

Abstract
Neutrophils are potent immune effectors against bacterial infections. Macrophages are important in infections as effectors and regulators, but their exact roles, phenotypic characterization and their relation to neutrophils is incompletely understood. Here we report in a model of bacterial urinary tract infection, one of the most prevalent bacterial infections that tissue-resident Ly6C− macrophages recruited circulating neutrophils and inflammatory Ly6C+ macrophages through chemokines. Neutrophils were primarily recruited through ligands of the chemokine receptor CXCR2, in particular by CXCL1 and less by macrophage migration inhibitory factor (MIF), but not through CXCL5 and CXCL2. Neutrophils, but not Ly6C+ macrophages, cleared the bacteria by phagocytosis. Ly6C+ macrophages instead performed a regulatory function: in response to the infection, they produced the cytokine tumor necrosis factor (TNF), which in turn caused the resident macrophages to secrete CXCL2. This chemokine induced the secretion of matrix metalloproteinase-9 (MMP-9) in neutrophils and allowed these cells to degrade the uroepithelial basement membrane, in order to enter the uroepithelium, the mucosal interface from where the bacteria invade the bladder. Thus, the phagocyte response against bacteria is a highly coordinated event, in which Ly6C− macrophages act as sentinels and Ly6C+ macrophages as innate helper cells. In analogy with T helper cells (Th), we propose to name these helper macrophages (Ph) as they provide a second signal on whether to unleash the principal effector phagocytes, the neutrophils. This cellular triage may prevent ‘false-positive’ immune responses. The role of TNF as innate ‘licensing’ factor contributes to its central role in antibacterial immunity.
Keywords: Macrophages, chemokines, bacterial infection, neutrophils, matrix metalloproteinase, MIF, immunologic help
There are two types of phagocytes: macrophages and neutrophils. Both cell types are critical for the defense against bacterial infections and form partnerships, but the exact nature of their collaboration is incompletely understood1. Neutrophils are bone marrow-derived, circulate through the blood and must be recruited into infected or inflamed tissues1b. They are attracted by ligands of the chemokine receptor CXCR1 or CXCR2, which include CXCL1, CXCL2, CXCL51c and macrophage inhibitory receptor (MIF)xy. By contrast, all organs harbor a dense network of resident macrophages, which has been proposed to partially originate from yolk sac progenitors2,2b. In infections, the bone marrow produces inflammatory monocytes, which travel through the blood and emigrate to sites of infection, where they differentiate into macrophages3. The functions of inflammatory macrophages are thought to overlap with those of resident macrophages.
Urinary tract infections (UTI) are highly prevalent, affecting more than 25% of the population in developed countries, especially young females4,5. They mainly result from uropathogenic E. coli (UPEC) ascending through the urethra into the bladder, where they invade the bladder uroepithelium6. UPECs can persist intracellularly and cause relapsing infections7. In humans, their ascension into the kidney causes pyelonephritis that may progress to renal failure8. The defense against UTI depends on TLR/MyD88 signaling9, the CXC chemokine receptors CXCR1 and CXCR21c and on neutrophils10, which phagocytose the UPEC. The role of macrophages in UTI is unclear.
Here we studied the roles of macrophages and of neutrophils in a mouse model of UTI induced by transureteral instillation of UPECs. Bladders of uninfected mice contained only resident macrophages defined by expression of the marker F4/80 and by the lack of Ly6C (Fig. 1a, Supplementary Fig. 1). Already 2 hours after infection, F4/80− Ly6C+ neutrophils started infiltrating the bladder and F4/80+ Ly6C+ inflammatory macrophages followed at 6 hours after infection (Fig. 1b). Ly6C− macrophage numbers remained largely constant during the course of infection (Fig. 1b). Histological analysis demonstrated neutrophils, and some Ly6C− macrophages within the uroepithelium, whereas Ly6C+ macrophages were confined to the lamina propria (Fig. 1c, d). When we infected mice with UPECs expressing recombinant green fluorescent protein (GFP) and determined bacterial uptake by flow cytometry, almost 90% of the phagocytosed UPECs were detected within neutrophils (Fig. 1e), consistent with their known critical role in UTI10.
Figure 1. Recruitment and positioning of macrophages and neutrophils in bacterial UTI.
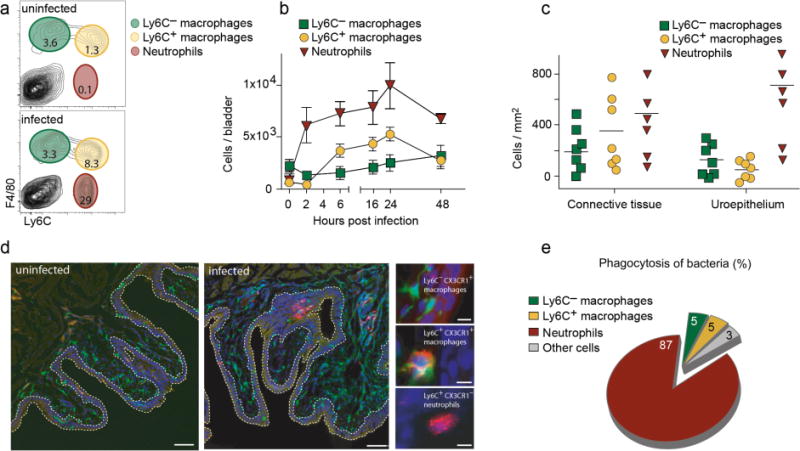
a, Flow cytometry analysis of bladder homogenates of uninfected and infected mice 24 hours after infection. Neutrophils (red), Ly6C+ (yellow) and Ly6C− (green) macrophages were distinguished by their expression of Ly6C and F4/80 as indicated. b, Numbers of neutrophils, Ly6C+ and Ly6C− macrophages in single cell suspensions of bladders determined by flow-cytometry. c, Numbers of neutrophils, Ly6C+ and Ly6C− macrophages in bladder cryosections 24 hours after infection. d, Cryosections from bladders of uninfected and infected CX3CR1+/GFP mice, which express green fluorescent protein (GFP) in macrophages31, were stained for Ly6C (red) and for cellular nuclei (DAPI-blue). The white dashed line highlights the uroepithelial border. The small images show representative stainings of the three phagocyte types. e, Contribution of phagocyte subsets to phagocytosis of GFP-expressing UPECs, calculated by the formula ((mean fluorescence intensity of GFP+ cells in UPEC-GFP infected mice – mean fluorescence intensity of GFP+ cells in UPEC infected mice) × number of UPEC-GFP+ cells)). Data are means ± s.e.m. and represent five (a, b, e) and three (c, d) independent experiments in groups of five mice.
