

Abstract
There have been significant advances in the past several years with regard to targeted therapy and immunotherapy for cancer. This is highlighted in melanoma, where treatment with targeted therapy (against the BRAF oncogene) results in responses in the majority of patients, though duration of response is limited. In contrast, treatment with immunotherapy results in a lower response rate, but ones that tend to be more durable. Insights regarding mechanisms of response and potential synergy between these treatment strategies for melanoma are a focus of this review, with opportunities to extend these insights to the treatment of other cancers.
Introduction
Within the past decade, key oncogenic mutations have been identified in melanoma and other cancers that contribute to their malignant potential, but may also serve as targets for therapy. Mutations in the BRAF gene occur in about half of melanomas (1) and lead to constitutive signaling through the MAPK pathway. Treatment of BRAF-mutant melanoma with agents targeting this oncogenic mutation represents one of the most significant advances in melanoma care in decades. Results from a phase III trial of single agent BRAF inhibitor versus standard of care chemotherapy showed a significant improvement in progression-free and overall survival (2), leading to FDA approval in 2011. However responses are generally short-lived (5-7 months) (2-4), and novel approaches are needed. A small subset of patients maintain responses beyond one year with little being known about what makes these cases unique.
There is intense research underway to identify strategies to improve durability of response to BRAF-targeted therapy. This has led to therapeutic strategies combining BRAF-targeted therapy with other treatment modalities. One example of this is the concurrent administration of BRAF and MEK inhibitors, thus targeting two nodes within the same pathway. When combining dabrafenib and trametinib, progression free survival is extended, though most patients still progress on therapy within 10 months (5), yet again reinforcing the need for improving the durability of response.
Another area of success in the treatment of melanoma involves the use of immunotherapy. In particular, the use of immune checkpoint inhibitors has shown clear promise with the FDA approval of ipilimumab, a monoclonal antibody that blocks the immuno-modulatory molecule Cytotoxic T-Lymphocyte Antigen-4 (CTLA-4) on the surface of T lymphocytes, in 2011 (6). However, only 10% of patients achieve objective responses with no more than 22% surviving beyond 3 years (7). Anti-Programed Death Receptor-1 (PD-1; CD279) and anti-Programmed Death Receptor Ligand-1 (PD-L1; B7-H1; CD274) represent a further advance derived from inhibiting an immune checkpoint, with objective response rates ranging between 17-28% amongst the most promising agents (8, 9).
There is accumulating evidence indicating that the therapeutic efficacy of several agents relies on their ability to influence the tumor-host interaction, including the activation of an immune response specific for malignant cells (10). In this group, there is mounting data that oncogenic BRAF contributes to immune escape, and that targeting this mutation may increase the immunogenicity of melanoma (11). Treatment with a BRAF inhibitor in patients with metastatic melanoma has a profound effect on the tumor and its microenvironment, with increased melanoma antigen expression, infiltration of CD8+ T cells, and a decrease in immunosuppressive cytokines and VEGF (12-15). However within 2 weeks of therapy, the expression of immuno-modulatory molecules on the surface of tumor and T cells is increased (12) which likely contributes to resistance to therapy. This provides evidence for combining BRAF-targeted therapy with immune checkpoint blockade, and this concept is being empirically investigated in clinical trials combining BRAF inhibitors with immune checkpoint blockade. Results from these trials are not yet mature, and critical questions remain regarding strategies combining these agents.
Successes and limitations of BRAF-targeted therapy
The oncogenic BRAF mutation was originally described in melanoma (1), but has subsequently been identified as a driver mutation in several other cancers, including colon cancer, thyroid cancer, and hairy cell leukemia (1, 16, 17). Selective agents targeting this mutation were first tested in clinical trials for melanoma in 2008, demonstrating high response rates (∼70-80%) (18) and a significant increase in survival when compared to then standard of care dacarbazine (leading to the FDA approval of vemurafenib in 2011) (2). However these high response rates were tempered with a short duration of response, with a median time to progression of ∼ 6 months (2). Intense research efforts have focused on understanding resistance to BRAF inhibitors, with numerous resistance mechanisms identified in melanoma (19-26) and other cancers (27). Resistance is often mediated through reactivation of the MAPK pathway (e.g. via BRAF amplification, splice variants, MEK mutations) but may also be mediated through stromal interactions (e.g. over-production of HGF in the tumor microenvironment)(21). Concomitant mutations in other signaling pathways have also been implicated in melanoma (such as PTEN)(28, 29) and in other histologies (such as EGFR in colon cancer and thyroid cancer) (27, 30), providing rationale for combining BRAF-targeted therapy with other signal transduction inhibitors. These combination strategies have shown some success in improving responses to therapy, though resistance is still a major issue. This is highlighted by the combination of dabrafenib (a BRAF inhibitor) with trametinib (a MEK inhibitor), which was FDA-approved in early 2014 based on progression-free and overall survival benefit in comparison to either BRAF inhibitor alone or MEK inhibitor alone (5). Median progression free survival was extended from under 6 months for BRAF inhibitor monotherapy to 9.4 months with combination BRAF + MEK inhibition (5), and the percentage of patients alive at one year increased from 10% to 40% (BRAF inhibitor monotherapy vs. combined BRAF + MEK inhibition)(5). However the majority of patients still progress within a year and there are very few complete responders (5). Nonetheless this incremental benefit in survival provides a window of opportunity for treatment with other promising forms of therapy (such as immunotherapy) and there is emerging evidence that these strategies may be synergistic.
