

Abstract
Clostridium difficile is a Gram-positive, spore forming bacillus and the most common cause of antibiotic-associated diarrhea in the United States. Clinical outcomes of C. difficile infection (CDI) range from asymptomatic colonization to pseudomembranous colitis, sepsis and death. Disease is primarily mediated by the action of the Rho-glucosylating toxins A and B, which induce potent pro-inflammatory signaling within the host. The role of this inflammatory response during infection is just beginning to be appreciated, with recent data suggesting inflammatory markers correlate closely with disease severity. In addition to the toxins, multiple innate immune signaling pathways have been implicated in establishing an inflammatory response during infection. In intoxication-based models of disease, inflammation typically enhances pathogenesis, while protection from infection seems to require some level of inflammatory response. Thus, the host immune response plays a key role in shaping the course of infection and a balanced inflammatory response which eradicates infection without damaging host tissues is likely required for successful resolution of disease.
1. Introduction
Clostridium difficile is a Gram-positive spore forming bacillus and an obligate anaerobe. It is currently the most common cause of hospital acquired antibiotic-associated diarrhea in the United States [1]. Disease is primarily mediated by the action of the Rho-glucosylating toxins toxin A (TcdA) and toxin B (TcdB), and clinical outcomes of CDI vary between asymptomatic colonization, pseudomembranous colitis, toxic megacolon, sepsis and death. Throughout the last ten years, incidence of C. difficile infection (CDI) has increased dramatically in developed countries, including the United States, Europe and Canada. Much of the increase in disease frequency and severity has been linked to the emergence of a hypervirulent strain known as PCR ribotype 027 [2]. C. difficile has an enormous economic inpact, and is estimated to account for over 1 billion dollars in excess medical costs per year in the U.S. alone [1]. The most common cause of susceptibility to CDI is antibiotic treatment, including exposure to clindamycin, aminopenicillins, cephalosporins and fluoroquinolones. Almost all broad-spectrum antibiotics have been implicated in disruption of the intestinal microbiome, a condition coined as “dysbiosis” which is the underlying cause of increased susceptibility to CDI [3].
Current treatments involve administration of vancomycin or metronidazole. However, recurrent infection is seen in 20-30% of patients, and 15% of individuals eventually succumb to disease [1,5-6]. Newer therapies have been developed with the goal of diminishing microbiome disruption or restoring healthy microbiota, including the narrow spectrum antibiotic Fidaxomicin, as well as fecal microbiota transplant. [7]. Simultaneously, understanding of the factors that influence disease severity has also evolved. Recent data suggest that the host immune response to C. difficile plays a large role in determining the eventual outcome of disease. This includes evidence that point mutations in the gene encoding IL-8, a cytokine responsible for neutrophil recruitment in humans, results in increased IL-8 production during CDI and predisposes individuals to infection [8]. These data suggest that the disease is partially mediated by host factors, and indeed, inflammatory markers correlate more closely to disease severity than pathogen burden [9]. Additionally, increased IL-8 protein levels and CXCL5 and IL-8 message levels have been associated with prolonged disease [10]. The role the host immune response plays during infection has just begun to be explored, and many fascinating questions remain.
