 nm long and can easily span the distance to the thin filament approximately 8–12 nm away (Luther et al., 2011; Oshima et al., 2012). Even with a maximum oscillatory excursion of 2.6 nm of the thin filament relative to the thick filament, cMyBP-C attached to the thin filament would not be expected to experience a significant strain and might be slack throughout. Thus, the concept that cMyBP-C contributes a viscous load to slow loaded sarcomere shortening (Korte et al., 2003), the rate of force development (Stelzer et al., 2006), or unloaded shortening velocity and actin velocity (Hofmann et al., 1991; Previs et al., 2012; Razumova et al., 2006; Saber et al., 2008) has limited relevance in interpreting the kinetic measurements of the current study. Furthermore, our observation that α-MyHC detachment rate at 100 μM [MgATP] is faster in the presence of cMyBP-C (Fig. 4) does not agree with isolated myosin or thick filament assays demonstrating that cMyBP-C slows unregulated actin sliding velocity (Previs et al., 2012; Razumova et al., 2006; Weith et al., 2012). Based on the above observations, we therefore speculate that cMyBP-C influences strain-dependent cross-bridge kinetics within the intact myofilament lattice through its effects on myofilament lattice stiffness and not necessarily though its interaction with the thin filament. Still, cMyBP-C may also contribute a viscous load to actin filaments when significant translation occurs via dynamic changes in sarcomere length during shortening and relaxation.
cMyBP-C as structural modulator of MyHC kinetics
Mechanical characteristics of the sarcomere influence myosin cross-bridge kinetics and thereby play a role in dictating contraction dynamics of striated muscle (Hunter et al., 1998; Land et al., 2012; Tanner et al., 2007). We had previously observed that a loss of cMyBP-C leads to a collapsed myofilament lattice in the t/t compared to controls (Palmer et al., 2011), which would be expected to reduce k−ADP and prolong ton (Tanner et al., 2012). Therefore, any collapse in lattice spacing with the loss of cMyBP-C in the current study is unlikely to underlie the enhanced k−ADP observed in the β-MyHC in the t/tPTU. We would instead suggest that, because cMyBP-C accounts for ~40% of the flexural rigidity of thick filament in part through its binding to titin (Nyland et al., 2009) and contributes to the radial rigidity of the myofilament lattice (Palmer et al., 2011), the enhanced structural stiffness of the myofilaments afforded by cMyBP-C would be expected to influence the distribution of strain throughout the sarcomere and alter the load borne by bound cross-bridges throughout their attached lifetime. The reduction in stiffness properties of these structures with a loss of cMyBP-C will result in greater deformation of the cMyBP-C-dependent structures and a commensurately reduced deformation of the myosin head, including the nucleotide binding pocket, thereby affecting the rates k−ADP and k+ATP.
We speculate at this time that the observed changes in k−ADP and k+ATP due to loss of cMyBP-C reflect stiffness-dependencies of k−ADP and k+ATP in the respective myosin isoforms. Specifically, the absence of cMyBP-C may lead to greater deformation of the myofilaments and less deformation of the myosin head, thus underlying the increased k−ADP in β-MyHC and reduced k+ATP in the α-MyHC. We would not expect cMyBP-C to interact with the subfragment-2 or light-meromyosin portions of MyHC differently between the two cardiac MyHC isoforms, as these myosins reportedly bear identical amino acid sequences in these binding regions (Bhuiyan et al., 2012; Gruen and Gautel, 1999).
Using skinned myocardial preparations, measures of myosin ATPase, loaded and unloaded shortening velocities, cross-bridge ton and cross-bridge detachment rate differ by roughly fourfold between cardiac α- and β-MyHC isoforms (Korte et al., 2005; Rundell et al., 2005; Wang et al., 2013), rather than the two-fold difference observed using isolated cardiac myosin (Palmiter et al., 1999; Pereira et al., 2001). These observations suggest that the intact myofilament lattice and sarcomeric structural proteins play important roles in dictating cross-bridge rates of nucleotide release and binding that vary with MyHC isoform, despite identical structures of the nucleotide binding pockets. The functional interchangeability of the structurally dissimilar loop-1 and loop-2 in the mouse cardiac isoforms (Krenz et al., 2003) underscores the likelihood that myofilament structure plays a significant role in governing myosin kinetics.