An important consideration in treatment with targeted therapy is the onset of tumor regression, which can be quite rapid. Metastatic lesions often demonstrate significant regression within 2 weeks of initiation of therapy, with associated symptomatic improvement. Tumor regression typically peaks by 4 months, with a median duration of response of 6 months. Thus the slope is steep but responses are not prolonged (Figure 1). Efforts to identify mechanisms to extend responses to targeted therapy are ongoing.
Figure 1. Representative response rates of targeted therapy, immunotherapy and combined targeted therapy and immunotherapy.

Plots representing the rate of response (left) and survival (right) after either targeted therapy (top), immunotherapy (middle) or the combination of the two (bottom). The response and survival plots on the top show typical responses to targeted therapy, with a deep but transient response rate and improved survival (compared to chemotherapy), but a “tail of the curve” approaching zero. This is in contrast to the response and survival plots in the middle for immunotherapy, with a more shallow but more durable response curve and improved survival (compared to chemotherapy) and a plateau in the “tail of the curve,” suggesting the potential for long-term disease control. The bottom plots show proposed response and survival curves for combined targeted therapy and immunotherapy, with a deep and prolonged response rate, resulting in higher survival than either therapy alone and a higher “tail of the curve” with disease control in a much greater proportion of patients.
Successes and limitations of immunotherapy
Immunotherapy is another treatment strategy for cancer having demonstrated dramatic advances over the past several years. This strategy is not new, with high dose interleukin-2 (IL-2) having been FDA-approved in 1998 based on its ability to produce durable responses in 6-10% of patients (31). Nonetheless, the use of high-dose IL-2 is limited to specialized centers given its toxicity profile (32), and has a low overall response rate.
Another form of immunotherapy receiving recent “breakthrough” status for the treatment of melanoma and other cancers involves the use of immune checkpoint inhibitors (33). One of these agents is ipilimumab, a monoclonal antibody directed against the CTLA-4 molecule on the surface of T lymphocytes. CTLA-4 is an immuno-modulatory molecule that functions to down-regulate an immune response (34). Treatment with a monoclonal antibody that blocks this interaction (ipilimumab) relieves cytotoxic T-lymphocytes from the inhibitory effects of CTLA-4, resulting in an enhanced immune response. Treatment with ipilimumab has shown an overall survival advantage in patients with advanced melanoma in a randomized, placebo-controlled trial (6) and received approval by the FDA in 2011. However, only 10% of patients obtain clear, objective responses, highlighting significant room for improvement. Other immune checkpoint inhibitors are in clinical trials and have shown promising results, including monoclonal antibodies blocking the immune-modulatory molecule PD-1 in the treatment of metastatic melanoma, which have shown response rates approaching 30% in a phase II clinical trial (8). Treatment with a monoclonal antibody blocking the ligand of this receptor, PD-L1, is also in clinical trials with similar response rates to those seen with PD-1 blockade (9). Interestingly, responses were also seen in other solid tumors, including renal cell carcinoma and non-small cell lung cancer (8). Mature response data was recently reported for PD-1 blockade with nivolumab demonstrating an estimated median overall survival of 16.8 months, with 1- and 2-year survival rates of 62% and 43%, respectively (35). Agents targeting PD-1 will likely be FDA approved in 2014.
What is unique about immunotherapeutic approaches (compared to targeted therapy) is the potential to achieve long-term disease control in a significant proportion of patients (∼6% for IL-2 (31, 32) and up to 22% for anti-CTLA-4 (7)). The mechanisms behind these durable responses are unclear, though are being actively investigated in pre-treatment and on-treatment tumor and blood samples of patients treated with several different forms of immunotherapy (adoptive immunotherapy, cytokine therapy, immune checkpoint blockade treatment).
To date, the main limitation of immunotherapy is the fact that only a minority of patients will respond to therapy. This is in contrast to targeted therapy, where a majority of patients achieve an objective response. However despite a lower overall response rate, responses to immunotherapy tend to be more durable than those seen with targeted therapy. Tumor regression peaks later, though the median duration of response can be much longer (i.e. up to 2 years in the case of treatment with PD-1 blockade). This has tremendous relevance, and brings into question what the proper sequence of therapy should be in patients with metastatic melanoma harboring a BRAF mutation. Of note, disease burden plays a significant role in determining which therapy to initiate first – as the tempo of response to targeted therapy is more rapid than to immunotherapy. However there is now growing evidence that immunotherapy may synergize with molecularly-targeted therapy suggesting that they be used in concert, with the potential for rapid tumor regression and durable response (Figure 1). A current question in melanoma is whether targeted therapy/immunotherapy combination regimens should simply incorporate the most active individual components, or whether there are certain points of intervention that would synergize.
