

Abstract
Pathway analysis can complement point-wise single nucleotide polymorphism (SNP) analysis in exploring genomewide association study (GWAS) data to identify specific disease-associated genes that can be candidate causal genes. We propose a straightforward methodology that can be used for conducting a gene-based pathway analysis using summary GWAS statistics in combination with widely available reference genotype data. We used this method to perform a gene-based pathway analysis of a type 1 diabetes (T1D) meta-analysis GWAS (of 7,514 cases and 9,045 controls). An important feature of the conducted analysis is the removal of the major histocompatibility complex gene region, the major genetic risk factor for T1D. Thirty-one of the 1,583 (2%) tested pathways were identified to be enriched for association with T1D at a 5% false discovery rate. We analyzed these 31 pathways and their genes to identify SNPs in or near these pathway genes that showed potentially novel association with T1D and attempted to replicate the association of 22 SNPs in additional samples. Replication P-values were skewed (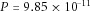 ) with 12 of the 22 SNPs showing
) with 12 of the 22 SNPs showing 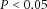 . Support, including replication evidence, was obtained for nine T1D associated variants in genes ITGB7 (rs11170466,
. Support, including replication evidence, was obtained for nine T1D associated variants in genes ITGB7 (rs11170466, 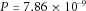 ), NRP1 (rs722988,
), NRP1 (rs722988, 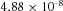 ), BAD (rs694739,
), BAD (rs694739, 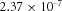 ), CTSB (rs1296023,
), CTSB (rs1296023, 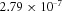 ), FYN (rs11964650,
), FYN (rs11964650, 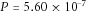 ), UBE2G1 (rs9906760,
), UBE2G1 (rs9906760, 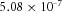 ), MAP3K14 (rs17759555,
), MAP3K14 (rs17759555, 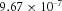 ), ITGB1 (rs1557150,
), ITGB1 (rs1557150, 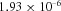 ), and IL7R (rs1445898,
), and IL7R (rs1445898, 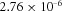 ). The proposed methodology can be applied to other GWAS datasets for which only summary level data are available.
). The proposed methodology can be applied to other GWAS datasets for which only summary level data are available.
Keywords: pathway analysis, genomewide association data, meta-analysis
Introduction
It is increasingly recognized that pathway analysis can complement point-wise single nucleotide polymorphism (SNP) analysis in exploring genomewide association study (GWAS) data, through the identification of pathways and SNPs (genes) associated with the tested phenotype. A number of pathway analysis methods have been proposed recently that incorporate biological knowledge about genes (or SNPs) to find pathways associated with the tested phenotypes [Carbonetto and Stephens, 2013; Eleftherohorinou et al., 2009; Evangelou et al., 2012, 2013; Holmans et al., 2009; O'Dushlaine et al., 2009; Schaid et al., 2012; Wang et al., 2007, 2011; Yu et al., 2009]. These methods can be characterized by a number of aspects, including the tested null hypothesis, the input data, their test statistics and the way of assessing the significance of each pathway. One of the two null hypotheses is the competitive (enrichment) one, that states that the pathway genes are no more associated with the phenotype than the nonpathway genes. Therefore, an enriched pathway, contains more significantly associated genes than would be expected by chance. Additionally, a number of studies have been published that compared some of these methods under different settings [Evangelou et al., 2012; Tintle et al., 2009]. Evangelou et al. [2012] showed that the Fisher's method and the adaptive rank truncated product method are the most powerful methods for testing the competitive null hypothesis, in agreement with the previous literature.
One of the crucial steps of a gene-based pathway analysis is the assignment of a gene statistic that represents the association of each gene with the tested trait. Two popular statistics are the minimum P-value statistic and the Fisher's method statistic [Chapman and Whittaker, 2008]. Permutation procedures are needed to adjust for gene size and linkage disequilibrium (LD) between the SNPs assigned to the gene, both of which are considered to be confounding factors of pathway analysis [Evangelou et al., 2012; Wang et al., 2007]. In many cases, only summary GWAS statistics are publicly available and therefore, in this present study, we propose using genotype data available from reference panels, for example, we have used the genotype data of the WTCCC controls, for generating the null distribution of the two aforementioned gene statistics.
Several statistical models have been proposed that incorporate the pathway membership of SNPs or genes for finding SNPs associated with complex traits. Examples include the Bayesian hierarchical models proposed by Evangelou et al. [2013] and Carbonetto and Stephens [2013], and the variable selection method applied by Eleftherohorinou et al. [2009]. In the present study, we used the enriched pathways to increase our prior belief for association of SNPs near pathway genes. Instead of applying a complex statistical model for finding SNPs associated with the tested phenotype, we propose a simple procedure for prioritizing SNPs. SNPs in or near genes in enriched pathways that have small P-values and that have not been reported previously as associated with the tested phenotype are selected for further analyses. We propose that by using additional samples for replicating the association, novel associations can be found. We argue that SNPs with combined P-values less than 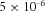 and with replication P-values less than 0.05 in additional cohorts are potential disease associated SNPs, because their membership of enriched pathways increases the prior belief of association.
and with replication P-values less than 0.05 in additional cohorts are potential disease associated SNPs, because their membership of enriched pathways increases the prior belief of association.
In this study, we explored the genetic architecture of type 1 diabetes (T1D) using pathway analysis, through which pathways statistically enriched for association with T1D are defined and used to identify additional T1D loci and candidate genes. T1D is a common autoimmune disease resulting from destruction of the insulin producing beta cells in the pancreas. Genetic predisposition to T1D has been explored through linkage and association studies. The strongest genetic risk factor for T1D lies in the major histocompatibility complex (MHC) region (chromosome 6p21), with 49 further loci showing association (T1DBase, 05/05/2014 - Burren et al. [2011]). T1D has a classic polygenic mode of inheritance and hence many more susceptibility loci remain to be mapped, as evident, for example, from the strong linear correlation between the number of samples analyzed and the number of loci reaching genome wide significance in published studies of genetic association in autoimmune diseases [Parkes et al., 2013].
Materials and Methods
Materials
GWAS Data
The Barrett et al. [2009] meta-analysis study includes three constituent studies: WTCCC, T1DGC, and GoKinD/NIMH. The standard quality control filters applied include SNPs with minor allele frequency (MAF) greater than 0.01, less than 5% missing data, and with the Z2-statistic for Hardy-Weinberg equilibrium within the controls smaller than 25. In total, 822,739 SNPs were retained for analysis.
Further, the available raw genotype data of the first two GWAS were analyzed. The WTCCC GWAS was described by The Wellcome Trust Case Control Consortium [2007]. The 2,000 WTCCC cases are part of the genetic resource for investigating diabetes (GRID) collection of the JDRF/wellcome trust diabetes and inflammation laboratory (DIL) [Todd et al., 2007]. One thousand and five hundred controls of this GWAS were recruited by the WTCCC in collaboration with the UK Blood Services and the other 1,868 controls are patients with bipolar disorder included in the WTCCC study. The individuals of this study were genotyped on the Affymetrix 500K Chip. The T1DGC GWAS was first presented in Barrett et al. [2009] and includes 4,000 British cases from the JDRF/Wellcome Trust DIL collection. In addition, 4,000 controls are included from the British 1958 Birth Cohort. The individuals of this GWAS were genotyped on the Illumina 550K platform. Barrett et al. [2009] analyzed both GWAS data using imputation to combine information across the different SNP content on the different chips. The samples that passed the quality control filters applied in Barrett et al. [2009] were included in the pathway analysis presented here. In total, the WTCCC GWAS includes 1,933 cases and 3,339 controls. The T1DGC GWAS includes 3,983 cases and 3,999 controls. Similar quality control filters as the ones applied in Barrett et al. [2009] were applied to the genotype data of both GWAS, except we set a more stringent threshold for missing data of 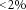 .
.
SNPs within an extended MHC gene region (chr6: 25,000,000–35,000,000) were removed for all three datasets. As discussed by Elbers et al. [2009], the MHC region should be removed from a pathway analysis as it is a region that could potentially bias the analysis by favoring pathways related with immune functions. In T1D the causal genes in the MHC region have been identified as the HLA class II and class I genes and hence exclusion of the MHC region does not compromise our study.