With this in mind, the current study suggests that β-MyHC is much more sensitive to structural perturbation in the myofilament lattice than α-MyHC, at least in mouse myocardium. This could follow from β-MyHC being attached ~four-times longer than α-MyHC, such that any reduced load borne due to diminished thick filament stiffness in the absence of cMyBP-C is manifest effectively as a more ‘assistive’ load resulting in less deformation of the nucleotide binding pocket in β-MyHC of the t/tPTU myocardium (Kad et al., 2007; Veigel et al., 2003). Consistently, β-MyHC in the NTGPTU mouse would experience a ‘resistive’ load due to the presence of cMyBP-C, less deformation of the myofilaments and greater deformation of the nucleotide binding pocket to reduce k−ADP and prolong ton. In contrast, the rates of nucleotide release and binding in the α-MyHC are so fast that at physiological [MgATP] it may complete the cross-bridge cycle in a manner that is relatively insensitive to the manner with which loads is borne or distributed through the myofilament lattice.
Detachment rate with increasing sarcomere length
The reduction in myosin cross-bridge detachment rate with increasing sarcomere lengths suggests increasing resistive loads on the myosin cross-bridge as the sarcomere lengthened, due to increasing passive tensions that are structurally supported by, and propagated throughout, the myofilament lattice as the sarcomere is stretched. Again, α-MyHC appears here to be much less sensitive to the changes in load that occur with myofilament structure than β-MyHC. We have reported previously a reduction in myosin detachment rate with compression of the myofilament lattice spacing in insect flight muscle (Tanner et al., 2012). We would expect, although we have not measured it directly, that the myofilament lattice is compressed with sarcomere lengthening as has been reported previously for cardiac muscle (Konhilas et al., 2002). The greater sensitivity of myosin detachment rate to sarcomere length in the t/tPTU may be due to a greater compressibility of myofilament distances when cMyBP-C is absent (Palmer et al., 2011).
It is also possible that the overlap of the thin filament with the C-zone of the thick filament could influence myosin detachment rate. Assuming that thick and thin filament structures provided by Luther et al. (Luther et al., 2008) and Burgoyne et al. (Burgoyne et al., 2008) are relevant to our experimental conditions, all 9 cMyBP-C stripes would overlap with the thin filament at 2.2 μm. At 2.6 to 2.8 μm ~6 to 4 cMyBP-C stripes overlap with the thin filament and there is no overlap at 3.3 μm (Pfuhl and Gautel, 2012). The finding that α-MyHC detachment rate is affected relatively little by sarcomere length and independent of cMyBP-C reinforces the concept that kinetics in the α-MyHC are rather insensitive to structural attributes of the sarcomere including overlap of the thin filament with cMyBP-C. The elevated sensitivity of the β-MyHC detachment rate to sarcomere length is consistent with the detachment rate being reduced with higher structural loads that would accompany lattice compression at longer sarcomere lengths. The persistence of an enhanced detachment rate for β-MyHC at 3.3 μm, where there is no overlap of the thin filament with the C-zone, suggests that interactions with the thin filament cannot be at play. Our data do not allow us to discount the possibility that kinetics of this isoform could be sensitive to cMyBP-C via its interaction with myosin heads on the thick filament as suggested by Pfuhl and Gautel (Pfuhl and Gautel, 2012).
Relevance to human cardiomyopathies
Humans express predominately β-MyHC in the ventricles. In light of our current results, we would expect the human ventricle is more sensitive to the effects of aberrant cMyBP-C. In particular, some human cardiomyopathies associated with truncation mutations of cMyBP-C are accompanied by a loss of 10–30% of cMyBP-C (Marston et al., 2009; van Dijk et al., 2009), which according to our results may lead to an abnormally high detachment rate in these ventricles. There are no reports of dysfunction in the atria, however, which express predominately α-MyHC and therefore, according to our results, are relatively insensitive to effects of low cMyBP-C incorporation.
As an illustration of this effect in the animal model, consider that mice lacking cMyBP-C or expressing a mutant non-phosphorylated cMyBP-C (AllP-) and simultaneously expressing ~70% β-MyHC in the ventricle do not survive more than 7 weeks after birth (Sadayappan et al., 2009), while homozygous mice completely lacking cMyBP-C and expressing predominately α-MyHC, even with up to ~20–50% β-MyHC, can live up to a year (Carrier et al., 2004; Harris et al., 2002; McConnell et al., 1999). These observations are consistent with the current findings that cMyBP-C incorporation into the cardiac sarcomere in vivo is an influential determinant of β-MyHC function, but of relatively little consequence to α-MyHC function.