We reasoned that most likely the bladder-resident Ly6C− macrophages recruited the circulating phagocytes. However, proving this hypothesis was difficult, because no techniques exist to selectively remove Ly6C− macrophages, and because vesical Ly6C− macrophages are not targeted by general macrophage depletion techniques like clodronate liposomes (Supplementary Fig. 2). Instead, we decided to block the chemokines that Ly6C− macrophages would predictably use to recruit inflammatory phagocytes. We first identified these mediators by intracellular flow cytometry. At 6 hours after infection, only Ly6C− macrophages produced the neutrophil attractors MIF, CXCL1, CXCL2 and CXCL5 (Fig. 2a, b). When we blocked these chemokines with neutralizing antibodies, CXCL1 was a critical chemokine and similarly important as CXCR2 for neutrophil recruitment, whereas CXCL2 and CXCL5 played minor roles (Fig. 2c). As a further control, Ly6C+ macrophage numbers were substantially reduced in Ccr2-deficient mice (Fig. 2d), whose ligand CCL2 was produced by Ly6C− macrophages as well (Fig. 2a,b). Also neutrophils produced some CCL2 (Fig. 2a,b) and consistently, Ly6C+ macrophage numbers were reduced also in Cxcr2-deficient mice (Fig. 2d). Bacterial clearance of the bladder was impaired in the absence of CXCL1, CXCL2, MIF, CXCR2, CCR2 and after depletion of Ly6C+ macrophages by clodronate-liposomes, but not after blocking CXCL5 (Fig. 2e). Thus, Ly6C− macrophages rapidly produced chemokines that attracted neutrophils and Ly6C+ macrophages into the infected bladder, and both cell types were important for antibacterial defense.
Figure 2. Chemokines produced by Ly6C− macrophages recruit Ly6C+ macrophages and neutrophils into the infected bladder.
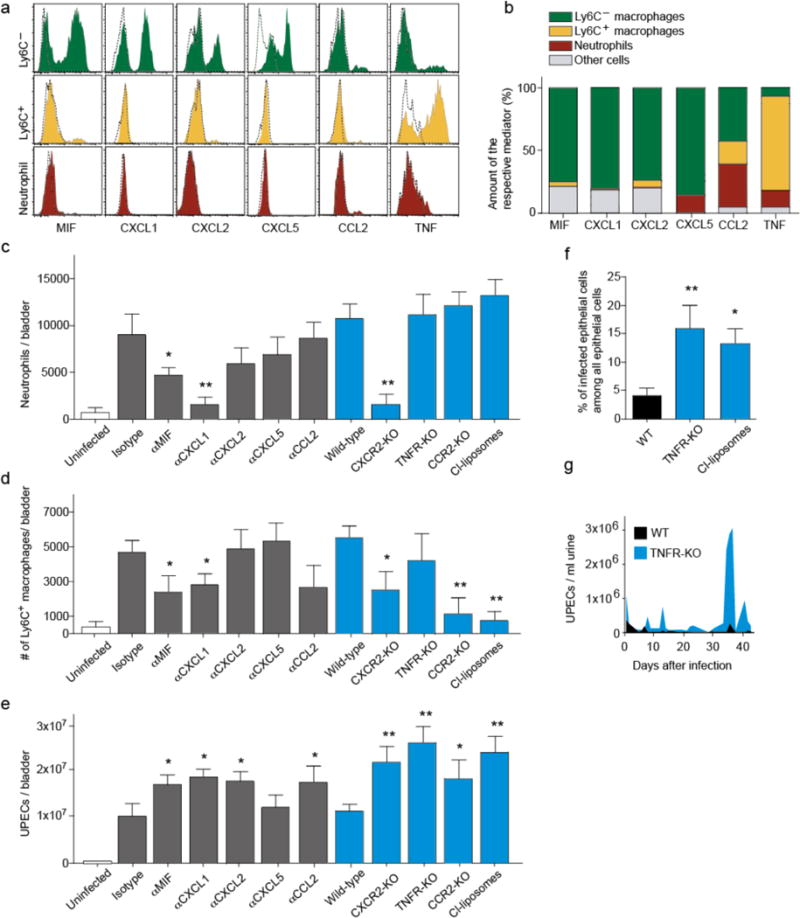
a, Intracellular flow-cytometric staining of Ly6C− (green) and Ly6C+ (yellow) macrophages and neutrophils (red) 6 hours after infection for immune regulatory molecules (solid line) and the respective isotype control (dashed line). b, Contribution of phagocyte subsets to the total production of immune regulatory molecules, calculated using the formula ((mean fluorescence intensity of antibody+ cells – mean fluorescence intensity of isotype+ cells) × number of cells positive for the molecule)) and giving the proportion within the total contribution of all bladder cells. c–e, Intravesical numbers of neutrophils (c), Ly6C+ macrophages (d) and UPECs (e) 6h after infection in mice treated with the isotype control or antibodies against the immune regulatory molecule (grey) and in wild-type controls or in deficient animals (blue). f, Mice were infected with GFP-expressing UPECs and the percentage of infected GFP+ epithelial cells were determined. g, Relapsing infections in wild-type (WT) and TNFR-KO mice analyzed by counting the number of UPECs within the urine. Data are means ± s.e.m. and represent three independent experiments in groups of three to five mice. *P < 0.05; **P < 0.01.
The importance of Ly6C+ macrophages was astonishing given that these cells hardly contributed to bacterial phagocytosis (Fig. 1e). We noted that these cells were the main producers of tumor necrosis factor (TNF) (Fig. 2a, b), a cytokine considered generally important in bacterial infections11. Several TNF functions have been reported in other models, for example inducing neutrophil degranulation12, phagocytosis12,13 and bactericidal activity13. The role of TNF in UTI is unknown. When we infected Tnfr-knockout (KO) mice deficient for both TNF receptors, the bacterial load in the bladder was much higher than in wild-type controls (Fig. 2e). Also the number of infected uroepithelial cells, which indicates persistence of UTI, was severely increased in Tnfr-KO mice and in the absence of the TNF-producing Ly6C+ macrophages (Fig. 2f). Moreover, over a period of 6 weeks, more relapses of higher severity occurred (Fig. 2g). But strikingly, the absolute number of neutrophils, was not reduced in Tnfr-KO mice (Fig. 2c), suggesting that the antibacterial function of TNF is distinct from recruiting neutrophils.
Since TNF was reported to activate neutrophils11, we first hypothesized that it might activate their antibacterial effector mechanisms. However, Tnfr-deficient neutrophils still phagocytosed UPECs in vitro (Supplementary Fig. 3) and maintained activity of the antibacterial effector molecules elastase and myeloid peroxidase (Supplementary Fig. 3). Moreover, adding TNF did not enhance the bactericidal activity of neutrophils (Supplementary Fig. 3). These findings argued against TNF controlling neutrophil activity. Given that TNF was important for antibacterial activity only in vivo but not in vitro, we next hypothesized that it might impact neutrophil positioning within the bladder. Indeed, histology revealed that neutrophils were excluded from the uroepithelium of Tnfr-KO mice, the location from where UPECs enter (Fig. 3a–c and Supplementary Fig. 4). Less neutrophils were detected also in the urine of Tnfr-KO mice and after depleting the TNF-producing Ly6C+ macrophages (Fig. 3d), consistent with their inability to enter and transmigrate through the uroepithelium into the bladder lumen. When we reconstituted Tnf-KO mice with the main TNF producers, Ly6C+ macrophages (Supplementary Fig. 4), neutrophils did enter the uroepithelium. This is a verification that these cells permitted epithelial migration of neutrophils.