Evidence for synergy of targeted therapy and immunotherapy
There have been numerous previous links between oncogene de-addiction and immunostimulation. In mouse models of T cell acute lymphoblastic lymphoma and pro-B cell leukemia, inactivation of MYC and BCR-ABL, respectively, required an intact immune system (specifically CD4+ T cells) for sustained tumor regression (36). Similarly in gastrointestinal stromal tumors, the KIT tyrosine kinase inhibitor imatinib mesylate prolongs the survival of patients through direct effects on tumor cells and by indirect immunostimulatory effects on T and NK cells (37, 38). Similar findings in models of hepatocellular, breast and liver cancer also support the role for immune-mediated tumor regression after gene de-addiction (39-42).
In melanoma, early evidence for potential synergy between targeted therapy and immunotherapy for melanoma was nested in the finding that oncogenic BRAF (BRAFV600E) can lead to immune escape in melanoma (43), and that blocking its activity via MAPK pathway inhibition leads to increased expression of melanocyte differentiation antigens (MDAs) (44). This was strengthened in the finding that targeted inhibition of the MAPK pathway leads to up to a 100-fold increase in expression of melanoma differentiation antigens in melanoma cell lines and fresh tumor digests in vitro, which conferred increased reactivity to antigen-specific T lymphocytes (11). The mechanism behind this seems to be related to transcriptional repression of MITF in the setting of oncogenic BRAF, with release of transcriptional repression when the MAPK pathway is blocked and subsequent expression of MITF targets (including the melanoma differentiation antigens MART-1, gp100, TRP-1 and TRP-2 (11)).
A critical component of this early work was the analysis of the effects of MAPK pathway inhibitors on T lymphocyte function, since the MAPK pathway is known to be critical in T cell activation and signaling. Importantly, “selective” inhibitors of BRAFV600E do not have any deleterious effect on T cell function (11), and may even augment T cell activation via paradoxical signaling through RAS-GTP pathways (45). In contrast, MEK inhibitors, demonstrate dose-dependent inhibition of T cell function in vitro (11). This has relevance when contemplating combination of BRAF-directed therapy with immunotherapy, as therapy including a MEK inhibitor may potentially have deleterious effects on T cells and thus may abrogate any potential synergy.
The first clinical evidence demonstrating potential synergy of BRAF-targeted therapy and immunotherapy was from 2 independent groups, which demonstrated enriched T cell infiltrates in tumors of patients with metastatic melanoma within 14 days of initiation of BRAF inhibitor therapy (13) (Figure 2). This was associated with an increase in melanoma antigen expression in tumors (Figure 2), and a decrease in immunosuppressive cytokines IL-6, IL-8 (12) and in vascular endothelial growth factor (VEGF) (46). The tumor stroma appears to play a critical role, as stromal cell-mediated immunosuppression via interleukin 1 (IL-1) is induced by oncogenic BRAF and blocked with BRAF inhibitors (14). Of note, these changes are lost at time of progression (12).
Figure 2. Changes in the tumor microenvironment with BRAF inhibition.

Patients with metastatic melanoma treated with BRAF inhibitors were biopsied pre-treatment and 10-14 days after treatment initiation. BRAF inhibition is associated with an increase in CD8+ T cells, melanoma differentiation antigen expression, and the immunomodulatory molecule PD-L1. Adapted from Frederick et al., 2013 (12).
Equally profound and clinically impactful is the observation of increased expression of immunomodulatory molecules (namely PD-1 and its ligand PD-L1) within 2 weeks of initiation of BRAF-targeted therapy (12) – a finding suggesting a potential immune-mediated resistance mechanism to BRAF inhibition. Namely, though the infiltrating T cells in tumors of patients being treated with BRAF inhibitors demonstrate an activated phenotype, they also express high levels of PD-1 (12). The PD-1 molecule is an immunomodulatory molecule found on the surface of T lymphocytes that serves to down-regulate an immune response after an initial period of activation, functioning in a physiologic sense to prevent autoimmunity (47). However within 2 weeks of initiation of treatment with a BRAF inhibitor, this increase in PD-1 expression is coupled with a significant increase in the expression of its ligand PD-L1 within the tumor stroma (Figure 2) (12). The up-regulation of PD-L1 may be due to interferon-gamma (IFN-γ) production by infiltrating T cells (48). Additional evidence supporting this as a mechanism of resistance is represented through in vitro studies demonstrating high PD-L1 expression in melanoma cell lines resistant to BRAF inhibition (49). Interestingly, in this model system, the addition of MEK inhibition abrogated the up-regulation of PD-L1 in cell lines (49), though the effects of combined BRAF and MEK inhibition in tumor biopsies of patients have not been thoroughly investigated. Nonetheless, these data suggest that immune checkpoint blockade (particularly blockade of the PD-1 pathway) may augment responses in the setting of treatment with a BRAF inhibitor in the treatment of metastatic melanoma (50). A critical gap in knowledge exists pertaining to similar effects on antigen presentation and immune cell dynamics in the setting of analogous targeted therapies in other oncogene-defined cancer populations (EGFR and ALK inhibitors in lung cancer and Her-2 targeted antibodies in breast cancer being notable examples).