2. Inflammatory Response to Toxins A and B
2.1 Intoxication by TcdA/B
Infection with Clostridium difficile spores can occur in the community as well as in the healthcare setting, although disease typically manifests following disruption of the intestinal microbiome with antibiotics [11]. Spores are transmitted by the fecal-oral route, and once ingested they are capable of passing through the gastric acid present in the stomach and germinating in the colon and cecum [12]. Once germination occurs, vegetative cells penetrate the mucus layer and colonize by adhering to the epithelial cells of the colon. Following successful colonization, C. difficile replicates and produces the enterotoxin TcdA and the cytotoxin TcdB. TcdA and B are primarily responsible for the abundant tissue damage, epithelial barrier disruption and fluid accumulation seen during disease. A hallmark of C. difficile infection is robust neutrophil infiltration, and the pseudomembranes seen in more severe disease are made up of these cells surrounded by mucin, fibrin and cellular debris [4]. Additionally, hypervirulent ribotype 027 strains produce a third toxin termed binary toxin, or C. difficile transferase (CDT). This toxin has been shown to increase colonization by the organism via induction of microtubule protrusions on host epithelial cells, providing one possible mechanism for increased virulence in 027 strains [13]. TcdA and TcdB lead to a robust inflammatory response from host epithelial cells, inducing the production of pro-inflammatory cytokines and chemokines which recruit additional immune cells. Intoxication occurs following toxin binding to host cell receptors and internalization via receptor-mediated endocytosis. Subsequent endosomal acidification triggers the insertion of the translocation domain into the endosomal membrane. This is thought to form a pore through which the glucosyltransferase domain is inserted into the cytoplasmic side of the endosomal membrane. The cysteine protease domain then cleaves off the glucosyltransferase domain, releasing it into the cytoplasm. There, the glucosyltransferase domain modifies Rho GTPases via the covalent attachment of a glucosyl residue, preventing the exchange of GDP for GTP and thereby blocking GTPase function. TcdA and TcdB have been specifically shown to glucosylate the Rho-family GTPases RhoA, Rac1 and Cdc42. This leads to a loss of integrity in the actin cytoskeleton, resulting in cell rounding and cytotoxicity. There is considerable debate other the type of cell death induced by TcdA and B, as characteristics of both apoptosis and necrosis have been observed [14]. However, cell death induced by Toxins A and B leads to the release of the pro-inflammatory danger signal uridine diphosphate (UDP). UDP signals through the P2Y6 receptor on host cells and enhances NFκB activation and IL-8 production, thereby contributing to the activation of the host immune response [15].
2.2 Toxin-induced Host Cell Signaling
Multiple Rho GTPase-independent pathways are also activated by TcdA and TcdB, and these are primarily involved in inflammatory gene expression. Toxins A and B are capable of inducing expression of numerous cytokines and chemokines, including IL-1, IL-6, IL-8, IL-12, IL-18, IFN-γ, TNF-α, macrophage inflammatory protein (MIP) 1α, CXCL2 as well as the adipocytokine leptin [16-19]. Although the exact pathways leading to pro-inflammatory gene expression are unknown, it has been demonstrated that intoxication of cells by TcdA and TcdB leads to intracellular calcium release and activation of multiple mitogen-activated protein kinase (MAPK) pathways, including p38 MAPK,c-Jun N-terminal Kinase (JNK), and extracellular signal-regulated protein kinase (Erk)1/2. These pathways result in subsequent activation of the transcription factors NFκB and AP-1, known inducers of chemokine and cytokine production [20]. In particular, p38 MAPK phosphorylation has been shown to be essential for NFκB activation in response to TcdA, and this is thought to occur as a result of mitochondrial oxygen radical generation [21-22]. Both toxins have also been shown to activate MAPK-activated protein kinase (MK2) downstream of p38 MAPK, and this pathway is essential for IL-8 expression [23]. Similarly, production of cyclooxygenase-2 (COX-2) and prostaglandin E2 (PEG2), thought to be responsible for the fluid accumulation seen in response to toxins, is also dependent on p38 MAPK activation and signaling via mitogen- and stress-activated protein kinase (MSK-1) [24]. TcdA has also been shown to promote dendritic cell maturation and induce expression of the monocyte and macrophage chemoattractant CX3CL1 via similar pathways, including p38 MAPK and NFκB [25].
Both toxins are also capable of activating the NLRP3 inflammasome, a cellular complex assembled in response PAMPs or danger signals which activates caspase 1, responsible for processing the cytokines IL-1β and IL-18 into their secreted forms [26]. NLRP3 inflammasome activation by TcdB occurs independently of its glucosylation activity, although the full length protein is required. Endocytosis and endosomal acidification are likewise necessary for inflammasome activation by TcdB [27]. Although the specific mechanism of toxin-induced NLRP3 activation is not known, the NLRP3 inflammasome complex has been shown to be activated by a variety of stimuli, including Reactive Oxygen Species (ROS), bacterial toxins, and extracellular ATP [28]. Interestingly, IL-β and IL-1 receptor signaling pathway genes were also found to be significantly upregulated after toxin challenge in a microarray-based study [19].