Figure 3. TNF from Ly6C+ macrophages enables neutrophils to enter the uroepithelium.
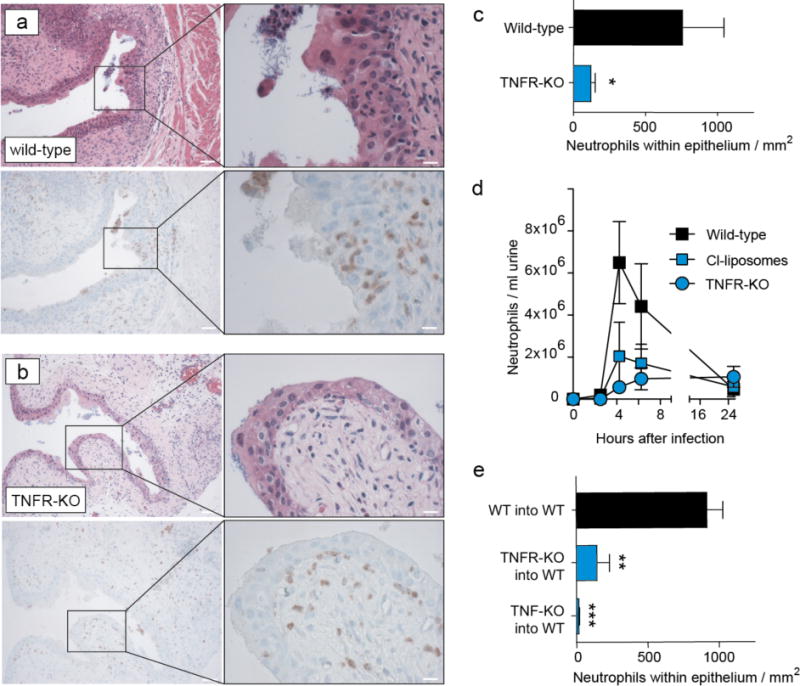
a, b, Presence of neutrophils in the urothelium in consecutive bladder sections one day after infecting wild-type (a) or TNFR-KO (b) mice, identified by HE (upper row) or Ly6G (brown staining in the lower panel row) staining. The right panels show fivefold magnifications of the inlay highlighted in the left column with a black box. White lines are 50μm (left) or 10μm (right). c, Quantitative analysis of the histology shown in (a) and (b). d, Kinetics of neutrophil numbers in the urine of wild-type or TNFR-KO mice or after depleting Ly6C+ macrophages with clodronate liposomes (Cl-liposomes) determined by flow-cytometry. e, Neutrophil numbers in bladder sections 1 day after infection in bone marrow chimeric mice expressing TNF or TNFR in all cells (WT into WT) or only in non-hematopoietic cells (TNFR-KO into WT, TNF into WT, respectively). Data are means ± s.e.m. and represent four (a–d) and three (e) independent experiments in groups of five to eight mice. *P < 0.05; **P < 0.01, ***P < 0.01.
We next asked whether such migration required TNF to directly act on neutrophils, by cotransferring neutrophils from Tnfr-deficient and –competent mice into infected wild-type mice. However, Tnfr-KO neutrophils entered the bladder as efficiently as the controls, and appeared in similar numbers in the uroepithelium (Supplementary Fig. 5), indicating that TNF acted on TNFR+ cells other than neutrophils. To identify this cell, we first generated bone marrow chimeras in which hematopoietic cells lacked either the TNFR or TNF (Supplementary Fig 6). Transepithelial neutrophil migration required TNFR expression and TNF secretion by bone marrow-derived cells (Fig. 3e and Supplementary Fig. 6). Therefore, we next examined whether the expression of molecules known to affect neutrophil migration are expressed in a TNF-dependent manner. We found that CXCR2, CXCL1, MIF, GM-CSF, IFNγ, IL-1a, IL-4, IL-5, L-6, IL-17, CCL2, CCL3, CCL4, CCL5, CCL7, CD11b, CD47, CD49b, ICAM-1 (Fig. 4a, b, c and Supplementary Fig. 7) were similarly expressed in infected Tnfr-KO and wild-type mice. Only CXCL2 was markedly reduced in Tnfr-KO mice (Fig. 4d). This is consistent with a previous report that this chemokine is required for uroepithelial neutrophil migration14. As only Ly6C− macrophages produced CXCL2 in the infected bladder (Fig. 2a, b), we concluded that TNF acted on these macrophages, causing these to produce CXCL2. If so, then the absence of TNF or of its source, the Ly6C+ macrophages, should abrogate CXCL2 production. Indeed, this chemokine was almost undetectable in the infected bladder after depleting Ly6C+ macrophages and in Tnf or Tnfr-deficient mice (Supplementary Fig. 8). Furthermore, transurethral inoculation of CXCL2 into the bladder of infected Tnf-KO mice restored the ability of neutrophils to enter the uroepithelium (Fig. 4e and Supplementary Fig. 9), confirming that CXCL2 can induce such migration and that it acted downstream of TNF. At later time points, CXCL2 might also be produced by neutrophils, consistent with previous reports15. Our findings at early time points suggest this might represent an autocrine feedback loop induced after neutrophils sensed CXCL2 from Ly6C− macrophages.
Figure 4. TNF induces CXCL2 in Ly6C− macrophages, which activates MMP9 in neutrophils.