For the purposes of this study, the genotype data of 1,350 controls recruited by the WTCCC in collaboration with the UK Blood Services, were used as the reference genotype panel for estimating the null distributions of the computed gene statistics. As discussed earlier, these controls were genotyped both on the WTCCC chip (Affymetrix 500K chip) and on the T1DGC chip (Illumina 550K platform).
GWAS Genes
One of the major steps of conducting a gene-based pathway analysis is the assignment of SNPs to genes. Our assignment was based on autosomal protein coding genes downloaded from Ensembl (Flicek et al. [2013], October, 2012) human assembly build GRCh37.
SNPs were mapped to genes according to their physical distance: a SNP was mapped to every gene whose coding sequence had an overlap with a 50 kb range around the SNP. In total, 18,528 overlapping genes were identified in the meta-analysis dataset. The WTCCC and T1DGC GWAS genes included were 18,353 and 18,477, respectively.
Pathway Databases
Three hundred and fourteen BioCarta and 1,272 Reactome [Croft et al., 2011; Matthews et al., 2009] pathways were downloaded (October, 2012). Three of the Reactome pathways did not have any of our GWAS genes. The downloaded BioCarta pathways have annotations for 1,572 genes. An average BioCarta pathway contains 17 genes and the largest pathway contains 84 genes. On the other hand, the Reactome pathways have annotations for 6,497 genes. An average Reactome pathway contains 46 genes and the largest Reactome pathway contains 1,740 genes. The two databases share 1,132 genes. Not all pathway genes are included in the lists of GWAS genes, and vice-versa. The three datasets have very similar presentation of genes for either database (Table1).
Table 1.
Summary statistics of the database genes within the two GWAS and the meta-analysis data of Barrett et al. [2009]. The “Theoretical” represents the genes of each pathway database as these were downloaded. These numbers are reduced when SNP coverage within the studies is taken into account
| Database | Study | Minimum | Median | Mean | Maximum |
|---|---|---|---|---|---|
| BioCarta | Theoretical | 1 | 15 | 16.97 | 84 |
| Meta-analysis | 1 | 13 | 14.79 | 76 | |
| WTCCC | 1 | 13 | 14.75 | 76 | |
| T1DGC | 1 | 13 | 14.70 | 76 | |
| Reactome | Theoretical | 1 | 16.50 | 46.31 | 1,740 |
| Meta-analysis | 0 | 15 | 37.23 | 1,506 | |
| WTCCC | 0 | 14 | 36.75 | 1,497 | |
| T1DGC | 0 | 15 | 37.15 | 1,496 |
Methods
Gene Statistics
The measure that summarises the association between disease and all the SNPs assigned to a gene into a single statistic is a crucial step in a gene-based pathway analysis. A number of different gene statistics have been proposed over the years. One popular choice is the minimum P-value of all the SNPs assigned to the gene, i.e. the P-value of the most significant SNP. Chapman and Whittaker [2008] discussed that the minimum P-value has very good performance in cases of both low and high LD between the SNPs mapped to the gene.
An alternative, also presented in Chapman and Whittaker [2008] is the Fisher's statistic
| 1 |
where 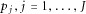 denote the single-SNP analysis P-values of association of the SNPs assigned to the gene with the studied phenotype. The Fisher's statistic has very good performance in cases where LD is high, but has low power in cases with no LD between the SNPs [Chapman and Whittaker, 2008]. The two tested gene statistics are denoted by (A) and (B), respectively. Gene size, i.e. the number of SNPs mapped to a gene, and the LD between the SNPs mapped to a gene can be confounding factors in a gene-based pathway analysis. In order to correct for both gene size and LD between the SNPs assigned to each gene the phenotype permutation procedure discussed in Evangelou et al. [2012] can be used.
denote the single-SNP analysis P-values of association of the SNPs assigned to the gene with the studied phenotype. The Fisher's statistic has very good performance in cases where LD is high, but has low power in cases with no LD between the SNPs [Chapman and Whittaker, 2008]. The two tested gene statistics are denoted by (A) and (B), respectively. Gene size, i.e. the number of SNPs mapped to a gene, and the LD between the SNPs mapped to a gene can be confounding factors in a gene-based pathway analysis. In order to correct for both gene size and LD between the SNPs assigned to each gene the phenotype permutation procedure discussed in Evangelou et al. [2012] can be used.
Here, the phenotypes were permuted 1,000 times and single-SNP analysis was redone. The two aforementioned gene statistics were then recomputed for each gene. The adjusted minimum P-value statistic of each gene is given by
| 2 |
where 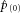 corresponds to the minimum P-value of the gene calculated using the observed data and
corresponds to the minimum P-value of the gene calculated using the observed data and 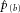 corresponds to the minimum P-value of the gene computed using the bth permuted dataset.
corresponds to the minimum P-value of the gene computed using the bth permuted dataset.
The corresponding adjusted statistic using the Fisher's statistic is given by
| 3 |
where, similarly, 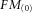 is the Fisher's method statistic calculated using the observed data and
is the Fisher's method statistic calculated using the observed data and 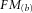 is the Fisher's method statistic computed using the bth permuted dataset.
is the Fisher's method statistic computed using the bth permuted dataset.
Often, only summary statistics are available for published GWAS, preventing the null distribution of the two gene statistics from phenotype or SNP permutations to be computed. In this study, we propose an alternative way of computing the null distribution of the gene statistics by using the genotype data from a reference panel, motivated by the work of Liu et al. [2010] and Swanson et al. [2013] who used genotype data from the HapMap reference panels [The International HapMap Consortium, 2003] for estimating the null distribution for gene-based association tests. We, on the other hand, used the genotype data of the WTCCC controls, to secure that all GWAS genotyped SNPs are included in the study.
We repeatedly generated Z-statistics for all the SNPs assigned to a gene from the multivariate normal distribution with mean zero and variance Σ, where Σ is the covariance matrix of the SNP genotype data estimated from the reference genotype panel. The diagonal of Σ is set equal to 1. Subsequently, SNP P-values were calculated by comparing the Z-statistics with a Normal (0,1) distribution. Both the minimum P-value statistic and the Fisher's method statistic were computed for each gene. This process was repeated 10,000 times. Finally, the simulated gene statistics were compared to the original gene statistics, and adjusted P-values for both gene statistics were generated as described in Equations2 and 3. Here, we would like to note, that both the gene statistics were computed based on the SNPs shared between the GWAS platforms and the reference panel.
For the purposes of distinction between the statistics, the adjusted P-values of the gene statistics computed using the reference genotype data will be referred to as the simulated gene statistics and indicated with subscript S.
Pathway Analysis Methods
As discussed by Evangelou et al. [2012] the Fisher's method and the adaptive rank truncated product method can be adapted to test the competitive null hypothesis by using the gene statistics  . The
. The 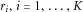 statistics equal the ranks of the genes of the study divided by the total number of genes in the study (K). The distribution of the gene statistics is a Uniform (0,1) and deviation from uniformity suggests enrichment of the pathway. By using the proposed gene statistics the analytic distribution of the Fisher's method and the empirical distribution of the adaptive rank truncated product method are used for testing the significance of the pathways [Evangelou et al., 2012].
statistics equal the ranks of the genes of the study divided by the total number of genes in the study (K). The distribution of the gene statistics is a Uniform (0,1) and deviation from uniformity suggests enrichment of the pathway. By using the proposed gene statistics the analytic distribution of the Fisher's method and the empirical distribution of the adaptive rank truncated product method are used for testing the significance of the pathways [Evangelou et al., 2012].
Fisher's method (FM). The FM statistic equals
| 4 |
where m is the pathway size. The significance of the computed FM statistic is compared with its exact χ2 distribution with 2m degrees of freedom.
Adaptive rank truncated product method (ARTP). The ARTP method is a generalization of the FM where only the best H gene statistics within each pathway are considered for computing the rank truncated product given by
| 5 |
with the gene statistics ranked from the smallest to the largest  . The rank truncated product combines the H smallest gene statistics of the tested pathway. The truncation point H as well as the significance of the P-value of the ARTP statistic were calculated using the empirical distribution of ARTP proposed by Evangelou et al. [2012].
. The rank truncated product combines the H smallest gene statistics of the tested pathway. The truncation point H as well as the significance of the P-value of the ARTP statistic were calculated using the empirical distribution of ARTP proposed by Evangelou et al. [2012].