Limitations
It is important to note that our choices for control mice were based solely on the intended use of thyroid hormone to control for myosin heavy chain isoform. We expect the mice lacking cMyBP-C will carry some secondary effects due to compensatory mechanisms that likely arise in response to the primary insult. We cannot know all of the secondary effects on the sarcomeric proteins, although we have tried to minimize any effects of potentially mismatched phosphorylation profiles with pretreatment by phosphatases (Wang et al., 2013). It is expected, although not guaranteed, that the resulting secondary effects are negligible compared to those effects that are directly associated with the loss of cMyBP-C from the cardiac sarcomere. Fibrosis and myocyte disarray were present in the t/tT4 mouse, but not in its WTt/tT4 control. These morphological differences may have affected the magnitudes of elastic and viscous moduli in the t/t, but it would not be expected to influence the frequency characteristics of the moduli, and therefore, would not affect the measure of myosin detachment rates that were central to this study, showing similar k−ADP rates for α-MyHC with and without cMyBP-C. We also report fibrosis in both the t/tPTU and NTGPTU control. In this pair expressing β-MyHC, the morphology was better matched yet the k−ADP rates were significantly different. We would infer then that any differences in structural morphology between the mice lacking cMyBP-C and their respective controls are negligible in affecting the myosin detachment rates compared to the effects of the lack of cMyBP-C in the cardiac sarcomere.
We must also note that there may have been some irreversible damage done to the sarcomere and lattice structure at sarcomere lengths longer than 2.8 μm. The characteristic peak of myosin cross-bridge kinetics in the viscous modulus (Fig. 5E–F), however, demonstrates that cross-bridges were binding and cycling despite myofilament disarray and a loss of overlap of the thin filament with the C-zone. According to Luther et al. (2008), we would expect ~300 nm of the ~730 nm force-producing length of the thick filament to form cross-bridges at 3.3 μm. Our results at 3.3 μm, therefore, represent the effects of cross-bridge cycling without the influence of cMyBP-C’s interaction with the thin filament.
Conclusion
Our findings suggest that cMyBP-C influences myosin cross-bridge kinetics in an isoform-dependent manner. Any other possible influence of cMyBP-C on sarcomere function, such as thin filament regulation or providing viscous drag, however, cannot be discerned from the current set of data. Our findings suggest that under normally high MgATP concentrations above 5 mM (Ingwall and Weiss, 2004) a loss of cMyBP-C as might underlie human familial cardiomyopathy (Marston et al., 2009; van Dijk et al., 2009) could shorten cross-bridge ton and enhanced mechanical performance in the slow β-MyHC typically expressed in human ventricles. Such an enhancement of cardiac contractile function might underlie the induction of hypertrophic cardiomyopathies in humans as has been suggested based on the enhanced functional effects of mutant myosins underlying hypertrophic cardiomyopathies (Debold et al., 2007; Marston et al., 2012);
Acknowledgments
This work was supported by a grant from the NIH P01 HL59408.
References
- Bhuiyan MS, Gulick J, Osinska H, Gupta M, Robbins J. Determination of the critical residues responsible for cardiac myosin binding protein C’s interactions. J Mol Cell Cardiol. 2012;53:838–847. doi: 10.1016/j.yjmcc.2012.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoyne T, Muhamad F, Luther PK. Visualization of cardiac muscle thin filaments and measurement of their lengths by electron tomography. Cardiovasc Res. 2008;77:707–712. doi: 10.1093/cvr/cvm117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell K, Chandra M, Kirkpatrick R, Slinker B, Hunter W. Interpreting cardiac muscle force-length dynamics using a novel functional model. Am J Physiol Heart Circ Physiol. 2004;286:H1535–H1545. doi: 10.1152/ajpheart.01029.2003. [DOI] [PubMed] [Google Scholar]