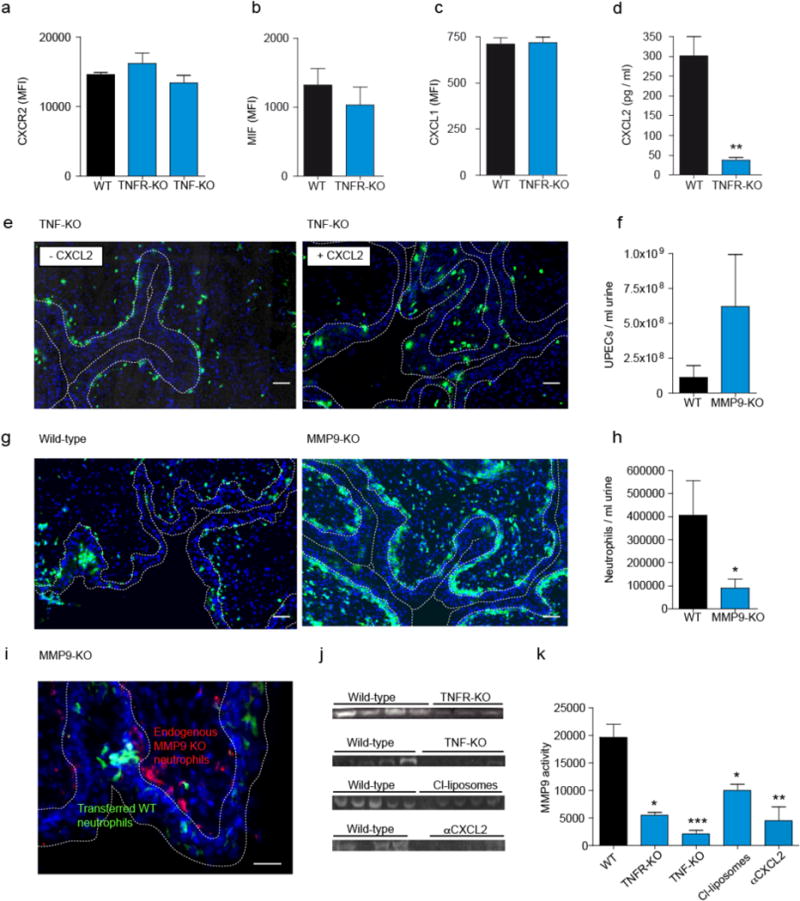
a–d, Expression of CXCR2 by neutrophils (a), and of MIF (b), CXCL1 (c) and CXCL2 (d) by Ly6C− macrophages in wild-type (WT), TNFR- or TNF-KO mice, determined by flow-cytometry (MFI=Mean fluorescence intensity) (a–c) or ELISA (d). e,g, Positioning of neutrophils in bladder sections of, wild-type TNF- (e) or MMP9-KO (g) mice one after infection, revealed by staining with Ly6G Alexa488 (green). The white dashed line highlights the uroepithelial border. f,h, Numbers of UPECs (f) and neutrophils (h) in the urine of WT and MMP9 KO mice. i, Inability of endogenous neutrophils in MMP-9-KO mice, stained with red Ly6G Alexa568, to enter the infected uroepithelium, in contrast to adoptively transferred MMP-9-competent neutrophils stained with CFSE (green). j,k, MMP-9 levels in bladder homogenates one day after infection determined by zymography (j) and numerically displayed after analysis by densitometry (k). Data are means ± s.e.m. and represent 3 independent experiments in groups of 5–8 mice. *P < 0.05; **P < 0.01; ***P < 0.001.
Next, we asked how CXCL2 enabled neutrophils to enter the uroepithelium. Epithelial basement membranes are comprised mainly of collagen IV, which is first cleaved in two fragments by e.g. neutrophil collagenase or matrix metalloproteinase-8 (MMP-8)15b. Thereafter, this denatured collagen is further cleaved by gelatinase B/MMP-915c,16. The role of MMP-9 in UTI is unknown. Mmp-9-KO mice showed a markedly higher bacterial load in the urine (Fig. 4f), indicating that MMP-9 is required for defense against UTI. Histological sections of infected Mmp-9-KO mice showed that neutrophils accumulated underneath the uroepithelium and failed to enter it (Fig. 4g and Supplementary Fig. 10) or to transmigrate into the bladder lumen (Fig. 4h). When we stained bladder cells for MMP-9 expression, more than 90% of the MMP-9+ cells were neutrophils (Supplementary Fig. 11). To test whether neutrophil-intrinsic MMP-9 was important, we transferred MMP-9-competent neutrophils into infected Mmp-9-KO mice. Only competent neutrophils were detected within the uroepithelium, but not the endogenous Mmp-9-KO neutrophils (Fig. 4i and Supplementary Fig. 12), confirming that neutrophil-intrinsic MMP-9 was required for crossing the uroepithelial basement membranes.
The migratory defect of neutrophils lacking MMP-9 was analogous to that in TNFR-KO (Fig. 3a, b), suggesting a direct or indirect causal connection between TNF and MMP-9. We tested this by measuring MMP-9 protein levels in neutrophils in TNFR-KO and wild-type mice. By ELISA for MMP-9 we only observed a trend, but no significant effect of TNF (Supplementary Fig. 13). We corroborated these findings by in gel zymography analysis16b. In line with the above observations MMP-9 levels were reduced by 70–80% in TNFR-KO and TNF-KO animals (Fig. 4j,k). A similar reduction was seen in mice depleted of Ly6C+ macrophages, the main TNF producers (Fig. 4j,k), indicating that TNF from Ly6C+ macrophages caused the induction of MMP-9. However, our finding that TNFR expression by neutrophils was dispensable for transepithelial migration (Supplementary Fig. 5) implied that a downstream mediator, such as CXCL2, induced the secretion of MMP-9 in neutrophils. We tested this by stimulating neutrophils directly with CXCL2 and measuring their gelatin zymolytic level16b. CXCL2 increased MMP-9 levels in wild-type and TNFR-KO, but not in Cxcr2-KO neutrophils (Supplementary Fig. 14). By contrast, TNF was unable to induce MMP-9 in neutrophils (Supplementary Fig. 14). This confirmed that CXCL2, and not TNF, activated neutrophils to release MMP-9. To test for in vivo relevance in UTI, we inhibited CXCL2 in infected mice with a blocking antibody. This reduced MMP-9 levels in the bladder substantially (Fig. 4j, k) and aggravated UTI (Fig. 2e). By contrast, levels of MMP-2, which has also been implicated in gelatin degradation16, was not regulated by TNF, Ly6C+ macrophages or by CXCL2 (Supplementary Fig. 15). Thus, TNF-induced CXCL2 specifically regulated MMP9 in UTI.
Taken together, our findings document the following sequence of events (Supplementary Fig. 16): 1) resident Ly6C− macrophages sense the infection and produce chemokines including CXCL1, MIF and CCL2, which recruit neutrophils and Ly6C+ macrophages; 2) recruited Ly6C+ macrophages produce TNF in response to the infection, 3) TNF induces CXCL2 production by Ly6C− macrophages, 4) CXCL2 causes MMP-9 secretion in neutrophils, which allows these cells to cross the epithelial basement membrane in order to combat the infection.
These results demonstrate that the antibacterial neutrophil response is coordinated by two macrophage subsets with distinct tasks: The Ly6C− macrophages act as tissue-resident sentinels and attract circulating phagocytes with chemokines. The Ly6C+ macrophages do not directly participate in bacterial elimination, but instead play a hitherto unknown regulatory role. In response to the infection, they license the Ly6C− macrophages to send neutrophils into the frontline of infection, the uroepithelium. The reliance on cues from helper cells is known from adaptive immunity. For example, dendritic cells can cross-prime cytotoxic CD8+ T lymphocytes (CTL) against viral infections only after having been licensed by CD4+ T helper cells or NKT cells17–19. This causes secretion of chemokines binding CCR5 or CCR4, respectively, which attract the CTL towards the licensed DCs. Later, in infected tissues, the crosstalk between T helper cells and dendritic cells results in the production of CXCR3-specific chemokines that direct CTL towards virus-infected cells20. Our findings reveal an analogous role for the crosstalk between Ly6C− and Ly6C+ macrophages in guiding neutrophils within bacterial infected tissues by coordinated chemokine production.
