

Abstract
Glycogen synthase kinase-3 (GSK3), particularly the isoform GSK3β, has been implicated in a wide range of physiological systems and neurological disorders including Alzheimer's Disease. However, the functional importance of GSK3α has been largely untested. The multifunctionality of GSK3 limits its potential as a drug target because of inevitable side effects. Due to its greater expression in the CNS, GSK3β rather than GSK3α has also been assumed to be of primary importance in synaptic plasticity. Here, we investigate bidirectional long-term synaptic plasticity in knockin mice with a point mutation in GSK3α or GSK3β that prevents their inhibitory regulation. We report that only the mutation in GSK3α affects long-term potentiation (LTP) and depression (LTD). This stresses the importance of investigating isoform specificity for GSK3 in all systems and suggests that GSK3α should be investigated as a drug target in cognitive disorders including Alzheimer's Disease. © 2014 The Authors. Hippocampus Published by Wiley Periodicals, Inc.
Keywords: glycogen synthase kinase-3, long-term potentiation, long-term depression, electrophysiology, knockin mice
INTRODUCTION
GSK3 is a highly conserved, multifunctional serine/threonine kinase; particularly abundant in the hippocampus (Grimes and Jope, 2001). Two isoforms of GSK3 are encoded by distinct genes, alpha and beta (Woodgett, 1990) and assumed to have similar function (Asuni et al., 2006). However, the isoform-specific actions are poorly understood. GSK3 is active under basal conditions but can be inhibited by N-terminal phosphorylation of Ser21 in GSK3α and Ser9 in GSK3β (Rubio-Moscardo et al., 2013). Many types of proteins are phosphorylated by GSK3 (Jope and Johnson, 2004) involving it in many intracellular pathways (Rayasam et al., 2009) and this, together with its prominent neuronal expression, leads GSK3 to be implicated in various neurological disorders including Alzheimer's Disease (Cohen and Goedert, 2004; Hernandez et al., 2009). Recent research has consequently focussed on its role in synaptic plasticity (Giese, 2009).
Consistent with a role in memory dysfunction, previous studies (Hooper et al., 2008; Peineau et al., 2008) indicate that GSK3:
inhibits long-term potentiation (LTP) when over-expressed
prevents long-term depression (LTD) but not LTP when inhibited and
is inhibited by LTP and activated by LTD.
Most authors have assumed this pivotal role to depend on GSK3β (Peineau et al., 2008), despite using general GSK3 inhibitors which cannot differentiate the functions of the GSK3 isoforms (Cohen and Goedert, 2004; Georgievska et al., 2013). Other studies have attempted to be more specific by using over- or under-expression models of GSK3β (Kimura et al., 2008; Kaidanovich-Beilin and Woodgett, 2011). However, nonphysiological levels of GSK3 may have various unforeseen knock-on effects, especially between the isoforms. Distinct cellular localisation of GSK3α has been demonstrated, which would be consistent with functions independent of the β isoform (Azoulay-Alfaguter et al., 2011). Yet, most previous studies have largely ignored the potential role of GSK3α, perhaps because its expression levels are lower than GSK3β and decrease with age (Giese, 2009).
One study demonstrated convincingly that GSK3β was the dominant isoform in heart and skeletal muscle (Mora et al., 2005) by developing GSK3 knockin mice (KIs) in which the inhibitory serine residue of the respective isoform is mutated to alanine, thereby creating two KI lines that lack the inhibitory regulation of either GSK3α or GSK3β (McManus et al., 2005). Both lines have normal expression of both GSK3 isoforms, but the activity-dependent inactivation of the relevant isoform is disrupted allowing differentiation of isoform-specific functions. To address the relative roles of the two isoforms in synaptic plasticity, we have compared basal synaptic transmission as well as LTP and LTD in hippocampal slices from these same KI mice, using wild-type (WT) litter mates as controls. Homozygous KI and WT littermates were kept together with heterozygous littermates so that mice used in experiments were never single housed. The genotype of all mice used for experimentation was confirmed by post hoc analysis of tail tissue using standard PCR techniques as previously described (McManus et al., 2005). As LTD is difficult to induce in older mice (Milner et al., 2004), two age groups of male KI and WT mice were used in these experiments: 16- to 21-day-old for LTD and 4- to 5-month-old for LTP.