In summary, there are eight combinations of pathway analysis methods and gene statistics (Table2). All methods were applied to the data of the T1DGC and WTCCC GWAS for comparison.
Table 2.
The names of the methods applied to the data. FM stands for Fisher's method and ARTP stands for adaptive rank truncated product method. The gene statistics computed are the minimum P-value statistic and the Fisher's method statistic, which were adjusted either using a phenotype permutation procedure or using the reference genotype data for generating the corresponding SNP P-values
| Name | Gene statistic | Procedure | Pathway analysis method |
|---|---|---|---|
| FM-(MIN) | Minimum P-value | Phenotype permutation | FM |
| FM-(FM) | Fisher's statistic | Phenotype permutation | FM |
| FM-(MINS) | Minimum P-value | Reference genotype data | FM |
| FM-(FMS) | Fisher's statistic | Reference genotype data | FM |
| ARTP-(MIN) | Minimum P-value | Phenotype permutation | ARTP |
| ARTP-(FM) | Fisher's statistic | Phenotype permutation | ARTP |
| ARTP-(MINS) | Minimum P-value | Reference genotype data | ARTP |
| ARTP(FMS) | Fisher's statistic | Reference genotype data | ARTP |
Simulation Study
A simulation study was also performed to examine the type-I error of the methods. We aimed to compare the type-I error of the methods as well as to test how pathway size affects their type-I error. For estimating the type-I error of the methods across different pathway sizes, 1,000 random pathways of different sizes were created from the list of T1DGC GWAS genes. A 95% confidence interval for a type-I error of 5% ranges between 0.0431 and 0.0569.
Extending Pathway Analysis
Pathway analysis was extended for identifying genes (and SNPs) potentially associated with T1D. We searched through the P-values of the genes within the enriched pathways and we selected the ones that had relatively small P-values but have not been reported previously as associated with T1D. The SNP with the strongest association with T1D within each of the selected genes was genotyped on additional case-control and family datasets either using TaqMan or the ImmunoChip platform, a custom Illumina chip designed for dense coverage of autoimmune and autoinflammatory associated regions (Cortes and Brown [2011], ImmunoBase). We performed an inverse variance meta-analysis for combining the results of the additional cohorts. Further, using Fisher's method we combined the meta-analysis P-values of Barrett et al. [2009] with the P-values of the additional cohorts. SNPs with combined P-values less than 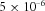 and with replication P-values less than 0.05 are highlighted in the Results Section. We ensured that there was no sample overlap between Barrett et al. [2009] and replication cohorts.
and with replication P-values less than 0.05 are highlighted in the Results Section. We ensured that there was no sample overlap between Barrett et al. [2009] and replication cohorts.
Results
Validation of Proposed Methodology
The permutation- and simulation- adjusted P-values were very similar for the minimum P-value gene statistic for both T1DGC and WTCCC GWAS, with their corresponding Spearman correlations equal to 0.9883 and 0.9690, respectively. Similarly, the Spearman correlations of the Fisher's method statistic were 0.9949 and 0.9896 for the two GWAS.
The eight methods were applied to both GWAS for comparing the agreement between the pathway P-values computed by each pair of competitive methods. Similarly to the observations of the gene statistics, a very high correspondence was observed between FM-(MIN) and FM-(MIN)S for both GWAS across all 1,583 tested pathways (Spearman correlations are 0.9892 and 0.9785 for T1DGC and WTCCC, respectively). The observed correlation between FM-(FM) and FM-(FM)S for both GWAS was similar (T1DGC ρ= 0.9823 and WTCCC ρ = 0.9735 across all 1,583 tested pathways), although lower correlations were observed between the P-values computed using ARTP method (Table3). Further, the simulation study showed that the type-I error of the gene statistics combined with Fisher's method is broadly maintained (Table4).
Table 3.
Spearman correlations of the P-values computed for each tested pathway, for each tested database for both T1DGC and WTCCC GWAS
| Database | Methods compared | Spearman correlation |
|---|---|---|
| T1DGC | ||
| BioCarta | FM-(MIN) vs FM-(MIN)S | 0.9939 |
| FM-(FM) vs FM-(FM)S | 0.9755 | |
| ARTP-(MIN) vs ARTP-(MIN)S | 0.9854 | |
| ARTP-(FM) vs ARTP-(FM)S | 0.9496 | |
| Reactome | FM-(MIN) vs FM-(MIN)S | 0.9878 |
| FM-(FM) vs FM-(FM)S | 0.8378 | |
| ARTP-(MIN) vs ARTP-(MIN)S | 0.9389 | |
| ARTP-(FM) vs ARTP-(FM)S | 0.8833 | |
| WTCCC | ||
| BioCarta | FM-(MIN) vs FM-(MIN)S | 0.9761 |
| FM-(FM) vs FM-(FM)S | 0.9739 | |
| ARTP-(MIN) vs ARTP-(MIN)S | 0.9793 | |
| ARTP-(FM) vs ARTP-(FM)S | 0.9303 | |
| Reactome | FM-(MIN) vs FM-(MIN)S | 0.9784 |
| FM-(FM) vs FM-(FM)S | 0.9729 | |
| ARTP-(MIN) vs ARTP-(MIN)S | 0.9638 | |
| ARTP-(FM) vs ARTP-(FM)S | 0.9210 | |
Table 4.
Type-I error of the gene statistics combined with Fisher's method for the different pathway analysis methods
| Method | ||||
|---|---|---|---|---|
| Pathway size | FM-(MIN) | FM-(FM) | FM-(MINS) | FM-(FMS) |
| 20 | 0.044 | 0.053 | 0.045 | 00057 |
| 50 | 0.043 | 0.041 | 0.043 | 0.044 |
| 100 | 0.059 | 0.071 | 0.060 | 0.058 |
| 200 | 0.053 | 0.054 | 0.048 | 0.049 |
| 500 | 0.042 | 0.053 | 0.046 | 0.055 |
| 1000 | 0.044 | 0.051 | 0.048 | 0.050 |
Given these results, we believe that the approximate null distribution is appropriate for both the FM-(MIN) and FM-(FM) methods, and we proceeded by analysing the Barrett et al. [2009] meta-analysis P-values using both methods.
Results of the Meta-Analysis Study
The complete lists of enriched pathways (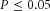 ) found by both methods are presented in Supplementary Tables S5–S8. We corrected for multiplicity of the tested pathways by computing the false discovery rate (FDR) P-values of the pathways [Benjamini and Hochberg, 1995]. Thirty-one BioCarta and Reactome pathways had FM-(MIN)S FDR P-values less than 0.05, whereas 21 pathways were identified by FM-(FM)S as enriched. As a larger number of pathways were identified by FM-(MIN)S method, we are presenting these enriched pathways in Table5. Four of the enriched pathways of Table5, the BioCarta pathways “Adhesion molecules on Lymphocyte,” “Antigen dependent B cell activation,” “B lymphocyte cell surface molecules,” and “Lck and Fyn tyrosine kinases in initiation of TCR activation” were previously
) found by both methods are presented in Supplementary Tables S5–S8. We corrected for multiplicity of the tested pathways by computing the false discovery rate (FDR) P-values of the pathways [Benjamini and Hochberg, 1995]. Thirty-one BioCarta and Reactome pathways had FM-(MIN)S FDR P-values less than 0.05, whereas 21 pathways were identified by FM-(FM)S as enriched. As a larger number of pathways were identified by FM-(MIN)S method, we are presenting these enriched pathways in Table5. Four of the enriched pathways of Table5, the BioCarta pathways “Adhesion molecules on Lymphocyte,” “Antigen dependent B cell activation,” “B lymphocyte cell surface molecules,” and “Lck and Fyn tyrosine kinases in initiation of TCR activation” were previously
Table 5.