The chemokines that bind CXCR2 are often considered redundant. Ly6C− macrophages produced several such chemokines, which played distinct chemotactic roles in regulating neutrophil migration in UTI. CXCL1, and to a lower extent MIF, caused endothelial migration, whereas CXCL2 played a non-redundant role in epithelial migration14. Epithelial immune cell migration is less well understood than endothelial migration21,22. In endothelial migration towards chemotactic cues, MMP-9 has been shown to be a determinant22b already long ago. Our knowledge about epithelial migration mostly stems from in vitro experiments, which do not mimic all aspects of the in vivo situation, especially barriers formed by basement membranes. The ability of CXCL2 to induce MMP-9, which assists in collagen IV degradation explains how neutrophil can penetrate such barriers. These findings identify MMP-9 as a CXCL2-regulated gate-keeper for tissue barriers.
The factor by which Ly6C+ macrophages licensed Ly6C− macrophages for CXCL2 production was TNF, a cytokine well known to be critical in bacterial infections11. Consequently, pharmacological TNF blockade in patients with rheumatoid arthritis or inflammatory bowel disease causes relapses of mycobacterium tuberculosis infection23, and is correlated with a higher incidence of pneumonia, and, notably, also of UTI24–28. Our findings in TNF-deficient mice can explain the latter association. Furthermore, these mice also showed more intraepithelial bacterial communities, which underlie the frequent relapses of bacterial cystitis, and more infectious relapses. Our findings show that TNF is required for their control. It remains to be seen whether CXCL2-induction is important also in other TNF-dependent bacterial infections, for example listeriosis29 or tuberculosis23.
The ability of intravesically applied CXCL2 to restore transepithelial migration of TNF-deficient neutrophils suggests therapeutic opportunities in patients with relapsing UTI, especially after treatment with TNF blockers. The topical application of chemokines has recently been shown to improve local CTL responses against genital herpes virus30. Accordingly, intravesical CXCL2 instillation might help eliminating bacteria and prevent their spreading into the kidney. Topical CXCL2 provision might also be effective in other infections that exacerbate during TNF inhibitor therapy.
In conclusion, our study demonstrates that the innate immune response against bacterial infections requires a coordinated triage between three phagocyte subsets with distinct tasks: 1. Ly6C− macrophages as sentinels/coordinators, 2. Ly6C+ macrophages as helpers/advisors, 3. Neutrophils as antibacterial effectors. The sentinels request a second opinion from the helpers, before unleashing the antibacterial effector cells. The reliance on the agreement between several immune cells can reduce the likelihood of inducing ‘false-positive’ immune responses and immunopathologies.
Supplementary Material
Supplementary Figure 1: Phenotype of neutrophils, Ly6C− and Ly6C+ macrophages. Flow cytometry analysis of bladder homogenates of infected CX3CR1+/GFP mice. Neutrophils, Ly6C+ and Ly6C− macrophages were distinguished by their expression of Ly6C and F4/80 (Fig. 1a) and analyzed for expression of CX3CR1, CD11c, Gr1, Ly6G, lin (CD3, B220, NK1.1) and CD115.
Supplementary Figure 2: Only Ly6C+ macrophages are sensitive to clodronate-liposome-mediated deletion. Numbers of Ly6C− (top) and Ly6C+ (bottom) macrophages determined at different time points by flow-cytometry in mice treated intravenously with clodronate-liposomes 6 hours prior to infection. Data represent three independent experiments with five mice per group.
Supplementary Figure 3: Antibacterial effector function of neutrophils is not influenced by TNF. a, Flow-cytometric quantitation of phagocytosis of UPECs expressing recombinant GFP by bone marrow-derived neutrophils from wild-type (WT) or TNFR-KO mice. b,c, Elastase (b) and MPO (c) content in neutrophils from infected WT or TNFR-KO mice, determined by flow cytometry 6 hours after infection. d, Bacterial uptake by bone marrow-derived neutrophils after incubation with GFP-expressing UPECs in the presence of 10 ng/ml TNF (rectangles), TNF and 20 μg/ml etanercept (triangles) or left untreated (circles), determined by flow cytometry for GFP content. Extracellular bacteria were killed with 100 μg/ml gentamycin after 100 minutes to prevent interference with the assay for intracellular bactericidal activity. Data are means ± s.e.m. and represent two to three independent experiments with three mice per group.
Supplementary Figure 4: Transfer of Ly6C+ monocytes into TNF-deficient mice restores transepithelial neutrophil migration. Paraffin sections one day after infection of wild-type or TNF-KO mice that had received Ly6C+ bone marrow monocytes from wild-type mice 10 minutes prior to infection. The bar graph indicates 100μm. Data are means ± s.e.m. and represent two independent experiments with three mice per group. Where are the bar graphs ?
Supplementary Figure 5: TNF acts in trans to enable transepithelial neutrophil migration. Neutrophils from the bone marrow of wild-type and TNFR-KO mice were labelled with CFSE and adoptively transferred into wild-type animals. One day after transfer CFSE+ neutrophils were detected by Ly6G staining in bladder cryosections. Data are means ± s.e.m. and represent three independent experiments in groups of five mice.
Supplementary Figure 6: TNF by bone marrow-derived cells is essential for transepithelial migration of neutrophils. a, b, Efficient reconstitution of CD45.1 donor neutrophils, Ly6C+ and Ly6C− macrophages within the bladder 8 (a) and 12 (b) weeks after irradiation of CD45.2 wild-type mice and transfer of CD45.1 bone marrow from wild-type animals. c, Histological sections of one day infected bone marrow chimeric mice expressing TNF or TNFR in all cells (WT into WT) or only in non-hematopoietic cells (TNFKO into WT, TNFR-KO into WT, respectively) by Ly6G. Quantification is shown in Fig. 3e.
Supplementary Figure 7: Expression of molecules in the infected bladder. a, Chemokine and cytokine levels in homogenates from infected (UPEC +) and uninfected (UPEC +) wild-type (WT) and TNFR-KO bladders, determined by FlowCytomix Multiplex assay 6 hours after infection. b, Expression of CD47, CD11b, CD49b and ICAM-1 in the infected bladder of WT, TNF-KO and TNFR-KO mice. Data are means ± s.e.m. and represent three independent experiments with 3–4 mice per group. a and b are not indicated.
Supplementary Figure 8: Lack of CXCL2 release in TNF- and TNFR KO mice and after depleting Ly6C+ macrophages. CXCL2 expression was determined by ELISA in bladder homogenates of wild-type, TNFR-KO, TNF-KO and clodronate-liposome-treated animals (Cl-liposomes) by ELISA 6 and 24 hours after infection.