Hippocampal brain slices were prepared using standard procedures modified for mice (Parsley et al., 2007) and left to recover for at least half an hour before stimulating the Schaffer collaterals and recording in the stratum radiatum of the CA1 field of hippocampal slices to obtain pairs of field excitatory postsynaptic potentials (fEPSPs). Once a field response was obtained, standard paired-pulse stimuli (stimulus duration: 100 µs, interstimulus interval: 50 ms) were applied at 0.1 Hz and averaged over 1 min across a range of voltages (10–60 V) to produce an input–output relationship. Thereafter, the stimulus intensity of evoked fEPSPs was set to ∼50% of the voltage required to evoke postsynaptic action potentials (“population spike”) and paired-pulse stimuli were applied at 0.1 Hz and averaged over 1 min. fEPSPs baseline was recorded for at least 15 min or until a stable response was obtained using standard paired-pulse stimuli and the final 10 min taken for analysis. The slope of averaged fEPSPs was determined with WinWCP v.4.1.5 using a linear curve fitting function and data transferred to SPSS 21.0 (IL) for statistical analysis using generalized linear mixed models, which have fewer test-specific assumptions and are more robust than standard analysis of variance (Krueger and Tian, 2004). As there were no statistical differences in the response of the respective WT litter mates of the two KI mice, all data from WT mice were pooled in the analyses.
The input/output ratio was unaffected by genotype at either of the ages studied (Figs. 1A,B) but GSK3αKIs showed significantly lower paired-pulse ratios (PPR) than the other genotypes at both ages (Figs. 1C,D) suggesting an increase in release probability in GSK3αKIs compared with WT mice.
FIGURE 1.
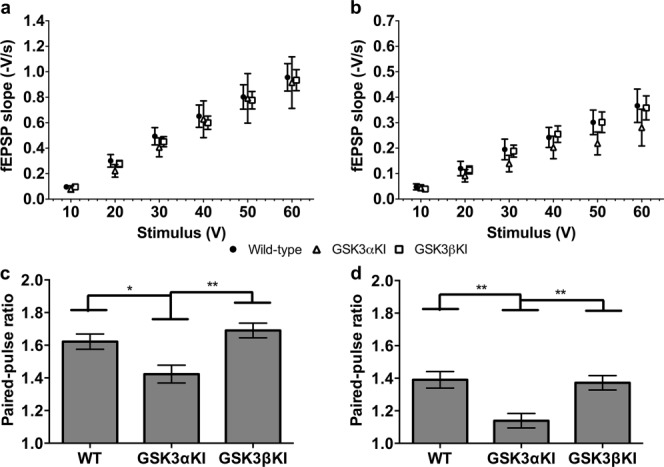
Basal synaptic transmission. (a) I/O curve; 19–21 days; WT: data from 32(12) slices(animals); GSK3αKI: 11(5); GSK3βKI: 33(8). No significant difference by genotype. (b) I/O curve; 4–6 months; WT: 17(9), GSK3αKI: 7(6), GSK3βKI: 15(6). No significant difference by genotype. Symbols on I/O curves are offset for purpose of visualisation but all genotypes were recorded at the same voltages as indicated on the axis below. (c) PPR(50 ms); 19–21 days. Significant difference between genotypes [F(2,73) = 6.5, P = 0.002]; post hoc comparison (adjusted using Sidak correction) indicated lower PPR in GSK3αKI than WT or GSK3βKI mice. (d) PPR(50 ms); 4–6 months. Significant difference between genotypes [F(2,37) = 7.6, P = 0.002]; post hoc comparison indicated lower PPR in GSK3αKI than WT or GSK3βKI mice. **P < 0.01; *P < 0.05. (Data expressed as mean ± SEM).
To induce LTP, a modified version of an established tetanus protocol (Emptage et al., 2001) consisting of a series of 5 tetani (100 Hz for 1s; 1.5s intertetanus interval) was applied to the Schaffer collaterals. This resulted in robust LTP in GSK3βKIs and WT slices (Fig. 2). In contrast, GSK3αKIs did not show significant potentiation and this was significantly different from the other genotypes (Fig. 2 inset). There was no change in PPR between baseline and the last 10 min of recording in any of the genotypes. Moreover, there was no significant difference in maximum level of post-tetanic potentiation nor its time course, suggesting that the difference in LTP was not due to the difference in basal release probability of the GSK3αKIs.