Pathways with FDR P-values of FM-(MIN)S method less than 0.05
| Number | Pathway | FDR p-value FM-(MIN)S |
|---|---|---|
| i | Activation of Csk by cAMP-dependent Protein Kinase Inhibits Signaling through the T Cell Receptor | 0.0436 |
| ii | IL-2 Receptor Beta Chain in T cell Activation | 0.0293 |
| iii | HIV Induced T Cell Apoptosis | 0.0106 |
| iv | CTL mediated immune response against target cells | 0.0323 |
| v | Antigen Dependent B Cell Activation | 0.0364 |
| vi | IL-10 Anti-inflammatory Signaling Pathway | 0.0115 |
| vii | Stathmin and breast cancer resistance to antimicrotubule agents | 0.0460 |
| viii | T Helper Cell Surface Molecules | 0.0021 |
| ix | NO2-dependent IL 12 Pathway in NK cells | 0.0372 |
| x | T Cytotoxic Cell Surface Molecules | 0.0014 |
| xi | IL 17 Signaling Pathway | 0.0387 |
| xii | The Co-Stimulatory Signal During T-cell Activation | 0.0003 |
| xiii | Lck and Fyn tyrosine kinases in initiation of TCR Activation | 0.0012 |
| xiv | Role of Tob in T-cell activation | 0.0414 |
| xv | T Cell Receptor and CD3 Complex | 0.0414 |
| xvi | Selective expression of chemokine receptors during T-cell polarization | 0.0395 |
| xvii | B Lymphocyte Cell Surface Molecules | 0.0375 |
| xviii | Monocyte and its Surface Molecules | 0.0460 |
| xix | Adhesion Molecules on Lymphocyte | 0.0429 |
| xx | Double Stranded RNA Induced Gene Expression | 0.0375 |
| xxi | IFN alpha signaling pathway | 0.0375 |
| xxii | Immune System | 0.0216 |
| xxiii | Adaptive Immune System | 0.0216 |
| xxiv | Integrin cell surface interactions | 0.0216 |
| xxv | Semaphorin interactions | 0.0299 |
| xxvi | Immunoregulatory interactions between a Lymphoid and a non-Lymphoid cell | 0.0012 |
| xxvii | Effects of PIP2 hydrolysis | 0.0216 |
| xxviii | Interleukin-6 signaling | 0.0445 |
| xxix | Signal regulatory protein (SIRP) family interactions | 0.0216 |
| xxx | Catecholamine biosynthesis | 0.0299 |
| xxxi | GRB7 events in ERBB2 signaling | 0.0264 |
identified by Peng et al. [2010] as enriched with T1D by analysing only the WTCCC GWAS dataset.
Using Enriched Pathways to Prioritize Potentially Novel T1D SNPs
We explored the gene members of the 31 enriched pathways listed in Table5 by looking at their unadjusted minimum P-values. We selected the genes with minimum meta-analysis SNP P-values less than 10−4 that have not been reported previously as associated with T1D (Table6).
Table 6.
Genes of the enriched pathways that have not been reported previously as associated with T1D, but have case-control meta-analysis minimum SNP P-values less than 10−4. The most significant SNP assigned to each gene with its meta-analysis P-value are given in columns 3 and 4. Columns 5 and 6 present the meta-analysis P-value of additional cohorts if available and the combined P-value with Barrett et al. [2009] P-value, respectively. The seventh column presents any other immune (either autoimmune or autoinflammatory) disease(s) that the genes are associated with (ImmunoBase, 26/08/2014). The disease symbols correspond to: UC, Ulcerative colitis; CEL, Celiac disease; PSO, psoriasis; IBD, inflammatory bowel disease; ALO, Alopecia; and CRO, Crohn's disease. The last column presents the pathway number that each gene belongs to (Table5)
| Gene | Chr | Most significant SNP | Barrett et al. [2009] meta-analysis SNP P-value | Additional cohorts meta-analysis SNP P-value | Combined P-value | Gene is located or is a candidate gene within an immune disease region | Pathway membership |
|---|---|---|---|---|---|---|---|
| PSMB2 | 1p34.3 | rs6703605 | 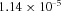 |
xxii, xxiii | |||
| IL6R | 1q21.3 | rs6427658 | 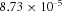 |
0.3260 | 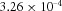 |
AS, JIA, RA | xxii, xxviii |
| FASLG | 1q24.3 | rs10912276 | 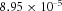 |
0.0290 | 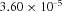 |
CEL, CRO, IBD | ii, iii, iv, v |
| PTPRC | 1q31.3 | rs2182419 | 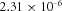 |
0.9593 | 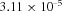 |
RA | i, viii, x, xiii, xvii, xxii, xxiii, xxv |
| ITGA6 | 2q31.1 | rs16860458 | 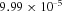 |
xxiv | |||
| RAF1 | 3p25.1 | rs2450855 | 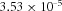 |
0.7642 | 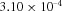 |
ii, xxii, xxiii | |
| TLR6 | 4p14 | rs4321646 | 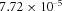 |
xxii | |||
| TLR10 | 4p14 | rs4321646 | 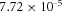 |
xxii | |||
| TRPC3 | 4q27 | rs4502701 | 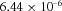 |
0.3697 | 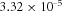 |
xxvii | |
| IL7R | 5p13.2 | rs1445898 | 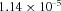 |
0.0146 | 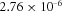 |
MS, PBC, UC, T1D | xxii |
| DCTN4 | 5q33.1 | rs4246045 | 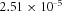 |
0.8456 | 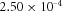 |
CRO, UC, IBD | xxii, xxiii |
| MAPK14 | 6p21.31 | rs2237093 | 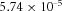 |
xxii | |||
| IRF4 | 6p25.3 | rs2048698 | 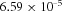 |
0.0296 | 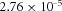 |
CEL, PSO, RA | xxii |
| FYN | 6q21 | rs11964650 | 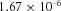 |
0.0183 | 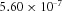 |
UC, CRO, IBD | xiii, xxii, xxiii, xxv |
| CTSB | 8p23.1 | rs1296023 | 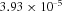 |
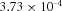 |
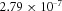 |
xxii, xxiii | |
| ITGB1 | 10p11.22 | rs1557150 | 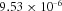 |
0.0119 | 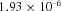 |
xviii, xix, xxii, xxiii, xxiv, xxv, xxvi | |
| NRP1 | 10p11.22 | rs722988 | 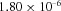 |
0.0013 | 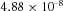 |
xxv | |
| PSMC3 | 11p11.2 | rs2293576 | 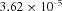 |
0.7537 | 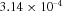 |
MS | xxii, xxiii |
| BAD | 11q13.1 | rs694739 | 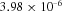 |
0.0031 | 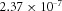 |
CRO, MS, UC, ALO, IBD | ii, xxii, xxiii |
| AMICA1 | 11q23.3 | rs11216829 | 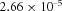 |
0.5807 | 0.0002 | xxii, xxiii, xxiv, xxvi | |
| ITGB7 | 12q13.13 | rs11170466 | 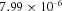 |
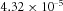 |
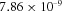 |
xxii, xxiii, xxiv, xxvi | |
| DGKA | 12q13.2 | rs11171710 | 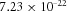 |
xxvii | |||
| DNAJC3 | 13q32.1 | rs9302086 | 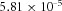 |
xx | |||
| HMGB1 | 13q12.3 | rs1360485 | 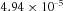 |
0.3819 | 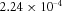 |
xxii | |
| PRKCH | 14q23.1 | rs1111107 | 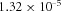 |
RA | xxvii | ||
| SOCS1 | 16p13.13 | rs149310 | 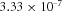 |
0.0064 | 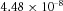 |
CEL, CRO, JIA, MS, PBC, PSO, UC, IBD | ii, xxii, xxiii |
| TP53 | 17p13.1 | rs16956936 | 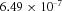 |
0.0834 | 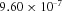 |
xx | |
| UBE2G1 | 17p13.2 | rs9906760 | 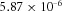 |
0.0047 | 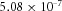 |
xxii, xxiii | |
| MAP3K14 | 17q21.31 | rs17759555 | 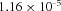 |
0.0047 | 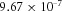 |
MS | xx, xxii, xxiii |
| CDC34 | 19p13.3 | rs12982646 | 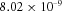 |
0.2323 | 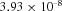 |
xxii, xxiii | |
| MADCAM1 | 19p13.3 | rs12982646 | 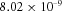 |
0.2323 | 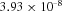 |
xxii, xxiii, xxiv, xxvi | |
| SAE1 | 19q13.32 | rs411560 | 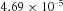 |
MS | xxii, xxiii |
Almost 50% of our selected genes have been associated with other immune diseases. For example, FYN lies in regions that are associated with Crohn's disease, inflammatory bowel disease (IBD) and ulcerative colitis [Jostins and et al., 2012].FASLG lies in a region known to be associated with Crohn's disease and IBD [Franke et al., 2010; Jostins and et al., 2012] as well as with celiac disease [Trynka et al., 2011]. Moreover, SNP rs10912276 of FALSG is in perfect LD (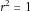 ) with the index SNP rs12068671 of the gene region for celiac disease (
) with the index SNP rs12068671 of the gene region for celiac disease (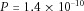 , Trynka et al. [2011]).