Supplementary Figure 9: TNF-dependent CXCL2 enables transepithelial migration of neutrophils. Position of neutrophils revealed by green Ly6G staining of bladder sections from infected TNF-KO mice transurethrally injected after 24 and 27 hours with CXCL2 (+CXCL2; lower row) or not injected (−CXCL2; upper row) and mice were sacrificed 28 hours post infection. Depicted are extracted bright field and fluorescence images (DAPI in blue, Ly6G in green) of the image shown in Fig. 4e.
Supplementary Figure 10: MMP-9 is required for neutrophil migration into the uroepithelium. a, Position of neutrophils revealed by green Ly6G staining of bladder sections from wild-type (upper row) and MMP-9-KO mice (lower row) 24 hours after infection, stained with Ly6G (green) and nuclear DAPI counterstaining (blue). b, Quantitative analysis of experiments as depicted in panel (a).
Supplementary Figure 11: MMP-9 is mainly expressed by neutrophils and is independent of TNFR. a, Coexpression of the neutrophil marker Ly6G in MMP9+ cells in infected wild-type and TNFR-KO mice 6 hours after infection by flow-cytometry. b, MMP9 expression levels in Ly6G+ neutrophils determined by flow-cytometry (MFI=mean fluorescence intensity) 6 hours after infection.
Supplementary Figure 12: MMP-9-competent neutrophils migrate into the uroepithelium in MMP-9-KO mice. Representative staining of Ly6G-AlexaFluor568-stained neutrophils in cryosections of bladders of infected MMP-9 mice that had been injected with CFSE-labelled bone marrow from wild-type mice.
Supplementary Figure 13: Expression of MMP-9 in homogenates is not regulated by TNF. MMP-9 protein expression in bladder homogenates from uninfected and infected WT and TNFR-KO mice, determined by ELISA 6 and 24 hours after infection. Data are means ± s.e.m. and represent two independent experiments with 3–5 mice per group.
Supplementary Figure 14: MMP-9 levels are increased by CXCL2. Supernatants were obtained from cell cultures containing bone marrow-derived neutrophils from WT (a), TNFR-KO (b) and CXCR2-KO (c) mice. Neutrophils were stimulated with TNF and/or CXCL2 as indicated or left untreated and MMP-9 levels were determined by zymography. Data are means ± s.e.m. and represent four independent experiments with four (a) and three (b, c) mice per group.
Supplementary Figure 15: MMP-2 levels are influenced neither by TNF nor CXCL2. MMP-2 in bladder homogenates from WT, TNFR-KO, TNF-KO, macrophage-depleted (Cl-liposomes) and anti-CXCL2 treated (αCXCL2) mice, determined by zymography 6 hours after infection. Data are means ± s.e.m. and represent four independent experiments in four (a), three (b, c) groups of 3–5 mice.
Supplementary Figure 16: Graphical summary of the sequence of events. 1) Resident Ly6C− macrophages sensed the infection and produced chemokines including CXCL1, MIF and CCL2, which recruited neutrophils and Ly6C+ macrophages, 2) recruited Ly6C+ macrophages produced TNF, 3) TNF induced CXCL2 production by Ly6C− macrophages, 4) CXCL2 caused MMP9 activation in neutrophils, which allowed them to cross the epithelial basement membrane in order to combat the infection.
Acknowledgments
The authors thank N. Garbi and W. Kastenmüller for helpful discussions, Juliane Maurer, Christina Ohliger, Tanja Töpfer, Chrystel Llanto, Peter Wurst, Greet Thijs, Manfred Dewor, and Hongqi Lue, and Andreas Dolf for technical assistance and H. Blüthmann for TNFR-KO mice. We acknowledge support by the Central Animal Facilities of the Medical Faculty Bonn and the Flow-Cytometry Core Facility at the Institutes of Molecular Medicine and Experimental Immunology Bonn. This work was supported by the Deutsche Forschungsgemeinschaft (Grants En984/1-1, KFO228, SFBTR57 TP07, TP10, and TP21, Excellence Cluster ImmunoSensation) and the EU Consortium INTRICATE and the Geconcerteerde Onderzoeksacties (GOA 2013/014) at the KU Leuven.
Footnotes
Author Contributions
M.S., C.W., L.F. and D.R.E. designed and performed the experiments. S.G. A.D., S.T., T.Q., and M.F. performed further experiments. U.P., H.-J. G., W.K. G.O, J.B., H.-J. G., P.A.K. and R.B. contributed essential analytic tools and edited the manuscript. C.K. and D.R.E conceived and supervised the study, interpreted the results and wrote the paper. All authors discussed and interpreted results.
References
- 1.Soehnlein O, Lindbom L. Phagocyte partnership during the onset and resolution of inflammation. Nat Rev Immunol. 2010;10:427–439. doi: 10.1038/nri2779. [DOI] [PubMed] [Google Scholar]
- 1b.Borregaard N. Neutrophils, from marrow to microbes. Immunity. 2010;33:657–670. doi: 10.1016/j.immuni.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 1c.Fan X, Patera AC, Pong-Kennedy A, Deno G, Gonsiorek W, Manfra DJ, Vassileva G, Zeng M, Jackson C, Sullivan L, Sharif-Rodriguez W, Opdenakker G, Van Damme J, Hedrick JA, Lundell D, Lira SA, Hipkin RW. Murine CXCR1 is a functional receptor for GCP-2/CXCL6 and interleukin-8/CXCL8. J Biol Chem. 2007;282:11658–11666. doi: 10.1074/jbc.M607705200. [DOI] [PubMed] [Google Scholar]
- 2.Schulz C, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 2b.Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, Wieghofer P, Heinrich A, Riemke P, Hölscher C, Müller DN, Luckow B, Brocker T, Debowski K, Fritz G, Opdenakker G, Diefenbach A, Biber K, Heikenwalder M, Geissmann F, Rosenbauer F, Prinz M. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci. 2013;16:273–80. doi: 10.1038/nn.3318. [DOI] [PubMed] [Google Scholar]
- 3.Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. 2006;7:311–317. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- 4.Foxman B. Epidemiology of urinary tract infections: incidence, morbidity, and economic costs. Am J Med. 2002;113(Suppl 1A):5S–13S. doi: 10.1016/s0002-9343(02)01054-9. [DOI] [PubMed] [Google Scholar]
- 5.Russo TA, Johnson JR. Medical and economic impact of extraintestinal infections due to Escherichia coli: focus on an increasingly important endemic problem. Microbes Infect. 2003;5:449–456. doi: 10.1016/s1286-4579(03)00049-2. [DOI] [PubMed] [Google Scholar]
- 6.Mulvey MA, et al. Induction and evasion of host defenses by type 1-piliated uropathogenic Escherichia coli. Science. 1998;282:1494–1497. doi: 10.1126/science.282.5393.1494. [DOI] [PubMed] [Google Scholar]