FIGURE 2.
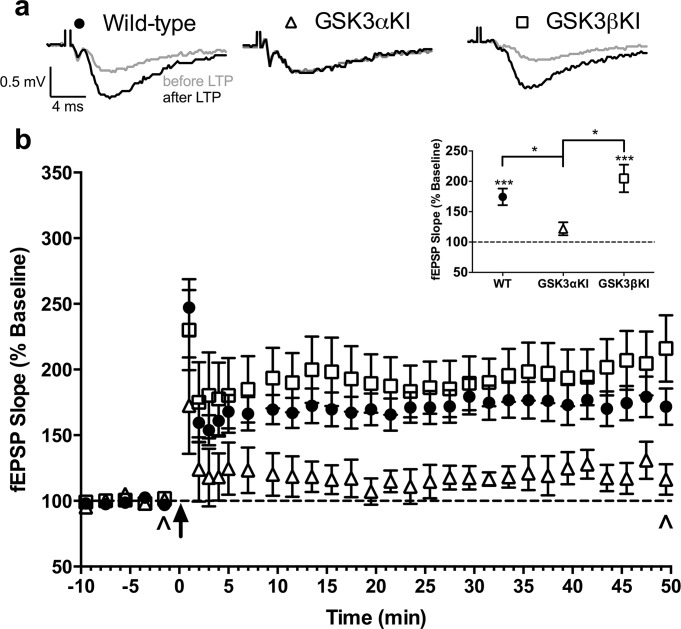
Long-term potentiation (LTP). (a) Representative fEPSP traces (average of 6; sampled at times indicated by ^ in b). (b) fEPSP slope before and after LTP induction (averaged over 1 min for first 5 min following LTP; remainder averaged over 2 min): WT 18(10) slices(animals); GSK3αKI: 7(6), GSK3βKI: 15(6). Arrow: tetanic stimulus. Inset: mean of last 10 min. There was a significant difference between genotypes, (41–50 min, F(2,36) = 7.4, P = 0.002): post hoc comparison (adjusted using Sidak correction) indicated that GSK3αKI but not GSK3βKI mice differed significantly from WT mice (line shows between-genotype differences, asterisks above genotype indicate baseline vs last 10 min). ***P < 0.001, *P < 0.05. (Data are mean ± SEM).
Robust LTD was induced following an established protocol (Milner et al., 2004) of low-frequency stimulation (900 single pulses at 1Hz) in slices from GSK3βKIs, which was indistinguishable from WT experiments (Fig. 3). Again, GSK3αKIs were significantly different from the other genotypes, failing to show depression (Fig. 3 inset).
FIGURE 3.
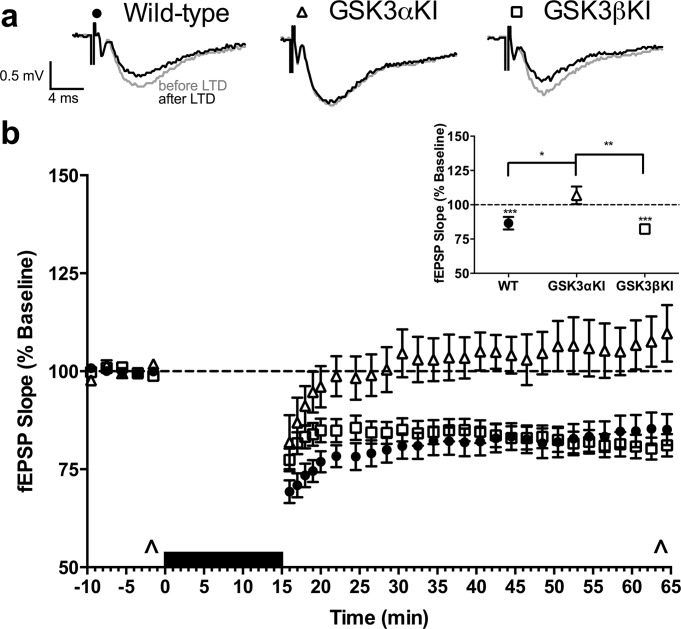
Long-term depression (LTD). (a) Representative fEPSP traces (average of 6; sampled at times indicated by ^ in b). (b) fEPSP slope before and after LTD induction (averaged over 1 min for first 5 min following LTD; remainder averaged over 2 min): WT: data from 32(12) slices (animals); GSK3αKI: 11(5), GSK3βKI: 33(8). Black bar: low-frequency stimulation. Inset: mean of last 10 min. There was a significant difference between genotypes (56–65 min, F(2,73) = 7.3, P < 0.001): post hoc comparison (adjusted using Sidak correction) indicated that GSK3αKI but not GSK3βKI mice differed significantly from WT mice (line shows between-genotype differences, asterisks above genotype indicate baseline vs last 10 min). ***P < 0.001, **P < 0.01, *P < 0.05. (Data are mean ± SEM).