, Trynka et al. [2011]).
The last column of Table6 shows the pathways that the genes belong to. The Reactome pathways “Immune system” and “Adaptive immune system” contain 24 and 17 genes of Table6, respectively, and they share 17 genes. On the other hand, 12 of the enriched pathways do not contain any of the genes presented in Table6. This suggests that the enrichment of these pathways was driven by genes already known to be associated with T1D. For example, one of them the “The Co-stimulatory signal during T-cell activation” pathway contains four genes that lie in regions associated previously with T1D: CTLA4, IL2, ICOS, and PTPN11, where CTLA4 and IL2 are likely causal gene candidates.
We tested whether the SNPs in Table6 were associated with T1D in additional case-control and family data. In total, genotype data for additional samples existed for 22 out of 30 selected SNPs (Supplementary Table S1). Twelve of the 22 SNPs had replication P-values less than 0.05 in the additional samples, an event with associated probability 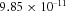 , whereas just one of them would be expected to have a P-value less than 0.05 by chance. The skewing of the replication P-values toward smaller values for the SNPs of Table6 suggests that more of these SNPs are expected to replicate with larger cohorts. We identified ten SNPs with combined P-values less than
, whereas just one of them would be expected to have a P-value less than 0.05 by chance. The skewing of the replication P-values toward smaller values for the SNPs of Table6 suggests that more of these SNPs are expected to replicate with larger cohorts. We identified ten SNPs with combined P-values less than 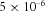 and with replication P-values less than 0.05 in the additional cohorts in or near the genes IL7R, FYN, CTSB, ITGB1, NRP1, BAD, ITGB7, SOCS1, UBE2G1, and MAP3K14. Although not all of these SNPs have reached genomewide significance (P
and with replication P-values less than 0.05 in the additional cohorts in or near the genes IL7R, FYN, CTSB, ITGB1, NRP1, BAD, ITGB7, SOCS1, UBE2G1, and MAP3K14. Although not all of these SNPs have reached genomewide significance (P
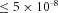 ), their membership of enriched pathways increases the prior for association.
), their membership of enriched pathways increases the prior for association.
SNP rs11170466 of ITGB7 reached genomewide significance (combined P-value=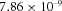 , Table6) and the signal is independent of the neighboring T1D region 12q13.3 (Supplementary Table S2). SNP rs11170466 is also a cis-eQTL in blood cells for ITGB7 (unadjusted P-value
, Table6) and the signal is independent of the neighboring T1D region 12q13.3 (Supplementary Table S2). SNP rs11170466 is also a cis-eQTL in blood cells for ITGB7 (unadjusted P-value 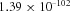 ), where the minor allele is associated both with increased risk of T1D and also with increased gene expression (Supplementary Table S1, [Westra et al., 2012]). The minor allele of SNP rs722988 of NRP1 reached genomewide significance with combined P-value =
), where the minor allele is associated both with increased risk of T1D and also with increased gene expression (Supplementary Table S1, [Westra et al., 2012]). The minor allele of SNP rs722988 of NRP1 reached genomewide significance with combined P-value = 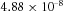 .
.
SNP rs193779 near SOCS1 showed a strong association with T1D (combined P-value= 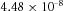 ). However, SOCS1 is located very close to an established T1D locus, with SNPs in intron 19 of CLEC16A reported to alter T1D risk through their effect on expression of DEXI [Davison et al., 2011]. We conditioned on the most associated SNP in the CLEC16A gene region and found the association with rs193779 was considerably attenuated (
). However, SOCS1 is located very close to an established T1D locus, with SNPs in intron 19 of CLEC16A reported to alter T1D risk through their effect on expression of DEXI [Davison et al., 2011]. We conditioned on the most associated SNP in the CLEC16A gene region and found the association with rs193779 was considerably attenuated ( ; Supplementary Table S4).
; Supplementary Table S4).
SNP rs1296023 of CTSB showed evidence of association with T1D (combined P-value=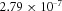 ; Table6). This is a novel association with T1D and the first association of T1D on chromosome 8 (T1DBase). SNP rs694739 of BAD also showed a strong association with T1D (combined P-value =
; Table6). This is a novel association with T1D and the first association of T1D on chromosome 8 (T1DBase). SNP rs694739 of BAD also showed a strong association with T1D (combined P-value = 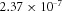 ). BAD is an interesting causal candidate gene as it overlaps with a region on chromosome 11q13.1 known to be associated with multiple sclerosis, ulcerative colitis, IBD, alopecia, and Crohn's disease (ImmunoBase). Our tested SNP rs694739 has shown convincing evidence of association with multiple sclerosis, Crohn's disease and alopecia and it is considered to be the index SNP for these three diseases in this gene region (ImmunoBase).
). BAD is an interesting causal candidate gene as it overlaps with a region on chromosome 11q13.1 known to be associated with multiple sclerosis, ulcerative colitis, IBD, alopecia, and Crohn's disease (ImmunoBase). Our tested SNP rs694739 has shown convincing evidence of association with multiple sclerosis, Crohn's disease and alopecia and it is considered to be the index SNP for these three diseases in this gene region (ImmunoBase).
SNPs rs9906760, rs11964650, and rs17759555 of UBE2G1, FYN, and MAP3K14 showed association with T1D with combined P-values less than 10−6. FYN has also been highlighted by Carbonetto and Stephens [2013] as a novel candidate T1D gene. FYN lies in a region of chromosome 6q21 known to be associated with Crohn's disease and ulcerative colitis [Jostins and et al., 2012]. SNP rs11964650 is not in LD with the index Crohn's disease and ulcerative colitis SNP of the region (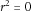 with rs3851228, ImmunoBase). The UBE2G1 SNP rs9906760 is related with decreased expression of cis-eQTL in blood cells with an unadjusted P-value
with rs3851228, ImmunoBase). The UBE2G1 SNP rs9906760 is related with decreased expression of cis-eQTL in blood cells with an unadjusted P-value 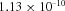 [Westra et al., 2012].
[Westra et al., 2012].
SNP rs1557150 near ITGB1 also showed association with T1D, with combined P-value 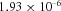 (Table6). We confirmed that the signal of SNP rs1557150 near ITGB1 is independent of the signal of SNP rs722988 of NRP1 (Supplementary Table S3). SNP rs1445898 of IL7R with combined P-value =
(Table6). We confirmed that the signal of SNP rs1557150 near ITGB1 is independent of the signal of SNP rs722988 of NRP1 (Supplementary Table S3). SNP rs1445898 of IL7R with combined P-value = 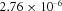 is a novel T1D association on chromosome 5.
is a novel T1D association on chromosome 5.
FYN, CTSB, BAD, ITGB7, UBE2G1, and MAP3K14 are members of the Reactome “Immune system” and “Adaptive immune system” pathways and are six of the 17 genes shared between the two pathways.
One SNP highlighted by our analysis is rs12982646 in CDC34/MADCAM1. Both genes are members of the reactome pathways “Immune system” and “Adaptive immune system,” and MADCAM1 member of the enriched reactome pathways “Integrin cell surface interaction” and “Immunoregulatory interactions between a lymphoid and a nonlymphoid cell” (with combined P-value = 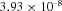 ). Eleftherohorinou et al. [2009] also identified MADCAM1 as associated with T1D. rs12982646 exceeded the genomewide significance threshold in Barrett et al. [2009] but at the time were unable to test for replication of the SNP because reliable TaqMan data were not available. The replication genotype data used here, both from ImmunoChip and a new and robust TaqMan assay, did not show any evidence of association (Table6). All our samples were subsequently genotyped on TaqMan, which confirmed the fidelity of the genotype calls in the Barrett et al. [2009] study and ImmunoChip (with 99.8% genotype agreement across 6,258 samples), but also showed no overall replication in the independent samples (combined P-value = 0.2323). Therefore, we conclude, that this SNP is either not associated with T1D, or associated with too small an effect to be detected in our available replication samples.