- Carrier L, Knöll R, Vignier N, Keller DI, Bausero P, Prudhon B, Isnard R, Ambroisine ML, Fiszman M, Ross J, Schwartz K, Chien KR. Asymmetric septal hypertrophy in heterozygous cMyBP-C null mice. Cardiovasc Res. 2004;63:293–304. doi: 10.1016/j.cardiores.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Debold EP, Schmitt JP, Patlak JB, Beck SE, Moore JR, Seidman JG, Seidman C, Warshaw DM. Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse alpha-cardiac myosin in the laser trap assay. Am J Physiol Heart Circ Physiol. 2007;293:H284–H291. doi: 10.1152/ajpheart.00128.2007. [DOI] [PubMed] [Google Scholar]
- Freiburg A, Gautel M. A molecular map of the interactions between titin and myosin-binding protein C. Implications for sarcomeric assembly in familial hypertrophic cardiomyopathy. Eur J Biochem. 1996;235:317–323. doi: 10.1111/j.1432-1033.1996.00317.x. [DOI] [PubMed] [Google Scholar]
- Godt RE, Lindley BD. Influence of temperature upon contractile activation and isometric force production in mechanically skinned muscle fibers of the frog. J Gen Physiol. 1982;80:279–297. doi: 10.1085/jgp.80.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruen M, Gautel M. Mutations in beta-myosin S2 that cause familial hypertrophic cardiomyopathy (FHC) abolish the interaction with the regulatory domain of myosin-binding protein-C. J Mol Biol. 1999;286:933–949. doi: 10.1006/jmbi.1998.2522. [DOI] [PubMed] [Google Scholar]
- Harris SP, Bartley CR, Hacker TA, McDonald KS, Douglas PS, Greaser ML, Powers PA, Moss RL. Hypertrophic cardiomyopathy in cardiac myosin binding protein-C knockout mice. Circ Res. 2002;90:594–601. doi: 10.1161/01.res.0000012222.70819.64. [DOI] [PubMed] [Google Scholar]
- Hofmann PA, Greaser ML, Moss RL. C-protein limits shortening velocity of rabbit skeletal muscle fibres at low levels of Ca2+ activation. J Physiol. 1991;439:701–715. doi: 10.1113/jphysiol.1991.sp018689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter PJ, McCulloch AD, ter Keurs HE. Modelling the mechanical properties of cardiac muscle. Prog Biophys Mol Biol. 1998;69:289–331. doi: 10.1016/s0079-6107(98)00013-3. [DOI] [PubMed] [Google Scholar]
- Ingwall JS, Weiss RG. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ Res. 2004;95:135–145. doi: 10.1161/01.RES.0000137170.41939.d9. [DOI] [PubMed] [Google Scholar]
- Kad NM, Patlak JB, Fagnant P, Trybus KM, Warshaw DM. Mutation of a conserved glycine in the SH1–SH2 helix affects the load-dependent kinetics of myosin. Biophys J. 2007;92:1623–1631. doi: 10.1529/biophysj.106.097618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai M, Halvorson H. Two step mechanism of phosphate release and the mechanisms of force generation in chemically skinned fibers of rabbit psoas muscle. Biophys J. 1991;59:329–342. doi: 10.1016/S0006-3495(91)82227-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konhilas JP, Irving TC, de Tombe PP. Myofilament calcium sensitivity in skinned rat cardiac trabeculae: role of interfilament spacing. Circ Res. 2002;90:59–65. doi: 10.1161/hh0102.102269. [DOI] [PubMed] [Google Scholar]
- Korte FS, Herron TJ, Rovetto MJ, McDonald KS. Power output is linearly related to MyHC content in rat skinned myocytes and isolated working hearts. Am J Physiol Heart Circ Physiol. 2005;289:H801–H812. doi: 10.1152/ajpheart.01227.2004. [DOI] [PubMed] [Google Scholar]
- Korte FS, McDonald KS, Harris SP, Moss RL. Loaded shortening, power output, and rate of force redevelopment are increased with knockout of cardiac myosin binding protein-C. Circ Res. 2003;93:752–758. doi: 10.1161/01.RES.0000096363.85588.9A. [DOI] [PubMed] [Google Scholar]
- Krenz M, Sanbe A, Bouyer-Dalloz F, Gulick J, Klevitsky R, Hewett TE, Osinska HE, Lorenz JN, Brosseau C, Federico A, Alpert NR, Warshaw DM, Perryman MB, Helmke SM, Robbins J. Analysis of myosin heavy chain functionality in the heart. J Biol Chem. 2003;278:17466–17474. doi: 10.1074/jbc.M210804200. [DOI] [PubMed] [Google Scholar]