3. Host Recognition of C. difficile
3.1 Role of Pattern Recognition Receptors
The innate immune system is the first responder to the presence of pathogenic microbes throughout the body, and plays a crucial role in shaping the adaptive response to come. The innate response is influential during CDI, as multiple innate signaling pathways have been shown to play a role in disease susceptibility. Pattern Recognition Receptors (PRRs) are present on host cells and recognize conserved bacterial signatures (Pathogen Associated Molecular Patterns, or PAMPs) to initiate the immune response [29]. A subset of PRRs, the Tolllike Receptors (TLRs) have been shown to recognize C. difficile PAMPs and contribute to the initiation of the host inflammatory response. Specifically, the TLR adaptor protein MyD88 has been shown to be involved in host defense. Mice lacking this molecule and thus, the majority of TLR signaling, show decreased survival during CDI [30-31]. In this context, MyD88-mediated signaling is essential for the production of the chemokine CXCL1, responsible for recruiting neutrophils to the colonic lamina propria. These cells play an important role in preventing the dissemination of commensal microbes to other organs [32]. Several specific PRRs have also been implicated in recognition and response to the pathogen. C. difficile has been shown to signal through nucleotide-binding oligomerization domain 1 (Nod1), an intracellular Nod-like Receptor (NLR) known to recognize diaminopimelic acid derived from peptidoglycan (PGN) [33]. Although Nod1 does not signal via MyD88, deletion of this receptor likewise impairs production of the neutrophil chemoattractant CXCL1, decreases neutrophil recruitment and results in more severe disease [34]. C. difficile infected Nod1-/- mice also displayed elevated levels of lipopolysaccharide from translocating commensals as well as the pyrogenic cytokine IL-1β in their sera, possibly due to reduced bacterial clearance [33]. Toll-like Receptor 4 (TLR4) has also been implicated in recognition of C. difficile. Purified surface layer protein (SLP) from C. difficile can activate NFκB downstream of TLR4 and induce TLR4 dependent dendritic cell (DC) maturation. SLP-treated DCs secrete IL-12, IL-23, TNFα, and IL-10, and are able to induce co-cultured T cells to secrete IL-17 and IFN-γ. Additionally, deletion of TLR4 in vivo causes an increase in disease severity [31].
C. difficile flagellin has also been shown to stimulate TLR5, resulting in NFκB and p38 MAP kinase activation and IL-8 secretion. Although large quantities of flagellin are required compared to the more potent Salmonella typhimurium flagellin, this effect can be prevented using neutralizing antibodies directed against TLR5 and is augmented by pre-treatment with Toxin B [35]. Interestingly, administration of flagellin from S. typhimurium prior to infection with C. difficile attenuates disease by delaying both growth and toxin production by C. difficile. However, deletion of TLR5 in mice does not result in more severe infection, suggesting that this signaling pathway may be less essential for recognition of the pathogen [36].
4. Host Inflammatory Response to C. difficile
4.1 Host Response to Intoxication
The role of inflammation in response to C. difficile infection is controversial and multifaceted. Prevailing thought derived from toxin-based models of infection reflects the idea that inflammation is deleterious, as blocking inflammatory responses can prevent some of the tissue damage usually seen after intoxication. Preventing inflammasome activation via deletion of the adaptor protein apoptosis-associated speck-like protein (ASC), present in multiple inflammasomes, prevents tissue inflammation and damage following challenge of mice with purified toxins. Similarly, blocking IL-1β and IL-1α signaling with the IL-1 receptor antagonist Anakinra ameliorates toxin damage [27]. Preventing neutrophil recruitment via antibody depletion prior to toxin treatment reduces fluid accumulation, cell death, permeability and histological damage in a rabbit model of intoxication [37]. Neutralization of the pro-inflammatory cytokine IFN-γ has also been shown to protect against TcdA-induced enteritis in a mouse model, and IFN-γ-/- mice are protected from tissue damage and show decreased cytokine production after challenge [38]. Mast cells have also been implicated in a damaging inflammatory response after intoxication, as mast cell deficient mice show decreased inflammation after treatment with TcdA [39]. The toxins are also able to induce significant levels of the neutrophil chemoattractant CXCL2 in rat ileal loops, and blocking this signal reduces histological damage following intoxication with TcdA. Similarly, genetic knockout of chemokine MIP1α, or of its receptor CCR1, decreased the damage associated with intoxication by TcdA [40-41]. Interestingly, the adipokine leptin also appears to play a role in the inflammatory response to TcdA, as ob/ob leptin deficient mice show reduced pathology after challenge with TcdA, including less severe fluid secretion and inflammation [42].