Thus, contrary to previous assumptions, a mutation limiting the dynamic range of GSK3β not only has no effect on basal synaptic transmission but also affects neither LTP nor LTD. In contrast, preventing inhibition of GSK3α prevents induction of plasticity in either direction. Although it is clear that neither direction of plasticity can be induced when inactivation of GSK3α is prevented, it would be premature to conclude that inactivation of GSK3α would be essential for induction of both LTP and LTD in a wild type mouse. While we can be confident that the mutation of GSK3β is ineffective in preventing either form of plasticity, caution is necessary as to whether the GSK3α mutation really affects both LTP and LTD directly. Previous studies inhibiting both isoforms of GSK3 (Peineau et al., 2007; Bradley et al., 2012), would suggest that preventing inactivation of the effective isoform could enhance LTD while inhibiting LTP. Under these conditions synapses would tend to become increasingly depressed in response to activity. Hence, GSK3αKI mice, in which GSK3α cannot be inhibited throughout life, may reach a floor effect where their synapses cannot be any further depressed. This would be entirely compatible with the observations here. Under such conditions, induction of LTP would be inhibited because GSK3α could not be inactivated while LTD would not be induced because the synapses were already depressed as a result. Moreover, the observation that the GSK3αKI mice show no change in input/output ratio but decreased paired pulse ratio could be explained by a homeostatic increase in release probability, compensating for chronic postsynaptic depression.
What is clear is that the dynamic nature of GSK3α rather than GSK3β is essential for bidirectional synaptic plasticity in the Schaffer collateral to CA1 synapse. This opens up the possibility that GSK3α could be targeted in neurodegenerative and cognitive disorders such as Alzheimer's disease, without the side effects that manipulating the ubiquitous GSK3β would inevitably incur.
REFERENCES
- Asuni AA, Hooper C, Reynolds CH, Lovestone S, Anderton BH, Killick R. GSK3alpha exhibits beta-catenin and tau directed kinase activities that are modulated by Wnt. Eur J Neurosci. 2006;24:3387–3392. doi: 10.1111/j.1460-9568.2006.05243.x. [DOI] [PubMed] [Google Scholar]
- Azoulay-Alfaguter I, Yaffe Y, Licht-Murava A, Urbanska M, Jaworski J, Pietrokovski S, Hirschberg K, Eldar-Finkelman H. Distinct molecular regulation of glycogen synthase kinase-3alpha isozyme controlled by its N-terminal region: functional role in calcium/calpain signaling. J Biol Chem. 2011;286:13470–13480. doi: 10.1074/jbc.M110.127969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley CA, Peineau S, Taghibiglou C, Nicolas CS, Whitcomb DJ, Bortolotto ZA, Kaang BK, Cho K, Wang YT, Collingridge GL. A pivotal role of GSK-3 in synaptic plasticity. Front Mol Neurosci. 2012;5:13. doi: 10.3389/fnmol.2012.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Goedert M. GSK3 inhibitors: development and therapeutic potential. Nat Rev Drug Discov. 2004;3:479–487. doi: 10.1038/nrd1415. [DOI] [PubMed] [Google Scholar]
- Emptage NJ, Reid CA, Fine A. Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron. 2001;29:197–208. doi: 10.1016/s0896-6273(01)00190-8. [DOI] [PubMed] [Google Scholar]
- Georgievska B, Sandin J, Doherty J, Mortberg A, Neelissen J, Andersson A, Gruber S, Nilsson Y, Schott P, Arvidsson PI, Hellberg S, Osswald G, Berg S, Falting J, Bhat RV. AZD1080, a novel GSK3 inhibitor, rescues synaptic plasticity deficits in rodent brain and exhibits peripheral target engagement in humans. J Neurochem. 2013;125:446–456. doi: 10.1111/jnc.12203. [DOI] [PubMed] [Google Scholar]