). Eleftherohorinou et al. [2009] also identified MADCAM1 as associated with T1D. rs12982646 exceeded the genomewide significance threshold in Barrett et al. [2009] but at the time were unable to test for replication of the SNP because reliable TaqMan data were not available. The replication genotype data used here, both from ImmunoChip and a new and robust TaqMan assay, did not show any evidence of association (Table6). All our samples were subsequently genotyped on TaqMan, which confirmed the fidelity of the genotype calls in the Barrett et al. [2009] study and ImmunoChip (with 99.8% genotype agreement across 6,258 samples), but also showed no overall replication in the independent samples (combined P-value = 0.2323). Therefore, we conclude, that this SNP is either not associated with T1D, or associated with too small an effect to be detected in our available replication samples.
Discussion
Our results illustrate the additional biological understanding and novel genetic associations that can be revealed in existing GWAS data by pathway analysis. We have shown by comparative analysis of real datasets that our proposed methodology for obtaining the null distribution of gene statistics using reference genotype panels and summary GWAS statistics is comparable to that found by phenotype permutations when full GWAS data are available. Although motivated by published approaches to gene-based association testing, this idea has not, to our knowledge, been applied to pathway analysis. Given the difficulties that can arise accessing individual level genetic data, our method will allow broader application of pathway analyses to published GWAS. Instead of using the genotype data from the available controls, a potential alternative is the use of the freely available genotype data either from the 1,000 Genomes project [The 1000 Genomes Project Consortium, 2012] or from the HapMap project. A potential drawback of using such genotype data is the loss of some of the SNPs genotyped on the GWAS platform but not included in the reference panel.
Over the last few years, a number of pathway analyses of the WTCCC T1D GWAS data have been published [Carbonetto and Stephens, 2013; Eleftherohorinou et al., 2009; Peng et al., 2010; Wang et al., 2011]. Most of these studies reported pathways related with “Antigen processing and presentation,” “Jak-STAT signaling,” “MAPK signaling” and “Type 1 diabetes mellitus” as enriched with T1D. A number of our enriched pathways can be regarded as novel because none of the previous published pathway analyses of T1D identified them, as for example the BioCarta pathway “The Co-stimulatory signal during T-cell activation” and the Reactome pathway “Immunoregulatory interactions between a lymphoid and a nonlymphoid cell.” The differences between our results and the results of previous analyses can be characterized by the greater sample size we used and by the exclusion of the MHC region. Pathways such as “Antigen processing and presentation” are characterized by the inclusion of the MHC region. By taking into account the enrichment of MHC in their proposed statistical method, Carbonetto and Stephens [2013] reported the “IL-2 signaling pathway” [Geer et al., 2010; Schaefer et al., 2009] as enriched for T1D.
Carbonetto and Stephens [2013] used their model-based approach for prioritizing variants within the enriched “IL-2 signaling pathway,” their analysis of the WTCCC T1D GWAS showed seven regions of the genome to have strong evidence for association within the pathway. Three of these (RAF1, MAPK14, FYN) had not been reported as associated with T1D previously and were suggested by Carbonetto and Stephens [2013] as potential candidate causal genes for T1D. The three genes were also highlighted by our approach as all of them are members of the enriched Reactome pathway “Immune system.” FYN is a key molecule in T cells and consequently a key signaling functional candidate in the T cell mediated autoimmune process of T1D.
We extended this approach by searching SNPs assigned to genes within all enriched pathways and selected the genes with relatively small P-values that have not been associated with T1D previously. Equally importantly, we also genotyped the selected SNPs in additional case-control and families for finding potential new T1D associations. Through the analyses of the additional datasets we identified nine novel T1D associated genes and variants, SNP rs1111107 of ITGB7, SNP rs722988 of NRP1, SNP rs694739 of BAD, SNP rs1296023 of CTSB, SNP rs11964650 of FYN, SNP rs9906760 of UBE2G1, SNP rs17759555 of MAP3K14, SNP rs1557150 of ITGB1, and SNP rs1445898 of IL7R.
Both ITGB7 and ITGB1 encode proteins that function directly with each other in receptor–ligand interactions in the homing of T cells from blood to tissues such as the intestine and pancreas. Although we cannot be confident of the MADCAM1 T1D association, MADCAM1 encodes the pancreas expressed receptor for the 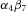 homing receptor on CD4+ T cells, encoded by genes ITGA4 and ITGB7, and hence is a highly plausible biological candidate. Interestingly, there is a peak of SNP association, with P-value
homing receptor on CD4+ T cells, encoded by genes ITGA4 and ITGB7, and hence is a highly plausible biological candidate. Interestingly, there is a peak of SNP association, with P-value 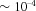 , ∼300 kb 5′ of ITGA4 in the ImmunoChip results (T1DBase, Supplementary Table S1), and the ITGB1 protein competes with ITGB7 in the
, ∼300 kb 5′ of ITGA4 in the ImmunoChip results (T1DBase, Supplementary Table S1), and the ITGB1 protein competes with ITGB7 in the 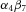 receptor [DeNucci et al., 2010]. Monoclonal antibodies against MADCAM1 and
receptor [DeNucci et al., 2010]. Monoclonal antibodies against MADCAM1 and 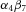 are showing clinical benefits in inflammatory bowel disease [Sheridan, 2014], and hence based on our genetic results presented here, investigation of the effects of these drugs in T1D is worth considering.
are showing clinical benefits in inflammatory bowel disease [Sheridan, 2014], and hence based on our genetic results presented here, investigation of the effects of these drugs in T1D is worth considering.
Furthermore, CTSB, encoding the lysosomal protease, cathepsin B, is included in the broadly defined Reactome pathways “Immune system” and “Adaptive immune system” and could participate in many processes such as apoptosis, autophagy and the NALP3 inflammsome. Nevertheless, it is not an obvious candidate gene. rs1296023 associates with CTSB expression in monocytes (with 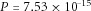 , Zeller et al. [2010]), with the T1D risk allele associating with increased expression. This adds support to the possibility that CTSB is a T1D causal gene. This SNP or region has not been associated with any other disease (http://www.genome.gov/) or with any immune disease (ImmunoBase), which makes it interesting in that it could be unique to T1D.
, Zeller et al. [2010]), with the T1D risk allele associating with increased expression. This adds support to the possibility that CTSB is a T1D causal gene. This SNP or region has not been associated with any other disease (http://www.genome.gov/) or with any immune disease (ImmunoBase), which makes it interesting in that it could be unique to T1D.
BAD is an obvious candidate gene, encoding a key proapoptopic protein, BCL2-associated agonist of cell death, associated previously with Crohn's disease, ulcerative colitis, IBD, alopecia and multiple sclerosis, and, for example, recently shown to function in TNF-a induced apoptosis [Yan et al., 2013]. The BAD SNP rs694739 is the same SNP as reported for Crohn's disease, multiple sclerosis and alopecia, but only 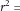 0.16 with the reported SNP for ulcerative colitis and IBD (ImmunoBase). This gene has also been associated with platelet count, via rs477895 [Qayyum et al., 2012], with very little LD between this and the T1D SNP (
0.16 with the reported SNP for ulcerative colitis and IBD (ImmunoBase). This gene has also been associated with platelet count, via rs477895 [Qayyum et al., 2012], with very little LD between this and the T1D SNP (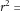 0.1, ImmunoBase). It appears that there may be multiple causal variants affecting BAD expression and/or function across a range of cell types. IL7R and NRP1 are obvious functional candidate genes, being key molecules in the adaptive immune response [Delgoffe et al., 2013; Jäger et al., 2013]. Note that the IL7R signal that we report, which peaks at rs1445898 is distinct from the exonic multiple sclerosis associated SNP rs6897932 which alters splicing (
0.1, ImmunoBase). It appears that there may be multiple causal variants affecting BAD expression and/or function across a range of cell types. IL7R and NRP1 are obvious functional candidate genes, being key molecules in the adaptive immune response [Delgoffe et al., 2013; Jäger et al., 2013]. Note that the IL7R signal that we report, which peaks at rs1445898 is distinct from the exonic multiple sclerosis associated SNP rs6897932 which alters splicing (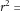 0.42) [Gregory et al., 2007].