4.2 Host Response to Infection
Many similar manipulations in infection models using live C. difficile have shown a lack of inflammatory response to be detrimental. Inflammasome deficient ASC-/- mice show decreased survival during infection, suggesting that some inflammation is necessary for bacterial clearance and disease resolution. Interestingly, ASC-/- mice show increased translocation of commensal microbes to organs such as the spleen, liver and lung, suggesting a role for inflammatory pathways in controlling bystander bacteria [43]. Preventing neutrophil recruitment via antibody depletion similarly worsens disease [32]. Inflammatory signaling by pattern recognition receptors has also been shown to be protective during CDI, as Nod1-/-, MyD88-/- and TLR4-/- mice all experience more severe disease [31-33]. Leptin signaling also appears to be protective, as leptin deficient mice show higher bacterial burdens [44]. The cytokine interleukin-23 (IL-23) stands out as an inducer of pathogenic inflammation during infection [45]. IL-23 has been implicated in multiple autoimmune diseases, and is best known for its ability to maintain TH17 cells and induce production of the cytokines IL-17 and IL-22. Inflammation in general has long been understood as a balance between eradicating infection while preventing destruction of host tissues, and it may be that IL-23 tips the balance towards pathogenic host damage rather than protective bacterial eradication.
5. Remaining Questions
Although understanding of the role of the host response to CDI is increasing, many questions remain. One prominent and fascinating area of inquiry involves the ability of the microbiome to influence host response during infection. In general, the ability of the host microbiota to prevent infection with pathogenic microbes is known as colonization resistance [46]. Direct colonization resistance refers to the ability of certain microbes to prevent pathogenic infection by competition for nutrients or by inhibiting pathogen growth via secretion of particular molecules. In the case of indirect colonization resistance, beneficial microorganisms prevent infection by activating or skewing host immune responses. It is widely appreciated that gut bacteria can influence local and systemic immune changes, and a prime example of this is the ability of segmented filamentous bacteria (SFB) to induce TH17 responses in the host gastrointestinal tract [47]. It remains to be seen what type of role indirect colonization resistance plays in protection from or susceptibility to CDI.
Understanding how the microbiome shapes the host immune response will be essential to fully appreciate the role of the immune system in infection, and may also lead to greater understanding of how the balance of inflammatory processes shapes the course of disease. This may provide clues towards preventing infection by reducing the immunological effects of microbiome disruption which lead to susceptibility and opens the door to numerous potential therapies targeting host signaling during CDI. To that end, identification of the cytokine IL-23 as a regulator of pathology presents an as-of-yet unique opportunity to determine how this cytokine is induced by C. difficile and identify particular cell subsets and their role in causing disease. This knowledge will increase our understanding of host recognition and response to C. difficile, as well as clarify the role of inflammation during CDI.
Fig. 1.
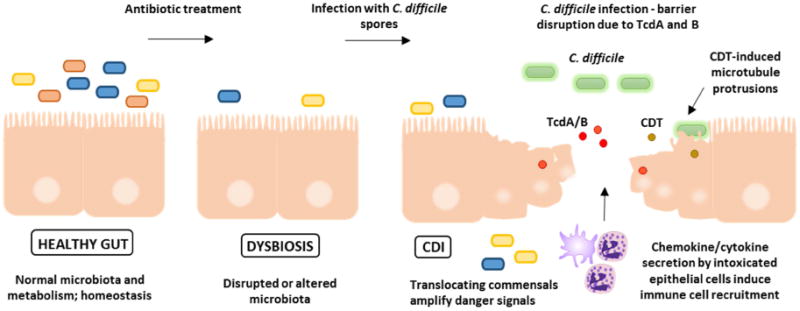
Healthy individuals possess normal microbiota in homeostasis with the host mucosal immune system. Antibiotics disrupt the microbiome and lead to dysbiosis. Ingested C. difficile spores germinate into vegetative cells which produce the major virulence factors, Toxins A and B. Ribotype 027 strains also produce a third toxin, C. difficile transferase (CDT), which enhances colonization by inducing microtubule protrusion formation on host cells. Toxins A and B further disrupt the epithelial barrier, trigger pro-inflammatory signaling from epithelial cells, and increased immune cell recruitment. Translocation of commensal microorganisms contributes to inflammatory signaling.