Several pre-clinical models have been used to study the hypothesis that BRAF-targeted therapy will synergize with immunotherapy. Results of these studies have been mixed, with all (46, 51-53) but one (54) showing synergy. Our own group has studied combined BRAF-targeted therapy and immune checkpoint blockade against the PD-1 pathway, and observed synergy when these 2 strategies are combined (53). These models are useful, and studies exploring multiple aspects of combination approaches (e.g. appropriate sequence and timing of therapy) should be performed to improve decision-making in design of clinical trials. The number of immune-competent melanoma models remains limited and this ultimately weakens the ability to thoroughly vet the most pressing questions regarding prioritization of agents, doses and schedules pre-clinically. There is growing data regarding the role of the tumor microenvironment in response and resistance to therapy (12, 14, 21), with the potential to target stromal cells (such as tumor-associated fibroblasts, endothelial cells, and macrophages or regulatory T cells) to improve responses. Additional insight regarding the contribution of each of these stromal components in response to monotherapy is critical in instructing how they may be used in combination for clinical trials, though pre-clinical in vitro models (such as high-throughput screening assays) and murine models may also be instructive.
Considerations in combining these strategies
Based on promising results from pre-clinical and clinical studies demonstrating potential synergy between immunotherapy and targeted therapy for melanoma (11, 12, 46, 51, 52), clinical trials are underway to investigate the efficacy and safety of combining targeted therapy and immunotherapy in patients with BRAF mutation-positive melanoma (55). In these trials, BRAF and/or MEK inhibitors are being combined with several forms of immunotherapy – including cytokines (IL-2), immune checkpoint inhibitors (against CTLA-4, PD-1, or PD-L1), and adoptive cell therapy (with tumor-infiltrating lymphocytes). Mature response and toxicity data are not yet available, however interesting data has been extracted from these trials (as well as from pre-clinical and clinical studies) that raises important considerations when combining these strategies.
One consideration is the issue of added toxicity in the setting of combined BRAF-targeted therapy and immunotherapy. This is relevant, as initial efforts in clinical trials using ipilimumab in combination with BRAF-targeted therapy were limited by this variable (56). In this trial, patients with metastatic melanoma with known BRAFV600E mutation received a one-month run in with a BRAF inhibitor (vemurafenib) alone followed by four infusions of ipilimumab. The primary goal of this trial was to assess safety and the target accrual was 50 patients, though the trial was stopped early due to toxicity. Specifically, hepatotoxicity was observed in a substantial proportion of patients, consisting mainly of grade 2 or 3 elevations in liver function tests (56). Though these elevations in liver function tests (LFTs) were completely asymptomatic and resolved when therapy was discontinued or with administration of systemic steroids, these data highlight the potential for unexpected toxicity and mandate that careful attention be paid when combining these agents. A similar trial is now underway with sequential (i.e. non-overlapping) administration of these agents. Trials combining other BRAF-targeted agents (e.g. dabrafenib) with immune checkpoint inhibitors are also underway, and have not shown the same toxicity profile though data is not mature. Toxicity with immune checkpoint blockade monotherapy targeting PD-1 and PD-L1 is significantly lower than that seen with therapy targeting CTLA-4 (6, 8, 9), but it is unclear if this will translate when used in combination with BRAFi therapy. Trials combining these agents are also underway.
Another important consideration is whether synergy will be seen when immunotherapy is combined with other forms of MAPK pathway blockade (e.g., MEK inhibitors), given that MAPK pathway activity is critical to T cell activation and may abrogate T cell responses (11). For each of the established targeted therapies relevant to other cancer subpopulations, similar concerns pertain to effect on the immune cell population, but have not been systemically investigated. We studied this in vitro, and demonstrated that though treatment with a MEK inhibitor resulted in increased melanoma antigen expression in both wild-type and mutant BRAF cell lines, it also resulted in impaired T cell proliferation and function. Namely, the increased reactivity to antigen-specific T cells conferred by increased antigen expression was completely abrogated with MEK inhibition (15). This issue is particularly relevant in light of recent data showing that responses to combined BRAF/MEK inhibition were superior to those seen with BRAF monotherapy (5), leading to the FDA approval of this combination regimen in 2014. However, it is important to note that no clear differences were observed with regard to the number of infiltrating T lymphocytes in patients receiving combined BRAFi/MEKi versus BRAFi monotherapy (15), suggesting that the MEK-mediated T cell inhibition observed in our in vitro studies may not be clinically-relevant. Studies are currently underway in murine models to help answer this question, and clinical trials combining BRAFi and MEKi with immune checkpoint inhibitors are also ongoing. Though the primary endpoints of these initial studies are safety and tolerability, secondary endpoints will focus on disease control rates and correlative studies in which a thoughtful analysis of pre-treatment and on-treatment tumor biopsy samples will be performed to gain insight into this important question.