- Giese KP. GSK-3: A key player in neurodegeneration and memory. IUBMB Life. 2009;61:516–521. doi: 10.1002/iub.187. [DOI] [PubMed] [Google Scholar]
- Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- Hernandez F, de Barreda EG, Fuster-Matanzo A, Goni-Oliver P, Lucas JJ, Avila J. The role of GSK3 in Alzheimer disease. Brain Res Bull. 2009;80:248–250. doi: 10.1016/j.brainresbull.2009.05.017. [DOI] [PubMed] [Google Scholar]
- Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer's disease. J Neurochem. 2008;104:1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Kaidanovich-Beilin O, Woodgett JR. GSK-3: Functional insights from cell biology and animal models. Front Mol Neurosci. 2011;4:40. doi: 10.3389/fnmol.2011.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Yamashita S, Nakao S, Park JM, Murayama M, Mizoroki T, Yoshiike Y, Sahara N, Takashima A. GSK-3beta is required for memory reconsolidation in adult brain. PLoS ONE. 2008;3:e3540. doi: 10.1371/journal.pone.0003540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger C, Tian L. A comparison of the general linear mixed model and repeated measures ANOVA using a dataset with multiple missing data points. Biol Res Nurs. 2004;6:151–157. doi: 10.1177/1099800404267682. [DOI] [PubMed] [Google Scholar]
- McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R, Alessi DR. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005;24:1571–1583. doi: 10.1038/sj.emboj.7600633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner AJ, Cummings DM, Spencer JP, Murphy KP. Bi-directional plasticity and age-dependent long-term depression at mouse CA3-CA1 hippocampal synapses. Neurosci Lett. 2004;367:1–5. doi: 10.1016/j.neulet.2004.04.056. [DOI] [PubMed] [Google Scholar]
- Mora A, Sakamoto K, McManus EJ, Alessi DR. Role of the PDK1-PKB-GSK3 pathway in regulating glycogen synthase and glucose uptake in the heart. FEBS Lett. 2005;579:3632–3638. doi: 10.1016/j.febslet.2005.05.040. [DOI] [PubMed] [Google Scholar]
- Parsley SL, Pilgram SM, Soto F, Giese KP, Edwards FA. Enriching the environment of alphaCaMKIIT286A mutant mice reveals that LTD occurs in memory processing but must be subsequently reversed by LTP. Learn Mem. 2007;14:75–83. doi: 10.1101/lm.356607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peineau S, Bradley C, Taghibiglou C, Doherty A, Bortolotto ZA, Wang YT, Collingridge GL. The role of GSK-3 in synaptic plasticity. Br J Pharmacol. 2008;153:S428–S437. doi: 10.1038/bjp.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J, Lo E, Wu D, Saule E, Bouschet T, Matthews P, Isaac JTR, Bortolotto ZA, Wang YT, Collingridge GL. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron. 2007;53:703–717. doi: 10.1016/j.neuron.2007.01.029. [DOI] [PubMed] [Google Scholar]
- Rayasam GV, Tulasi VK, Sodhi R, Davis JA, Ray A. Glycogen synthase kinase 3: More than a namesake. Br J Pharmacol. 2009;156:885–898. doi: 10.1111/j.1476-5381.2008.00085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Moscardo F, Seto-Salvia N, Pera M, Bosch-Morato M, Plata C, Belbin O, Gene G, Dols-Icardo O, Ingelsson M, Helisalmi S, Soininen H, Hiltunen M, Giedraitis V, Lannfelt L, Frank A, Bullido M, Combarros O, Sanchez-Juan P, Boada M, Tarraga L, Pastor P, Perez-Tur J, Baquero M, Molinuevo JL, Sanchez-Valle R, Fuentes-Prior P, Fortea J, Blesa R, Munoz FJ, Lleo A, Valverde MA, Clarimon J. Rare variants in calcium homeostasis modulator 1 (CALHM1) found in early onset Alzheimer's disease patients alter calcium homeostasis. PLoS ONE. 2013;8:e74203. doi: 10.1371/journal.pone.0074203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990;9:2431–2438. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]