Fig. 2.
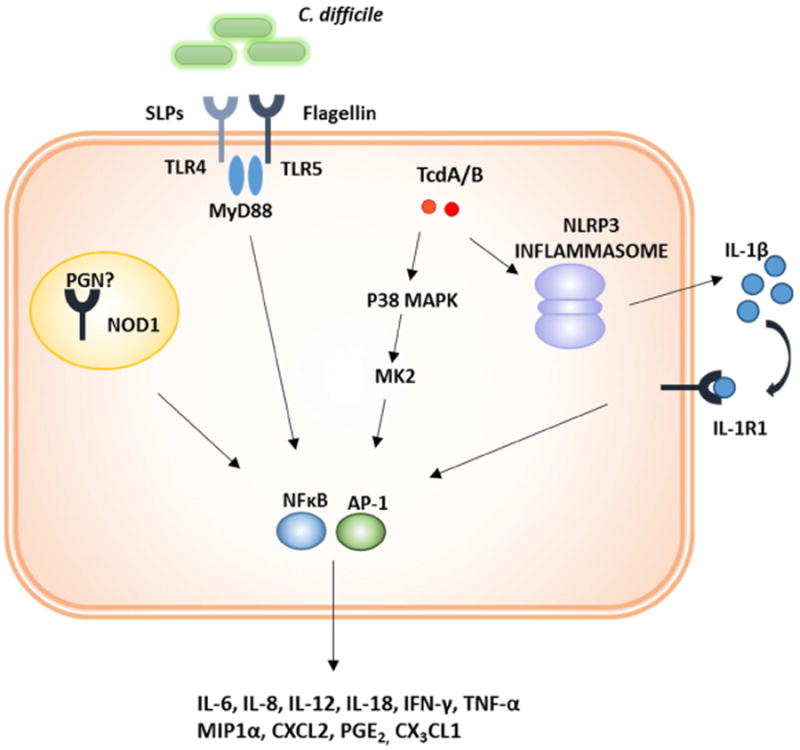
Multiple innate immune pathways contribute to inflammation during CDI. C. difficile PAMPs, including Surface Layer Proteins (SLPs) and flagellin activate Toll-like Receptor 4 (TLR4) and TLR5 respectively. C. difficile can likewise stimulate Nucleotide-binding oligomerization domain-containing protein 1 (Nod1) via an unidentified, secreted PAMP. Toxins A and B also activate inflammatory signaling cascades, including p38 mitogen-activated protein kinase (MAPK). P38 MAPK signals though MAPK-activated protein kinase (MK2) to activate the transcription factors nuclear factor κB (NFκB) and activator protein 1 (AP-1) to induce proinflammatory cytokine and chemokine production. The toxins can also activate the NLRP3 inflammasome, leading to IL-1β secretion and NFκB activation via the IL-1 receptor.
Table 1.
The role of inflammation in response to C. difficile toxins or infection depends on the type of challenge. Many studies using intoxication as a model (light gray boxes) report that inflammatory pathways are deleterious. However, infection based models (dark gray boxes) have found that certain inflammatory pathways are necessary for survival, with the exception of the pro-inflammatory cytokine IL-23. Thus, the role of inflammation during CDI is likely to be multifaceted and complex.