A related consideration in combining these strategies includes the proper sequence and timing of regimens. This is likely a critical factor, though we do not yet have a comprehensive understanding of the optimal sequence and timing when BRAF-targeted therapy is combined with immunotherapy. Nonetheless, insights do exist and additional studies are underway to illuminate this point. Early data suggest that the immune response to BRAF inhibitors happens fairly quickly (within 10-14 days of initiation of a BRAF inhibitor) but is transient, with very few T cells remaining a month after initiation of therapy (15). Thus there may be a narrow window to optimize recruitment and activation of T lymphocytes in the setting of BRAF-targeted therapy. The hypothesis based on this early work would propose that BRAF inhibitor therapy be initiated with a short lead-in (e.g. 2 weeks) followed by the addition of immunotherapy. A corollary to this hypothesis would suggest that an immune checkpoint inhibitor (specifically targeting the PD-1 pathway) should be included in this regimen, given up-regulation of PD-L1 in the tumor microenvironment within 2 weeks of initiation of BRAF-targeted therapy (15). Another finding from these studies suggests that the beneficial changes to the tumor microenvironment induced by treatment with a BRAF inhibitor are transient and are essentially absent at 4 weeks, with reversion to a more hostile microenvironment at time of progression (15). Thus the appropriate timing to add an immunomodulatory agent to the regimen is early during treatment rather than at the time of progression (Figure 3), a concept which is supported by the observation that patients who are treated with ipilimumab after progression on BRAF inhibitors exhibit a poor response rate to therapy (57). Nonetheless, these suggestions are based on limited data and speculative, and need to be validated with other targeted agents and combination regimens. Studies are currently underway to elaborate on this point, both in the context of in vivo murine models as well as in carefully planned correlative studies in combination clinical trials. Timing and sequence of therapy will both bead dressed in these studies.
Figure 3. Sequence and timing considerations for combining targeted therapy and immunotherapy.
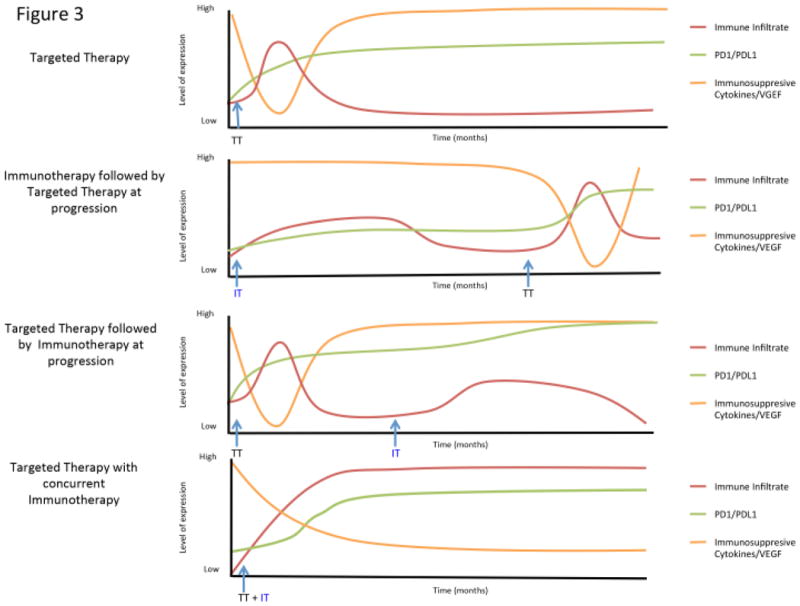
The proposed plots show changes in immune infiltrate and expression of immunomodulatory molecules / cytokines / VEGF depending on the sequence of treatment with targeted therapy and immunotherapy. With targeted therapy, there is an early but transient increase in immune infiltrate that is associated with a transient decrease in immunosuppressive cytokines, but an increase in PD-1 and PD-L1. Treatment with immunotherapy results in a more gradual and prolonged increase in immune infiltrate, and is also associated with an increase in PD-1 and PD-L1. The addition of targeted therapy at time of progression on immunotherapy may not lead to synergy given the kinetics of these changes in the tumor microenvironment. Likewise, the addition of immunotherapy at time of progression to targeted therapy may not lead to synergy for similar reasons. However, the concurrent use of targeted therapy and immunotherapy simultaneously promotes a favorable microenvironment along with T cell activation, with the potential for synergy and prolonged responses.
Another concept germane to this discussion involves the use of intermittent dosing in BRAF-targeted therapy. There is recent evidence that intermittent dosing of BRAF inhibitors may significantly improve responses to therapy, which appears to modulate the clonal evolution of resistant cells (58). This is relevant, as intermittent dosing may alter the immune effects of BRAF inhibitors and may also influence the toxicity of regimens combining these strategies.
Unanswered questions
A key question when examining the immune effects of BRAF inhibitors and their related potential synergy with immunotherapeutic strategies is whether or not this represents an antigen-specific response. There is data suggesting that this is at least in part the case, as treatment with BRAF inhibitors results in increased melanoma antigen expression by tumors (15) and a more clonal T cell response (59). However, it is very unlikely that this is purely due to a response to melanoma differentiation antigens, as multiple other changes in the tumor microenvironment (namely a decrease in immunosuppressive cytokines and VEGF) likely facilitate recruitment and proliferation of T cells in the context of treatment with a BRAF inhibitor (15).