- Land S, Niederer SA, Aronsen JM, Espe EKS, Zhang L, Louch WE, Sjaastad I, Sejersted OM, Smith NP. An analysis of deformation-dependent electromechanical coupling in the mouse heart. J Physiol. 2012;590:4553–4569. doi: 10.1113/jphysiol.2012.231928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther PK, Bennett PM, Knupp C, Craig R, Padrón R, Harris SP, Patel J, Moss RL. Understanding the organisation and role of myosin binding protein c in normal striated muscle by comparison with mybp-c knockout cardiac muscle. J Mol Biol. 2008;384:60–72. doi: 10.1016/j.jmb.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther PK, Winkler H, Taylor K, Zoghbi ME, Craig R, Padrón R, Squire JM, Liu J. Direct visualization of myosin-binding protein c bridging myosin and actin filaments in intact muscle. Proc Natl Acad Sci U S A. 2011;108:11423–11428. doi: 10.1073/pnas.1103216108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marston S, Copeland O, Gehmlich K, Schlossarek S, Carrier L. How do MYBPC3 mutations cause hypertrophic cardiomyopathy? J Muscle Res Cell Motil. 2012;33:75–80. doi: 10.1007/s10974-011-9268-3. [DOI] [PubMed] [Google Scholar]
- Marston S, Copeland O, Jacques A, Livesey K, Tsang V, McKenna WJ, Jalilzadeh S, Carballo S, Redwood C, Watkins H. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res. 2009;105:219–222. doi: 10.1161/CIRCRESAHA.109.202440. [DOI] [PubMed] [Google Scholar]
- McConnell BK, Jones KA, Fatkin D, Arroyo LH, Lee RT, Aristizabal O, Turnbull DH, Georgakopoulos D, Kass D, Bond M, Niimura H, Schoen FJ, Conner D, Fischman DA, Seidman CE, Seidman JG, Fischman DH. Dilated cardiomyopathy in homozygous myosin-binding protein-C mutant mice. J Clin Invest. 1999;104:1235–1244. doi: 10.1172/JCI7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyland LR, Palmer BM, Chen Z, Maughan DW, Seidman CE, Seidman JG, Kreplak L, Vigoreaux JO. Cardiac myosin binding protein-C is essential for thick-filament stability and flexural rigidity. Biophys J. 2009;96:3273–3280. doi: 10.1016/j.bpj.2008.12.3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima K, Sugimoto Y, Irving TC, Wakabayashi K. Head-head interactions of resting myosin crossbridges in intact frog skeletal muscles, revealed by synchrotron x-ray fiber diffraction. PLoS One. 2012;7:e52421. doi: 10.1371/journal.pone.0052421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer BM, Noguchi T, Wang Y, Heim JR, Alpert NR, Burgon PG, Seidman JG, Seidman CE, Maughan DW, LeWinter MM. Effect of cardiac myosin binding protein-C on mechanoenergetics in mouse myocardium. Circ Res. 2004;94:1615–1622. doi: 10.1161/01.RES.0000132744.08754.f2. [DOI] [PubMed] [Google Scholar]
- Palmer BM, Sadayappan S, Wang Y, Weith AE, Previs MJ, Bekyarova T, Irving TC, Robbins J, Maughan DW. Roles for cardiac MyBP-C in maintaining myofilament lattice rigidity and prolonging myosin cross-bridge lifetime. Biophys J. 2011;101:1661–1669. doi: 10.1016/j.bpj.2011.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmiter KA, Tyska MJ, Dupuis DE, Alpert NR, Warshaw DM. Kinetic differences at the single molecule level account for the functional diversity of rabbit cardiac myosin isoforms. Journal of Physiology. 1999;519:669–678. doi: 10.1111/j.1469-7793.1999.0669n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira JS, Pavlov D, Nili M, Greaser M, Homsher E, Moss RL. Kinetic differences in cardiac myosins with identical loop 1 sequences. J Biol Chem. 2001;276:4409–4415. doi: 10.1074/jbc.M006441200. [DOI] [PubMed] [Google Scholar]
- Pfuhl M, Gautel M. Structure, interactions and function of the N-terminus of cardiac myosin binding protein C (MyBP-C): who does what, with what, and to whom? J Muscle Res Cell Motil. 2012;33:83–94. doi: 10.1007/s10974-012-9291-z. [DOI] [PubMed] [Google Scholar]
- Previs MJ, Previs SB, Gulick J, Robbins J, Warshaw DM. Molecular mechanics of cardiac myosin-binding protein C in native thick filaments. Science. 2012;337:1215–1218. doi: 10.1126/science.1223602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razumova MV, Shaffer JF, Tu AY, Flint GV, Regnier M, Harris SP. Effects of the N-terminal domains of myosin binding protein-C in an in vitro motility assay: Evidence for long-lived cross-bridges. J Biol Chem. 2006;281:35846–35854. doi: 10.1074/jbc.M606949200. [DOI] [PubMed] [Google Scholar]