| Challenge | Model | Result | Reference |
|---|---|---|---|
| TcdA/B | ASC-/- Mice Anakinra (IL-1Ra) Mice |
Decrease in disease severity | Ng, 2010 |
| TcdA | MIP-2 Neutralized Rat | Decrease in disease severity | Castagliuolo, 1998 |
| TcdA | CCR1-/- Mice MIP1α -/- Mice |
Decrease in disease severity | Morteau, 2002 |
| TcdA | Anti-CD18 mAB (neutrophil depletion) Rabbit | Decrease in disease severity | Kelly, 1994 |
| TcdA | IFN-γ -/- Mice IFN-γ Neutralized Mice |
Decrease in disease severity | Ishida, 2004 |
| TcdA | Mast Cell Deficient Mice | Decrease in disease severity | Wershil, 1998 |
| TcdA | Ob/ob (Leptin Deficient) Mice | Decrease in disease severity | Mykoniatis, 2003 |
| VPI 10463 | Nod1-/- Mice | Decreased survival | Hasegawa, 2011 |
| VPI 10463 | ASC-/- Mice | Decreased survival | Hasegawa, 2012 |
| VPI 10463 | MyD88-/- Mice Neutrophil depleted Mice |
Decreased survival | Jarchum, 2012 |
| R13537 | TLR4-/- Mice | Decreased survival | Ryan, 2011 |
| VPI 10463 | IL-23p19-/- Mice | Increased survival | Buonomo, 2013 |
Highlights.
Clostridium difficile infection (CDI) is primarily mediated by Toxins A and B
Toxins A and B induce pro-inflammatory signaling within the host
Multiple Pattern Recognition Receptors (PRRs) also contribute to inflammation
Intoxication models show inflammatory signaling can be pathogenic
Infection models show that some level of inflammation is required for disease resolution
Acknowledgments
This work was supported by grants from the National Institutes of Health (5R01AI026649-25) and (5T32AI07046-38).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dubberke ER, Olsen MA. Burden of Clostridium difficile on the Healthcare System. Clin Infect Dis. 2012;55:S88–S92. doi: 10.1093/cid/cis335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bacci S, Mølbak K, Kjeldsen MK, Olsen KEP. Binary Toxin and Death after Clostridium difficile Infection. Emerging Infectious Diseases. 2011;17:976–982. doi: 10.3201/eid1706.101483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bignardi GE. Risk factors for Clostridium difficile infection. J Hosp Infect. 1998;40:1–15. doi: 10.1016/s0195-6701(98)90019-6. [DOI] [PubMed] [Google Scholar]
- 4.Poutanen SM, Simor AE. Clostridium difficile-associated diarrhea in adults. CMAJ. 2004;171:51–58. doi: 10.1503/cmaj.1031189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ananthakrishnan AN. Clostridium difficile infection: epidemiology, risk factors and management. Nature Reviews Gastroenterology & Hepatology. 2010;8:17–26. doi: 10.1038/nrgastro.2010.190. [DOI] [PubMed] [Google Scholar]
- 6.Loo VG, Poirier L, Miller MA, Oughton M, Libman MD, Michaud S, Bourgault AM, Nguyen T, Frenette C, Kelly M, Vibien A, Brassard P, Fenn S, Dewar K, Hudson T, Horn R, René P, Monczak Y, Dascal A. A Predominantly Clonal Multi-Institutional Outbreak of Clostridium difficile–Associated Diarrhea with High Morbidity and Mortality. New England Journal of Medicine. 2005;353:2442–2449. doi: 10.1056/NEJMoa051639. [DOI] [PubMed] [Google Scholar]
- 7.Bakken JS. Fecal bacteriotherapy for recurrent Clostridium difficile infection. Anaerobe. 2009;15:285–289. doi: 10.1016/j.anaerobe.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 8.Jiang ZD, et al. A common polymorphism in the interleukin 8 gene promoter is associated with Clostridium difficile diarrhea. Am J Gastroenterol. 2006;101:1112–1116. doi: 10.1111/j.1572-0241.2006.00482.x. [DOI] [PubMed] [Google Scholar]