One intriguing possibility is that T cell responses against other antigens (namely neoantigens) play a significant role in the response to BRAF inhibition. There is data to support the role of reactivity to neoantigens in melanoma, as T cells recognizing these antigens are associated with response to therapy in the context of treatment with tumor-infiltrating lymphocytes (TIL) (60) and immune checkpoint blockade (61). Though neoantigens have been studied in a small subset of patients, larger cohorts are clearly needed to better understand their role in response to therapy and the potential use of T cells targeting these antigens for personalized cancer therapy.
Another question is which form of immunotherapy should be used in combination with targeted therapy. Though early efforts have focused on combining BRAF-targeted therapy with FDA-approved agents (such as IL-2 and ipilimumab), subsequent trials have combined these strategies with immunotherapeutic strategies still in clinical trials (such as PD-1 and PD-L1). This adds a layer of complexity, as response rates and toxicity with these agents may be more difficult to interpret as definitive estimates of benefit from the newer agents are lacking. In addition, novel agents currently in development or in early stage clinical trials (such as monoclonal antibodies targeting TIM-3) will soon be added to the armamentarium for combination strategies and we have little insight into the optimal mode of stimulating immune effectors cells in the setting of “priming” by targeted therapies.
In addition to immunotherapy, numerous other strategies may be added to a backbone of BRAF-targeted therapy and immunotherapy with the goal of further enhancing responses. An example of this is radiation therapy – a modality reported to have potential synergy both with targeted therapy (62) as well as immunotherapy (63). The potential benefit of this strategy hinges on the abscopal effect –a phenomenon in which tumors regress at sites remote from an irradiated target (62, 64, 65). This is likely related to increased antigen presentation and enhanced killing by T cells (62). Radiation is being used in combination with immunotherapy in clinical trials, and holds potential to augment responses to combined BRAF-targeted therapy and immunotherapy as well.
Extending these concepts to other cancers
There is growing evidence that similar combination strategies may be used to enhance responses to therapy for non-melanoma malignancies. In order for these combinatorial strategies to work, the targeted therapy must be able to inhibit pertinent oncogenic events resulting in a more favorable tumor microenvironment for the anti-tumor immune response. In addition, targeted therapies shouldn't inhibit critical immune effector cells and should rather potentiate or stimulate an inactive effector immune population (66). Similar to BRAF inhibitors in melanoma, abl and c-kit inhibitors have had great success for the treatment of chronic myelogenous leukemia and gastrointestinal stromal tumors (67, 68). T cells have been found to be a crucial component of the anti-tumor effect after imatinib treatment in GIST, activating CD8+ T cells while inducing regulatory T cell apoptosis within the tumor (37). Another potential target is the PI3K pathway, as glioblastoma multiforme cells with a PTEN deficiency have an increase in PD-L1 expression that can be reversed following PI3K inhibition (69). In addition, EGFR-driven lung tumors have recently been shown to inhibit antitumor immunity by activation of the PD-1/PD-L1 pathway (70). Accordingly, using combination of PD-1 blockade and EGFR tyrosine kinase inhibitors has been suggested as a promising strategy in order to extend the durability of treatment and delay disease progression (70). As the genomics era and data of the first generation of targeted therapy matures, our ability to extend the concept of combined immunotherapy and targeted therapy to other histologies will continue to grow. The key now is to identify compounds that may show synergy with immunotherapy, and high throughput screening assays are currently in development.
Implications for research and clinical trials
The concept of potential synergy with BRAF-targeted therapy and immunotherapy is being empirically investigated in clinical trials, however much remains to be learned. Response data from these initial trials is not mature, and additional trials will be needed to determine the appropriate sequence, schedule, and duration of therapy if there is evidence of synergy. If not, then a careful analysis of data should be performed prior to discarding this concept (including a thoughtful analysis of tumor and blood samples) as dosing and schedule may factor into these early trials.
In addition to optimizing sequence schedule and duration, new agents are becoming available on both the targeted therapy and immunotherapy fronts. Novel targeted agents (e.g. MEK, CDK4, PI3K, MDM2, FGFR, c-MET) and immunotherapeutic approaches (e.g. against TIM-3, Lag-3, GITR, OX40, CD27) are now available and are in clinical trials, and could also be used in combination. In addition to combination “doublets”, trials are also now ongoing combining three different agents (such as BRAFi and MEKi with an immune checkpoint inhibitor). Thus these combination strategies are becoming increasingly complex, with a large number of potential combinations / schedules which must be carefully considered. Methods to predict success of potential combinations in the setting of an intact immune system are critically needed, and are in development.
An additional consideration in designing these trials is potential toxicity when used in combination. An example of this is hepatotoxicity, which was observed when patients were treated with vemurafenib and ipilimumab (56). The mechanism behind this is not entirely understood, though it mandates thorough investigation as insights gained may shed light on other potential toxicities and will help guide development of subsequent clinical trials.
Of paramount importance in the development of these trials is incorporation of longitudinal blood and tumor sampling into the treatment schema. This allows for validation of target inhibition as well as for critical correlative studies that will ultimately allow for biomarker discovery. Serial biopsies and blood draws are now often mandated by industry-sponsored trials to drive their own biomarker discovery, and provide an opportunity for additional research to better understand mechanisms of response and resistance to therapy.