- 7.Martinez JJ, Mulvey MA, Schilling JD, Pinkner JS, Hultgren SJ. Type 1 pilus-mediated bacterial invasion of bladder epithelial cells. The EMBO journal. 2000;19:2803–2812. doi: 10.1093/emboj/19.12.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Svensson M, et al. Acute pyelonephritis and renal scarring are caused by dysfunctional innate immunity in mCxcr2 heterozygous mice. Kidney Int. 2011;80:1064–1072. doi: 10.1038/ki.2011.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang D, et al. A toll-like receptor that prevents infection by uropathogenic bacteria. Science. 2004;303:1522–1526. doi: 10.1126/science.1094351. [DOI] [PubMed] [Google Scholar]
- 10.Frendeus B, et al. Interleukin 8 receptor deficiency confers susceptibility to acute experimental pyelonephritis and may have a human counterpart. J Exp Med. 2000;192:881–890. doi: 10.1084/jem.192.6.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6:173–182. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 12.Klebanoff SJ, et al. Stimulation of neutrophils by tumor necrosis factor. J Immunol. 1986;136:4220–4225. [PubMed] [Google Scholar]
- 13.Shalaby MR, et al. Activation of human polymorphonuclear neutrophil functions by interferon-gamma and tumor necrosis factors. J Immunol. 1985;135:2069–2073. [PubMed] [Google Scholar]
- 14.Hang L, et al. Macrophage inflammatory protein-2 is required for neutrophil passage across the epithelial barrier of the infected urinary tract. J Immunol. 1999;162:3037–3044. [PubMed] [Google Scholar]
- 15.Armstrong DA, Major JA, Chudyk A, Hamilton TA. Neutrophil chemoattractant genes KC and MIP-2 are expressed in different cell populations at sites of surgical injury. J Leukocyte Biol. 2004;75:641–648. doi: 10.1189/jlb.0803370. [DOI] [PubMed] [Google Scholar]
- 15b.Van den Steen PE, Proost P, Brand DD, Kang AH, Van Damme J, Opdenakker G. Generation of glycosylated remnant epitopes from human collagen type II by gelatinase B. Biochemistry. 2004;43:10809–10816. doi: 10.1021/bi0493665. [DOI] [PubMed] [Google Scholar]
- 15c.Rosenblum G, Van den Steen PE, Cohen SR, Bitler A, Brand DD, Opdenakker G, Sagi I. Direct visualization of protease action on collagen triple helical structure. PLoS One. 2010;5:e11043. doi: 10.1371/journal.pone.0011043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van den Steen PE, et al. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9) Crit Rev Biochem Mol Biol. 2002;37:375–536. doi: 10.1080/10409230290771546. [DOI] [PubMed] [Google Scholar]
- 16b.Vandooren J, Geurts N, Martens E, Van den Steen PE, Opdenakker G. Zymography methods for visualizing hydrolytic enzymes. Nat Methods. 2013;10:211–220. doi: 10.1038/nmeth.2371. [DOI] [PubMed] [Google Scholar]
- 17.Smith CM, et al. Cognate CD4(+) T cell licensing of dendritic cells in CD8(+) T cell immunity. Nat Immunol. 2004;5:1143–1148. doi: 10.1038/ni1129. [DOI] [PubMed] [Google Scholar]
- 18.Castellino F, et al. Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell-dendritic cell interaction. Nature. 2006;440:890–895. doi: 10.1038/nature04651. [DOI] [PubMed] [Google Scholar]
- 19.Semmling V, et al. Alternative cross-priming through CCL17-CCR4-mediated attraction of CTLs toward NKT cell-licensed DCs. Nat Immunol. 2010;11:313–320. doi: 10.1038/ni.1848. ni.1848 [pii] 10.1038/ni.1848. [DOI] [PubMed] [Google Scholar]
- 20.Nakanishi Y, Lu B, Gerard C, Iwasaki A. CD8(+) T lymphocyte mobilization to virus-infected tissue requires CD4(+) T-cell help. Nature. 2009;462:510–513. doi: 10.1038/nature08511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 22.Nourshargh S, Hordijk PL, Sixt M. Breaching multiple barriers: leukocyte motility through venular walls and the interstitium. Nat Rev Mol Cell Biol. 2010;11:366–378. doi: 10.1038/nrm2889. [DOI] [PubMed] [Google Scholar]
- 22b.D’Haese A, Wuyts A, Dillen C, Dubois B, Billiau A, Heremans H, Van Damme J, Arnold B, Opdenakker G. In vivo neutrophil recruitment by granulocyte chemotactic protein-2 is assisted by gelatinase B/MMP-9 in the mouse. J Interferon Cytokine Res. 2000;20:667–674. doi: 10.1089/107999000414853. [DOI] [PubMed] [Google Scholar]
- 23.Flynn JL, et al. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity. 1995;2:561–572. doi: 10.1016/1074-7613(95)90001-2. [DOI] [PubMed] [Google Scholar]
- 24.Wright HL, Moots RJ, Bucknall RC, Edwards SW. Neutrophil function in inflammation and inflammatory diseases. Rheumatology (Oxford) 2010;49:1618–1631. doi: 10.1093/rheumatology/keq045. [DOI] [PubMed] [Google Scholar]
- 25.Winthrop KL, Chang E, Yamashita S, Iademarco MF, LoBue PA. Nontuberculous mycobacteria infections and anti-tumor necrosis factor-alpha therapy. Emerg Infect Dis. 2009;15:1556–1561. doi: 10.3201/eid1510.090310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dixon WG, et al. Rates of serious infection, including site-specific and bacterial intracellular infection, in rheumatoid arthritis patients receiving anti-tumor necrosis factor therapy: results from the British Society for Rheumatology Biologics Register. Arthritis Rheum. 2006;54:2368–2376. doi: 10.1002/art.21978. [DOI] [PubMed] [Google Scholar]
- 27.Listing J, et al. Infections in patients with rheumatoid arthritis treated with biologic agents. Arthritis Rheum. 2005;52:3403–3412. doi: 10.1002/art.21386. [DOI] [PubMed] [Google Scholar]
- 28.Strangfeld A, Listing J. Infection and musculoskeletal conditions: Bacterial and opportunistic infections during anti-TNF therapy. Best Pract Res Clin Rheumatol. 2006;20:1181–1195. doi: 10.1016/j.berh.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 29.Pfeffer K, et al. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell. 1993;73:457–467. doi: 10.1016/0092-8674(93)90134-c. [DOI] [PubMed] [Google Scholar]
- 30.Shin H, Iwasaki A. A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature. 2012;491:463–467. doi: 10.1038/nature11522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zigmond E, et al. Ly6C hi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen-presenting cells. Immunity. 2012;37:1076–1090. doi: 10.1016/j.immuni.2012.08.026. [DOI] [PubMed] [Google Scholar]
- 32.Bernhagen, et al. Nat Med. 2007 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Phenotype of neutrophils, Ly6C− and Ly6C+ macrophages. Flow cytometry analysis of bladder homogenates of infected CX3CR1+/GFP mice. Neutrophils, Ly6C+ and Ly6C− macrophages were distinguished by their expression of Ly6C and F4/80 (Fig. 1a) and analyzed for expression of CX3CR1, CD11c, Gr1, Ly6G, lin (CD3, B220, NK1.1) and CD115.