- 9.Feghaly REE, et al. Markers of Intestinal Inflammation, Not Bacterial Burden, Correlate With Clinical Outcomes in Clostridium difficile Infection. Clin Infect Dis. 2013 doi: 10.1093/cid/cit147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.El Feghaly RE, Stauber JL, Tarr PI, Haslam DB. Intestinal Inflammatory Biomarkers and Outcome in Pediatric Clostridium difficile Infections. The Journal of Pediatrics. 2013;163:1697–1704.e2. doi: 10.1016/j.jpeds.2013.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eyre DW, et al. Diverse Sources of C. difficile Infection Identified on Whole-Genome Sequencing. New England Journal of Medicine. 2013;369:1195–1205. doi: 10.1056/NEJMoa1216064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Howerton A, Patra M, Abel-Santos E. Fate of Ingested Clostridium difficile Spores in Mice. PLoS ONE. 2013;8:e72620. doi: 10.1371/journal.pone.0072620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwan C, et al. Clostridium difficile toxin CDT induces formation of microtubule-based protrusions and increases adherence of bacteria. PLoS Pathog. 2009;5:e1000626. doi: 10.1371/journal.ppat.1000626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chumbler NM, et al. Clostridium difficile Toxin B Causes Epithelial Cell Necrosis through an Autoprocessing-Independent Mechanism. PLoS Pathog. 2012;8:e1003072. doi: 10.1371/journal.ppat.1003072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hansen A, et al. The P2Y6 Receptor Mediates Clostridium difficile Toxin-Induced CXCL8/IL-8 Production and Intestinal Epithelial Barrier Dysfunction. PLoS ONE. 2013;8:e81491. doi: 10.1371/journal.pone.0081491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun X, Savidge T, Feng H. The Enterotoxicity of Clostridium difficile Toxins. Toxins. 2010;2:1848–1880. doi: 10.3390/toxins2071848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flegel WA, et al. Cytokine response by human monocytes to Clostridium difficile toxin A and toxin B. Infect Immun. 1991;59:3659–3666. doi: 10.1128/iai.59.10.3659-3666.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee JY, et al. Clostridium difficile toxin A promotes dendritic cell maturation and chemokine CXCL2 expression through p38, IKK, and the NF-kappaB signaling pathway. J Mol Med. 2009;87:169–180. doi: 10.1007/s00109-008-0415-2. [DOI] [PubMed] [Google Scholar]
- 19.D'Auria KM, et al. In vivo physiological and transcriptional profiling reveals host responses to Clostridium difficile toxin A and toxin B. Infect Immun. 2013 doi: 10.1128/IAI.00869-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jefferson KK, Smith MF, Jr, Bobak DA. Roles of intracellular calcium and NF-κB in the Clostridium difficile toxin A-induced up-regulation and secretion of IL-8 from human monocytes. The Journal of Immunology. 1999;163:5183–5191. [PubMed] [Google Scholar]
- 21.Chumbler NM, et al. Clostridium difficile Toxin B Causes Epithelial Cell Necrosis through an Autoprocessing-Independent Mechanism. PLoS Pathog. 2012;8:e1003072. doi: 10.1371/journal.ppat.1003072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee JY, et al. Clostridium difficile toxin A promotes dendritic cell maturation and chemokine CXCL2 expression through p38, IKK, and the NF-kappaB signaling pathway. J Mol Med. 2009;87:169–180. doi: 10.1007/s00109-008-0415-2. [DOI] [PubMed] [Google Scholar]
- 23.Bobo LD, et al. MK2 Kinase Contributes to Clostridium difficile-Associated Inflammation. Infect Immun. 2012 doi: 10.1128/IAI.00186-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim H, et al. Clostridium difficile Toxin A Regulates Inducible Cyclooxygenase-2 and Prostaglandin E2 Synthesis in Colonocytes via Reactive Oxygen Species and Activation of p38 MAPK. J Biol Chem. 2005;280:21237–21245. doi: 10.1074/jbc.M413842200. [DOI] [PubMed] [Google Scholar]
- 25.Ko SH, et al. Mitogen-activated protein kinase/IκB kinase/NF-κB-dependent and AP-1-independent CX3CL1 expression in intestinal epithelial cells stimulated with Clostridium difficile toxin A. J Mol Med. 2013 doi: 10.1007/s00109-013-1117-y. [DOI] [PubMed] [Google Scholar]
- 26.Tschopp J, Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10:210–215. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 27.Ng J, et al. Clostridium difficile toxin-induced inflammation and intestinal injury are mediated by the inflammasome. Gastroenterology. 2010;139:542–552. 552.e1–3. doi: 10.1053/j.gastro.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 28.Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481:278–286. doi: 10.1038/nature10759. [DOI] [PubMed] [Google Scholar]