In addition to clinical trials, there are tremendous opportunities for research in pre-clinical studies using in vitro techniques and murine models. Importantly, there is now a shift towards incorporating immune cells for in vitro screening methods and incorporating murine models with intact immune systems in an effort to determine the anti-cancer and immune effects of single agents or combination therapies. These techniques and models have gained traction and should lead to improved prediction of efficacy in clinical trials.
Conclusions
There is evidence for potential synergy of targeted therapy and immunotherapy, however significant inroads must be made to optimize combination approaches to maximize responses and limit toxicity. A better understanding of mechanisms of response and resistance to each of these forms of therapy is critical to instruct how best to combine therapeutic agents. Central to this understanding is translational research conducted on patient samples, which may instruct (and be instructed by) parallel in vitro and murine studies. However this clearly represents a changing paradigm in cancer treatment, with a critical understanding of the interplay between genomics and anti-tumor immunity.
Statement of Significance.
Two major advances in melanoma have occurred concurrently and involve treatment with targeted therapy and immune checkpoint blockade. However, each of these approaches has limitations with regards to overall response rates or duration of response. To address this, investigators have proposed combining these strategies and this concept is being tested empirically in clinical trials. There is scientific rationale supporting the combination of targeted therapy and immunotherapy, and these concepts are discussed herein.
Acknowledgments
Grant support: JAW acknowledges NIH grants 1K08CA160692-01A1 and the generous philanthropic support of several families whose lives have been affected by melanoma. JAW and KTF would like to acknowledge NIH grant U54CA163125-01.
Footnotes
Disclosure of Potential Conflicts of Interest: K.T. Flaherty has served as a consultant to GlaxoSmithKline and Roche/Genentech. J.A. Wargo has honoraria from speakers' bureau of Dava Oncology and is an advisory board member for GlaxoSmithKline, Roche/Genentech, and Amgen. No potential conflicts of interest were disclosed by the other author.
References
- 1.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 2.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. The New England journal of medicine. 2011;364:2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–65. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 4.Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. The New England journal of medicine. 2012;366:707–14. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations. The New England journal of medicine. 2012;367:1694–703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. The New England journal of medicine. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in metastatic or locally advanced, unresectable melanoma. European journal of cancer. 2013 doi: 10.1200/JCO.2014.56.2736. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. The New England journal of medicine. 2012;366:2443–54. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. The New England journal of medicine. 2012;366:2455–65. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity. 2013;39:74–88. doi: 10.1016/j.immuni.2013.06.014. [DOI] [PubMed] [Google Scholar]
- 11.Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CN, Sloss CM, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer research. 2010;70:5213–9. doi: 10.1158/0008-5472.CAN-10-0118. [DOI] [PubMed] [Google Scholar]
- 12.Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:1225–31. doi: 10.1158/1078-0432.CCR-12-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:1386–94. doi: 10.1158/1078-0432.CCR-11-2479. [DOI] [PubMed] [Google Scholar]
- 14.Khalili JS, Liu S, Rodriguez-Cruz TG, Whittington M, Wardell S, Liu C, et al. Oncogenic BRAF(V600E) Promotes Stromal Cell-Mediated Immunosuppression Via Induction of Interleukin-1 in Melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:5329–40. doi: 10.1158/1078-0432.CCR-12-1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu C, Peng W, Xu C, Lou Y, Zhang M, Wargo JA, et al. BRAF Inhibition Increases Tumor Infiltration by T cells and Enhances the Antitumor Activity of Adoptive Immunotherapy in Mice. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:393–403. doi: 10.1158/1078-0432.CCR-12-1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tiacci E, Trifonov V, Schiavoni G, Holmes A, Kern W, Martelli MP, et al. BRAF mutations in hairy-cell leukemia. The New England journal of medicine. 2011;364:2305–15. doi: 10.1056/NEJMoa1014209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xing M. BRAF mutation in thyroid cancer. Endocrine-related cancer. 2005;12:245–62. doi: 10.1677/erc.1.0978. [DOI] [PubMed] [Google Scholar]
- 18.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. The New England journal of medicine. 2010;363:809–19. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–72. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–7. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–4. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montagut C, Sharma SV, Shioda T, McDermott U, Ulman M, Ulkus LE, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer research. 2008;68:4853–61. doi: 10.1158/0008-5472.CAN-07-6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:20411–6. doi: 10.1073/pnas.0905833106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whittaker SR, Theurillat JP, Van Allen E, Wagle N, Hsiao J, Cowley GS, et al. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer discovery. 2013;3:350–62. doi: 10.1158/2159-8290.CD-12-0470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi H, Moriceau G, Kong X, Lee MK, Lee H, Koya RC, et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012;3:724. doi: 10.1038/ncomms1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–90. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer discovery. 2012;2:227–35. doi: 10.1158/2159-8290.CD-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davies MA, Stemke-Hale K, Lin E, Tellez C, Deng W, Gopal YN, et al. Integrated Molecular and Clinical Analysis of AKT Activation in Metastatic Melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15:7538–46. doi: 10.1158/1078-0432.CCR-09-1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bucheit AD, Syklawer E, Jakob JA, Bassett RL, Jr, Curry JL, Gershenwald JE, et al. Clinical characteristics and outcomes with specific BRAF and NRAS mutations in patients with metastatic melanoma. Cancer. 2013;119:3821–9. doi: 10.1002/cncr.28306. [DOI] [PubMed] [Google Scholar]