- Reiser PJ, Kline WO. Electrophoretic separation and quantitation of cardiac myosin heavy chain isoforms in eight mammalian species. Am J Physiol. 1998;274:H1048–H1053. doi: 10.1152/ajpheart.1998.274.3.H1048. [DOI] [PubMed] [Google Scholar]
- Rundell VLM, Manaves V, Martin AF, de Tombe PP. Impact of beta-myosin heavy chain isoform expression on cross-bridge cycling kinetics. Am J Physiol Heart Circ Physiol. 2005;288:H896–H903. doi: 10.1152/ajpheart.00407.2004. [DOI] [PubMed] [Google Scholar]
- Saber W, Begin KJ, Warshaw DM, VanBuren P. Cardiac myosin binding protein-C modulates binding and kinetics in the in vitro motility assay. J Mol Cell Cardiol. 2008;44:1053–1061. doi: 10.1016/j.yjmcc.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadayappan S, Gulick J, Klevitsky R, Lorenz JN, Sargent M, Molkentin JD, Robbins J. Cardiac myosin binding protein-c phosphorylation in a beta-myosin heavy chain background. Circulation. 2009;119:1253–1262. doi: 10.1161/CIRCULATIONAHA.108.798983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadayappan S, Gulick J, Osinska H, Martin LA, Hahn HS, Dorn GW, Klevitsky R, Seidman CE, Seidman JG, Robbins J. Cardiac myosin-binding protein-c phosphorylation and cardiac function. Circ Res. 2005;97:1156–1163. doi: 10.1161/01.RES.0000190605.79013.4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzer JE, Fitzsimons DP, Moss RL. Ablation of myosin-binding protein-C accelerates force development in mouse myocardium. Biophys J. 2006;90:4119–4127. doi: 10.1529/biophysj.105.078147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner BCW, Daniel TL, Regnier M. Sarcomere lattice geometry influences cooperative myosin binding in muscle. PLoS Comput Biol. 2007;3:e115. doi: 10.1371/journal.pcbi.0030115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner BCW, Farman GP, Irving TC, Maughan DW, Palmer BM, Miller MS. Thick-to-thin filament surface distance modulates cross-bridge kinetics in Drosophila flight muscle. Biophys J. 2012;103:1275–1284. doi: 10.1016/j.bpj.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyska MJ, Warshaw DM. The myosin power stroke. Cell Motil Cytoskeleton. 2002;51:1–15. doi: 10.1002/cm.10014. [DOI] [PubMed] [Google Scholar]
- van Dijk SJ, Dooijes D, dos Remedios C, Michels M, Lamers JMJ, Winegrad S, Schlossarek S, Carrier L, ten Cate FJ, Stienen GJM, van der Velden J. Cardiac myosin-binding protein c mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation. 2009;119:1473–1483. doi: 10.1161/CIRCULATIONAHA.108.838672. [DOI] [PubMed] [Google Scholar]
- Veigel C, Molloy JE, Schmitz S, Kendrick-Jones J. Load-dependent kinetics of force production by smooth muscle myosin measured with optical tweezers. Nat Cell Biol. 2003;5:980–986. doi: 10.1038/ncb1060. [DOI] [PubMed] [Google Scholar]
- Wang Y, Tanner BCW, Lombardo AT, Tremble SM, Maughan DW, Vanburen P, Lewinter MM, Robbins J, Palmer BM. Cardiac myosin isoforms exhibit differential rates of MgADP release and MgATP binding detected by myocardial viscoelasticity. J Mol Cell Cardiol. 2013;54:1–8. doi: 10.1016/j.yjmcc.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weith A, Sadayappan S, Gulick J, Previs MJ, Vanburen P, Robbins J, Warshaw DM. Unique single molecule binding of cardiac myosin binding protein-C to actin and phosphorylation-dependent inhibition of actomyosin motility requires 17 amino acids of the motif domain. J Mol Cell Cardiol. 2012;52:219–227. doi: 10.1016/j.yjmcc.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Kawai M. The effect of the lattice spacing change on cross-bridge kinetics in chemically skinned rabbit psoas muscle fibers. II. Elementary steps affected by the spacing change. Biophys J. 1993;64:197–210. doi: 10.1016/S0006-3495(93)81357-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi ME, Woodhead JL, Moss RL, Craig R. Three-dimensional structure of vertebrate cardiac muscle myosin filaments. Proc Natl Acad Sci U S A. 2008;105:2386–2390. doi: 10.1073/pnas.0708912105. [DOI] [PMC free article] [PubMed] [Google Scholar]