0.42) [Gregory et al., 2007].
The skewing of the replication P-values toward smaller values for the SNPs of Table6 suggests that more of these SNPs are expected to replicate with larger cohorts, and emphasize the potential utility of applying our proposed pathway analysis method to GWAS for which only summary statistics are available. This is also supported by the fact that some of the genes that just miss reaching our statistical threshold do show intriguing and probably meaningful links to the mechanisms of T1D. A combination of increased sample size in T1D and further GWAS coupled to the pathway analysis described here will further increase our understanding of disease mechanisms, allowing the targeting of specific genes, molecules, and cells for functional studies. The greater the number of genes and pathways accurately identified, the greater the chance of selecting pathways that might be amenable to specific therapeutic modulation. The identification of the 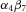 T cell adhesion pathway in T1D genetic etiology does provide a new target for potential therapeutic intervention in T1D.
T cell adhesion pathway in T1D genetic etiology does provide a new target for potential therapeutic intervention in T1D.
Web Resources
Reactome pathways: http://www.reactome.org/cgi-bin/mart
BioCarta pathways: http://cgap.nci.nih.gov/pathways/BioCarta_pathways
Ensembl: http://www.ensembl.org/index.html
T1DBase: http://www.t1dbase.org
ImmunoBase: http://www.immunobase.org
R package for pathway analysis: PAGWAS http://cran.r-project.org/web/packages/PAGWAS/index.html
Blood eQTL browser http://genenetwork.nl/bloodeqtlbrowser/
Author Contributions
Conceived and designed the experiments: ME CW. Performed the experiments: ME. Analyzed the data: ME MDF HG JAT. TaqMan genotyping: DJS. ImmunoChip genotyping: SOG WMC PC SSR. Prepared data: OSB NMW. Wrote the manuscript: ME JAT CW. All authors have read and approved the manuscript.
Acknowledgments
Chris Wallace and Hui Guo are funded by the Wellcome Trust (WT089989) and Mary Fortune is funded by the Wellcome Trust (WT099772/Z/12/Z).
This work was supported by the JDRF UK Centre for Diabetes Genes, Autoimmunity and Prevention (D-GAP; 4-2007-1003), the JDRF International, the Wellcome Trust (WT061858/091157), the National Institute for Health Research Cambridge Biomedical Research Centre (CBRC) and the Medical Research Council (MRC) Cusrow Wadia Fund. The Cambridge Institute for Medical Research (CIMR) is in receipt of a Wellcome Trust Strategic Award (100140).
We gratefully acknowledge the participation of all the patients, control subjects and family members. We would like to thank the UK Medical Research Council and Wellcome Trust for funding the collection of DNA for the British 1958 Birth Cohort (MRC grant G0000934, WT grant 068545/Z/02). DNA control samples were prepared and provided by S. Ring, R. Jones, M. Pembrey, W. McArdle, D. Strachan, and P. Burton.
We thank David Dunger, Barry Widmer, and the British Society for Paediatric Endocrinology and Diabetes for the TID case collection. We acknowledge use of DNA from The UK Blood Services collection of Common Controls (UKBS collection), funded by the Wellcome Trust grant 076113/C/04/Z, by the Wellcome Trust/JDRF grant 061858, and by the National Institute of Health Research of England. The collection was established as part of the Wellcome Trust Case Control Consortium.
We acknowledge use of DNA from the Human Biological Data Interchange and Diabetes UK for the USA and UK multiplex families, respectively; D. Savage, C. Patterson, D. Carson and P. Maxwell for the Northern Irish families; the Genetics of Type 1 Diabetes in Finland (GET1FIN); J. Tuomilehto, L. Kinnunen, E. Tuomilehto-Wolf, V. Harjutsalo and T. Valle for the Finnish families; and C. Guja and C. Ionescu-Tirgoviste for the Romanian families.
This research utilizes resources provided by the Type 1 Diabetes Genetics Consortium, a collaborative clinical study sponsored by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), National Institute of Allergy and Infectious Diseases (NIAID), National Human Genome Research Institute (NHGRI), National Institute of Child Health and Human Development (NICHD), and Juvenile Diabetes Research Foundation International (JDRF) and supported by U01 DK062418.
This study makes use of data generated by the Wellcome Trust Case Control Consortium. A full list of the investigators who contributed to the generation of the data are available from http://www.wtccc.org.uk/. Funding for the project was provided by the Wellcome Trust under award 076113.
We gratefully acknowledge the National Institute of Mental Health for generously allowing the use of their control CEL and genotype data. Control subjects from the National Institute of Mental Health Schizophrenia Genetics Initiative (NIMH-GI), data and biomaterials are being collected by the Molecular Genetics of Schizophrenia II (MGS-2) collaboration. The investigators and co-investigators are as follows: P.V. Gejman (Collaboration coordinator) and A.R. Sanders (ENH/Northwestern University, MH059571); F. Amin (Emory University School of Medicine, MH59587); N. Buccola (Louisiana State University Health Sciences Center, MH067257); W. Byerley (University of California-Irvine,MH60870); C.R. Cloninger (Washington University, St. Louis, U01, MH060879); R. Crowe (PI) and D. Black (University of Iowa, MH59566); R. Freedman (University of Colorado, MH059565); D. Levinson (University of Pennsylvania, MH061675); B. Mowry (University of Queensland, MH059588); and J. Silverman (Mt. Sinai School of Medicine, MH59586). The samples were collected by V.L. Nimgaonkar's group at the University of Pittsburgh as part of a multi-institutional collaborative research project with J. Smoller and P. Sklar (Massachusetts General Hospital) (grant MH 63420).
We acknowledge the National Institutes of Health for allowing the use of their control allele signal intensity and genotype data. The dataset(s) used for the analyses described in this manuscript were obtained from the GAIN Database, controlled through dbGaP accession number phs000018.v1.p1.
We gratefully acknowledge the participation of all Cambridge BioResource (CBR) volunteers. We thank staff of the CBR recruitment team for assistance with volunteer recruitment and K. Beer, T. Cook, S. Hall and J. Rice for blood sample collection. We thank M. Woodburn and T. Attwood for their contribution to sample management. We thank members of the Cambridge BioResource SAB and management committee for their support and the National Institute for Health Research Cambridge Biomedical Research Centre for funding.
The authors would like to thank Premanand Achuthan with his help regarding accessing the pathways data, Ellen Schofield and Ricardo Ferreira for helpful discussions.