Supplementary Figure 2: Only Ly6C+ macrophages are sensitive to clodronate-liposome-mediated deletion. Numbers of Ly6C− (top) and Ly6C+ (bottom) macrophages determined at different time points by flow-cytometry in mice treated intravenously with clodronate-liposomes 6 hours prior to infection. Data represent three independent experiments with five mice per group.
Supplementary Figure 3: Antibacterial effector function of neutrophils is not influenced by TNF. a, Flow-cytometric quantitation of phagocytosis of UPECs expressing recombinant GFP by bone marrow-derived neutrophils from wild-type (WT) or TNFR-KO mice. b,c, Elastase (b) and MPO (c) content in neutrophils from infected WT or TNFR-KO mice, determined by flow cytometry 6 hours after infection. d, Bacterial uptake by bone marrow-derived neutrophils after incubation with GFP-expressing UPECs in the presence of 10 ng/ml TNF (rectangles), TNF and 20 μg/ml etanercept (triangles) or left untreated (circles), determined by flow cytometry for GFP content. Extracellular bacteria were killed with 100 μg/ml gentamycin after 100 minutes to prevent interference with the assay for intracellular bactericidal activity. Data are means ± s.e.m. and represent two to three independent experiments with three mice per group.
Supplementary Figure 4: Transfer of Ly6C+ monocytes into TNF-deficient mice restores transepithelial neutrophil migration. Paraffin sections one day after infection of wild-type or TNF-KO mice that had received Ly6C+ bone marrow monocytes from wild-type mice 10 minutes prior to infection. The bar graph indicates 100μm. Data are means ± s.e.m. and represent two independent experiments with three mice per group. Where are the bar graphs ?
Supplementary Figure 5: TNF acts in trans to enable transepithelial neutrophil migration. Neutrophils from the bone marrow of wild-type and TNFR-KO mice were labelled with CFSE and adoptively transferred into wild-type animals. One day after transfer CFSE+ neutrophils were detected by Ly6G staining in bladder cryosections. Data are means ± s.e.m. and represent three independent experiments in groups of five mice.
Supplementary Figure 6: TNF by bone marrow-derived cells is essential for transepithelial migration of neutrophils. a, b, Efficient reconstitution of CD45.1 donor neutrophils, Ly6C+ and Ly6C− macrophages within the bladder 8 (a) and 12 (b) weeks after irradiation of CD45.2 wild-type mice and transfer of CD45.1 bone marrow from wild-type animals. c, Histological sections of one day infected bone marrow chimeric mice expressing TNF or TNFR in all cells (WT into WT) or only in non-hematopoietic cells (TNFKO into WT, TNFR-KO into WT, respectively) by Ly6G. Quantification is shown in Fig. 3e.
Supplementary Figure 7: Expression of molecules in the infected bladder. a, Chemokine and cytokine levels in homogenates from infected (UPEC +) and uninfected (UPEC +) wild-type (WT) and TNFR-KO bladders, determined by FlowCytomix Multiplex assay 6 hours after infection. b, Expression of CD47, CD11b, CD49b and ICAM-1 in the infected bladder of WT, TNF-KO and TNFR-KO mice. Data are means ± s.e.m. and represent three independent experiments with 3–4 mice per group. a and b are not indicated.
Supplementary Figure 8: Lack of CXCL2 release in TNF- and TNFR KO mice and after depleting Ly6C+ macrophages. CXCL2 expression was determined by ELISA in bladder homogenates of wild-type, TNFR-KO, TNF-KO and clodronate-liposome-treated animals (Cl-liposomes) by ELISA 6 and 24 hours after infection.
Supplementary Figure 9: TNF-dependent CXCL2 enables transepithelial migration of neutrophils. Position of neutrophils revealed by green Ly6G staining of bladder sections from infected TNF-KO mice transurethrally injected after 24 and 27 hours with CXCL2 (+CXCL2; lower row) or not injected (−CXCL2; upper row) and mice were sacrificed 28 hours post infection. Depicted are extracted bright field and fluorescence images (DAPI in blue, Ly6G in green) of the image shown in Fig. 4e.
Supplementary Figure 10: MMP-9 is required for neutrophil migration into the uroepithelium. a, Position of neutrophils revealed by green Ly6G staining of bladder sections from wild-type (upper row) and MMP-9-KO mice (lower row) 24 hours after infection, stained with Ly6G (green) and nuclear DAPI counterstaining (blue). b, Quantitative analysis of experiments as depicted in panel (a).
Supplementary Figure 11: MMP-9 is mainly expressed by neutrophils and is independent of TNFR. a, Coexpression of the neutrophil marker Ly6G in MMP9+ cells in infected wild-type and TNFR-KO mice 6 hours after infection by flow-cytometry. b, MMP9 expression levels in Ly6G+ neutrophils determined by flow-cytometry (MFI=mean fluorescence intensity) 6 hours after infection.
Supplementary Figure 12: MMP-9-competent neutrophils migrate into the uroepithelium in MMP-9-KO mice. Representative staining of Ly6G-AlexaFluor568-stained neutrophils in cryosections of bladders of infected MMP-9 mice that had been injected with CFSE-labelled bone marrow from wild-type mice.
Supplementary Figure 13: Expression of MMP-9 in homogenates is not regulated by TNF. MMP-9 protein expression in bladder homogenates from uninfected and infected WT and TNFR-KO mice, determined by ELISA 6 and 24 hours after infection. Data are means ± s.e.m. and represent two independent experiments with 3–5 mice per group.
Supplementary Figure 14: MMP-9 levels are increased by CXCL2. Supernatants were obtained from cell cultures containing bone marrow-derived neutrophils from WT (a), TNFR-KO (b) and CXCR2-KO (c) mice. Neutrophils were stimulated with TNF and/or CXCL2 as indicated or left untreated and MMP-9 levels were determined by zymography. Data are means ± s.e.m. and represent four independent experiments with four (a) and three (b, c) mice per group.
Supplementary Figure 15: MMP-2 levels are influenced neither by TNF nor CXCL2. MMP-2 in bladder homogenates from WT, TNFR-KO, TNF-KO, macrophage-depleted (Cl-liposomes) and anti-CXCL2 treated (αCXCL2) mice, determined by zymography 6 hours after infection. Data are means ± s.e.m. and represent four independent experiments in four (a), three (b, c) groups of 3–5 mice.
Supplementary Figure 16: Graphical summary of the sequence of events. 1) Resident Ly6C− macrophages sensed the infection and produced chemokines including CXCL1, MIF and CCL2, which recruited neutrophils and Ly6C+ macrophages, 2) recruited Ly6C+ macrophages produced TNF, 3) TNF induced CXCL2 production by Ly6C− macrophages, 4) CXCL2 caused MMP9 activation in neutrophils, which allowed them to cross the epithelial basement membrane in order to combat the infection.