- 29.Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 30.Lawley TD, et al. Antibiotic Treatment of Clostridium difficile Carrier Mice Triggers a Supershedder State, Spore-Mediated Transmission, and Severe Disease in Immunocompromised Hosts. Infect Immun. 2009;77:3661–3669. doi: 10.1128/IAI.00558-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ryan A, et al. A role for TLR4 in Clostridium difficile infection and the recognition of surface layer proteins. PLoS Pathog. 2011;7:e1002076. doi: 10.1371/journal.ppat.1002076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jarchum I, Liu M, Shi C, Equinda M, Pamer EG. Critical role for MyD88-mediated neutrophil recruitment during Clostridium difficile colitis. Infect Immun. 2012;80:2989–2996. doi: 10.1128/IAI.00448-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hasegawa M, et al. Nucleotide-binding oligomerization domain 1 mediates recognition of Clostridium difficile and induces neutrophil recruitment and protection against the pathogen. J Immunol. 2011;186:4872–4880. doi: 10.4049/jimmunol.1003761. [DOI] [PubMed] [Google Scholar]
- 34.Strober W, Murray PJ, Kitani A, Watanabe T. Signalling pathways and molecular interactions of NOD1 and NOD2. Nat Rev Immunol. 2006;6:9–20. doi: 10.1038/nri1747. [DOI] [PubMed] [Google Scholar]
- 35.Yoshino Y, et al. Clostridium difficile flagellin stimulates toll-like receptor 5, and toxin B promotes flagellin-induced chemokine production via TLR5. Life Sci. 2013;92:211–217. doi: 10.1016/j.lfs.2012.11.017. [DOI] [PubMed] [Google Scholar]
- 36.Jarchum I, Liu M, Lipuma L, Pamer EG. Toll-Like Receptor 5 Stimulation Protects Mice from Acute Clostridium difficile Colitis. Infect Immun. 2011;79:1498–1503. doi: 10.1128/IAI.01196-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kelly CP, et al. Neutrophil recruitment in Clostridium difficile toxin A enteritis in the rabbit. J Clin Invest. 1994;93:1257–1265. doi: 10.1172/JCI117080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ishida Y, et al. Essential involvement of IFN-gamma in Clostridium difficile toxin A-induced enteritis. J Immunol. 2004;172:3018–3025. doi: 10.4049/jimmunol.172.5.3018. [DOI] [PubMed] [Google Scholar]
- 39.Wershil BK, Castagliuolo I, Pothoulakis C. Direct evidence of mast cell involvement in Clostridium difficile toxin A-induced enteritis in mice. Gastroenterology. 1998;114:956–964. doi: 10.1016/s0016-5085(98)70315-4. [DOI] [PubMed] [Google Scholar]
- 40.Castagliuolo I, et al. Clostridium difficile toxin A stimulates macrophage-inflammatory protein-2 production in rat intestinal epithelial cells. J Immunol. 1998;160:6039–6045. [PubMed] [Google Scholar]
- 41.Morteau O, et al. Genetic deficiency in the chemokine receptor CCR1 protects against acute Clostridium difficile toxin A enteritis in mice. Gastroenterology. 2002;122:725–733. doi: 10.1053/gast.2002.31873. [DOI] [PubMed] [Google Scholar]
- 42.Mykoniatis A, et al. Leptin mediates Clostridium difficile toxin A-induced enteritis in mice. Gastroenterology. 2003;124:683–691. doi: 10.1053/gast.2003.50101. [DOI] [PubMed] [Google Scholar]
- 43.Hasegawa M, et al. Protective role of commensals against Clostridium difficile infection via an IL-1β-mediated positive-feedback loop. J Immunol. 2012;189:3085–3091. doi: 10.4049/jimmunol.1200821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Madan R, et al. Role of Leptin-Mediated Colonic Inflammation in Defense against Clostridium difficile Colitis. Infect Immun. 2014;82:341–349. doi: 10.1128/IAI.00972-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buonomo EL, et al. Role of IL-23 signaling in Clostridium difficile Colitis. J Infect Dis jit277. 2013 doi: 10.1093/infdis/jit277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buffie CG, Pamer EG. Microbiota-mediated colonization resistance against intestinal pathogens. Nat Rev Immunol. 2013;13:790–801. doi: 10.1038/nri3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ivanov II, et al. Induction of Intestinal Th17 Cells by Segmented Filamentous Bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]