References
- Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, Julier C, Morahan G, Nerup J, Nierras C. Genome-wide association study and meta-analysis finds over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41:703–707. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol B (Methodological) 1995;57:289–300. [Google Scholar]
- Burren OS, Adlem EC, Achuthan P, Christensen M, Coulson RMR, Todd JA. T1DBase: update 2011, organization and presentation of large-scale data sets for type 1 diabetes research. Nucleic Acids Research. 2011;39(Database Issue):D997–D1001. doi: 10.1093/nar/gkq912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbonetto P, Stephens M. Integrated enrichment analysis of variants and pathways in genome-wide association studies indicates central for for IL-2 signaling genes in type 1 diabets, and cytokine signaling genes in Crohn's disease. PLoS Genet. 2013;9:e1003770. doi: 10.1371/journal.pgen.1003770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman J, Whittaker J. Analysis of multiple SNPs in a candidate gene or region. Genet Epidemiol. 2008;32:560–566. doi: 10.1002/gepi.20330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes A, Brown MA. Promise and pitfalls of the ImmunoChip. Arthritis Res Ther. 2011;13:101. doi: 10.1186/ar3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft D, O'Kelly G, Wu G, Haw R, Gillespie M, Matthews L, Caudy M, Garapati P, Gopinath G, Jassal B. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res. 2011;39(Database issue):D691–D697. doi: 10.1093/nar/gkq1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davison LJ, Wallace C, Cooper JD, Cope NF, Wilson NK, Smyth DJ, Howson JM, Saleh N, Al-Jeffery A, Angus KL. Long-range dna looping and gene expression analyses identify dexi as an autoimmune disease candidate gene. Hum Mol Genet. 2011;21(2):322–333. doi: 10.1093/hmg/ddr468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgoffe GM, Woo SR, Turnis ME, Gravano DM, Guy C, Overacre AE, Bettini ML, Vogel P, Finkelstein D, Bonnevier J. Stability and function on regulatory T cells is maintained by a neuropilin-1-semaphorin-4a axis Nature. 2013;501:252–256. doi: 10.1038/nature12428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNucci CC, Pagán AJ, Mitchell JS, Shimizu Y. Control of alpha4beta7 integrin expression and cd4 t cell homing by the beta1 integrin subunit. J Immunol. 2010;184:2458–2467. doi: 10.4049/jimmunol.0902407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbers CC, van Eijk KR, Franke L, Mulder F, van der Schouw YT, Wijmenga C, Onland-Moret NC. Using genome-wide pathway analysis to unravel the etiology of complex diseases. Genet Epidemiol. 2009;33:419–430. doi: 10.1002/gepi.20395. [DOI] [PubMed] [Google Scholar]
- Eleftherohorinou H, Wright V, Hoggart C, Hartikainen AL, Jarvelin MR, Balding D, Coin L, Levinet M. Pathway analysis of GWAS provides new insights into genetic susceptibility to 3 inflammatory diseases. PLoS One. 2009;4:e8068. doi: 10.1371/journal.pone.0008068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evangelou M, Rendon A, Ouwehand WH, Wernisch L, Dudbridge F. Comparison of methods for competitive tests of pathway analysis. PLoS One. 2012;7:e41018. doi: 10.1371/journal.pone.0041018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evangelou M, Dudbridge F, Wernisch L. Two novel pathway analysis methods based on a hierarchical model. Bioinformatics. 2013;30(5):690–697. doi: 10.1093/bioinformatics/btt583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S 2013. Ensembl. Nucleic Acids Res. 2013;41(Database issue):D48–D55. doi: 10.1093/nar/gks1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geer LY, Marchler-Bauer A, Geer RC, Han L, He J, He S, Liu C, Shi W, Bryant SH. The NCBI BioSystems database. Nucleic Acids Res. 2010;38:D492–D496. doi: 10.1093/nar/gkp858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory SG, Schmidt S, Seth P, Oksenberg JR, Hart J, Prokop A. Ban M, Goris A, Barcellos LF. Interleukin 7 receptor α chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat Genet. 2007;39:1083–1091. doi: 10.1038/ng2103. Caillier SJ, and others. [DOI] [PubMed] [Google Scholar]
- Holmans P, Green EK, Pahwa JS, Ferreira MA, Purcell SM, Sklar P WellcomeTrust Case-Control Consortium. Owen MJ, MC'Donovan O. Craddock N. Gene ontology analysis of GWA study data sets provides insights into the biology of bipolar disorder. Am J Hum Genet. 2009;85:13–24. doi: 10.1016/j.ajhg.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäger J, Schulze C, Rösner S, Martin R. IL7RA haplotype-associated alteration in cellular immune function and gene expression patterns in multiple sclerosis. Genes Immun. 2013;14:453–461. doi: 10.1038/gene.2013.40. [DOI] [PubMed] [Google Scholar]
- Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY. Schumm LP, Sharma Y, Anderson CA. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. Lee JC, and others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JZ, Mcrae AF, Nyholt DR, Medland SE, Wray NR, Brown KM, Hayward NK, Montgomery GW, Visscher PM, Martin NG. A versatile gene-based test for genome-wide association studies. Am J Hum Genet. 2010;87:139–145. doi: 10.1016/j.ajhg.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews L, Gopinath G, Gillespie M, Caudy M, Croft D, de Bono B, Garapati P, Hemish J, Hermjakob H, Jassal B. Reactome knowledgebase of human biological pathways and processes. Nucleic Acids Res. 2009;37(Database issue):D619–D622. doi: 10.1093/nar/gkn863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Dushlaine C, Heron EA, Segurado R, Gill M, Morris DW, Corvin A. The SNP ratio test: pathway analysis of genome-wide association datasets. Bioinform, Appl Note. 2009;25:2762–2763. doi: 10.1093/bioinformatics/btp448. [DOI] [PubMed] [Google Scholar]
- Parkes M, Cortes A, van Heel DA, Brown MA. Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nat Rev, Genet. 2013;14:661–673. doi: 10.1038/nrg3502. [DOI] [PubMed] [Google Scholar]
- Peng G, Luo L, Siu H, Zhu Y, Hu P, Hong S, Zhao J, Zhou X. Jin L. Gene and pathway-based second-wave analysis of genome-wide association studies. Eur J Hum Genet. 2010;18:111–117. doi: 10.1038/ejhg.2009.115. Reveille JD, and others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qayyum R, Snively BM, Ziv E, Nalls MA, Liu Y, Tang W, Yanek LR, Lange L, Evans MK, Ganesh S. A meta-analysis and genome-wide association study of platelet count and mean platelet volume in African Americans. PLoS Genet. 2012;8:e1002491. doi: 10.1371/journal.pgen.1002491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer CF, Anthony K, Krupa S, Buchoff J, Day M, Hannay T. PID: the pathway interaction database. Nucleic Acids Research. 2009;37:D674–D679. doi: 10.1093/nar/gkn653. Buetow KH. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaid DJ, Sinnwell JP, Jenkins GD, McDonnell SK, Ingle JN, Kubo M, Goss PE, Costantino JP, Wickerham DL, Weinshilboum RM. Using the gene ontology to scan multilevel gene sets for associations in genome wide association studies. Genet Epidemiol. 2012;36:3–16. doi: 10.1002/gepi.20632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan C. First integrin inhibitor since tysabri nears approval for IBD. Nat Biotechnol. 2014;32:205–207. doi: 10.1038/nbt0314-205. [DOI] [PubMed] [Google Scholar]
- Swanson DM, Blacker D, AlChawa T, Ludwig KU, Mangold E, Lange C. Properties of permutation-based gene tests and controlling type 1 error using a summary statistic based gene test. BMC Genet. 2013;14:108. doi: 10.1186/1471-2156-14-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000GenomesProjectConsortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The InternationalHapMapConsortium. The international hapmap project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- The WellcomeTrustCaseControlConsortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tintle N, Borchers B, Brown M, Bekmetjev A. Comparing gene set analysis methods on single-nucleotide polymorphism data from genetic analysis workshop 16. BMC Proceedings. 2009;3:S96. doi: 10.1186/1753-6561-3-s7-s96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, Bailey R, Nejentsev S, Field SF, Payne F. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. 2007;39:857–884. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trynka G, Hunt KA, Bockett NA, Romanos J, Mistry V, Szperl A. Bardella MT. Castillejo G. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat Genet. 2011;43:1193–1201. doi: 10.1038/ng.998. Bakker SF,, Bhaw-Rosun L, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Bucan M. Pathway-based approaches for analysis of genomewide association studies. Am J Hum Genet. 2007;81:1278–1283. doi: 10.1086/522374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Jia P, Wolfinger RD, Chen X, Grayson BL, Aune TM, Zhao Z. An efficient hierarchical generalized linear mixed model for pathway analysis of genome-wide association studies. Bioinformatics. 2011;27:686–692. doi: 10.1093/bioinformatics/btq728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra H-J, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, Christiansen MW, Fairfax BP, Schramm K, Powelland JE. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet. 2012;45:1238–1243. doi: 10.1038/ng.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Xiang J, Lin Y, Ma J, Zhang J, Zhang H, Sun J, Danial NN, Liu J, Lin A. Inactivation of BAD by IKK inhibits TNF α-Induced apoptosis independently of NF-κB activation. Cell. 2013;152:304–315. doi: 10.1016/j.cell.2012.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Li Q, Bergen AW, Pfeiffer RM, Rosenberg PS, Caporaso N, Kraft P, Chatterjee N. Pathway analysis by adaptive combination of p-values. Genet Epidemiol. 2009;33:700–709. doi: 10.1002/gepi.20422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller T, Wild P, Szymczak S, Rotival M, Schillert A, Castagne R, Maouche S, Germain M, Lackner K, Rossmann H. Genetics and beyond the transcriptome of human monocytes and disease susceptibility. PLoS One. 2010;5:e10693. doi: 10.1371/journal.pone.0010693. [DOI] [PMC free article] [PubMed] [Google Scholar]