- 30.Fisher KE, Jani JC, Fisher SB, Foulks C, Hill CE, Weber CJ, et al. Epidermal growth factor receptor overexpression is a marker for adverse pathologic features in papillary thyroid carcinoma. The Journal of surgical research. 2013;185:217–24. doi: 10.1016/j.jss.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1999;17:2105–16. doi: 10.1200/JCO.1999.17.7.2105. [DOI] [PubMed] [Google Scholar]
- 32.Atkins MB, Kunkel L, Sznol M, Rosenberg SA. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: long-term survival update. Cancer J Sci Am. 2000;6(Suppl 1):S11–4. [PubMed] [Google Scholar]
- 33.Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science. 2013;342:1432–3. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- 34.Egen JG, Kuhns MS, Allison JP. CTLA-4: new insights into its biological function and use in tumor immunotherapy. Nature immunology. 2002;3:611–8. doi: 10.1038/ni0702-611. [DOI] [PubMed] [Google Scholar]
- 35.Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2014;32:1020–30. doi: 10.1200/JCO.2013.53.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rakhra K, Bachireddy P, Zabuawala T, Zeiser R, Xu L, Kopelman A, et al. CD4(+) T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer cell. 2010;18:485–98. doi: 10.1016/j.ccr.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nature medicine. 2011;17:1094–100. doi: 10.1038/nm.2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rusakiewicz S, Semeraro M, Sarabi M, Desbois M, Locher C, Mendez R, et al. Immune infiltrates are prognostic factors in localized gastrointestinal stromal tumors. Cancer research. 2013;73:3499–510. doi: 10.1158/0008-5472.CAN-13-0371. [DOI] [PubMed] [Google Scholar]
- 39.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–60. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park S, Jiang Z, Mortenson ED, Deng L, Radkevich-Brown O, Yang X, et al. The therapeutic effect of anti-HER2/neu antibody depends on both innate and adaptive immunity. Cancer cell. 2010;18:160–70. doi: 10.1016/j.ccr.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stagg J, Loi S, Divisekera U, Ngiow SF, Duret H, Yagita H, et al. Anti-ErbB-2 mAb therapy requires type I and II interferons and synergizes with anti-PD-1 or anti-CD137 mAb therapy. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:7142–7. doi: 10.1073/pnas.1016569108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hutcheson J, Bourgo RJ, Balaji U, Ertel A, Witkiewicz AK, Knudsen ES. Retinoblastoma protein potentiates the innate immune response in hepatocytes: Significance to hepatocellular carcinoma. Hepatology. 2014 doi: 10.1002/hep.27217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. The Journal of experimental medicine. 2006;203:1651–6. doi: 10.1084/jem.20051848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kono M, Dunn IS, Durda PJ, Butera D, Rose LB, Haggerty TJ, et al. Role of the mitogen-activated protein kinase signaling pathway in the regulation of human melanocytic antigen expression. Mol Cancer Res. 2006;4:779–92. doi: 10.1158/1541-7786.MCR-06-0077. [DOI] [PubMed] [Google Scholar]
- 45.Callahan MK, Masters G, Pratilas CA, Ariyan C, Katz J, Kitano S, et al. Paradoxical activation of T cells via augmented ERK signaling mediated by a RAF inhibitor. Cancer immunology research. 2014:2. doi: 10.1158/2326-6066.CIR-13-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu C, Peng W, Xu C, Lou Y, Zhang M, Wargo JA, et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:393–403. doi: 10.1158/1078-0432.CCR-12-1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–7. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 48.Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Science translational medicine. 2013;5:200ra116. doi: 10.1126/scitranslmed.3006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang X, Zhou J, Giobbie-Hurder A, Wargo J, Hodi FS. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:598–609. doi: 10.1158/1078-0432.CCR-12-2731. [DOI] [PubMed] [Google Scholar]
- 50.Cooper ZA, Frederick DT, Ahmed Z, Wargo JA. Combining checkpoint inhibitors and BRAF-targeted agents against metastatic melanoma. Oncoimmunology. 2013;2:e24320. doi: 10.4161/onci.24320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koya RC, Mok S, Otte N, Blacketor KJ, Comin-Anduix B, Tumeh PC, et al. BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer research. 2012;72:3928–37. doi: 10.1158/0008-5472.CAN-11-2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Knight DA, Ngiow SF, Li M, Parmenter T, Mok S, Cass A, et al. Host immunity contributes to the anti-melanoma activity of BRAF inhibitors. The Journal of clinical investigation. 2013;123:1371–81. doi: 10.1172/JCI66236. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 53.Cooper ZA, Juneja VR, Sage PT, Frederick DT, Piris A, Mitra D, et al. Response to BRAF inhibition in melanoma is enhanced when combined with immune checkpoint blockade. Cancer immunology research. 2014;2:643–54. doi: 10.1158/2326-6066.CIR-13-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hooijkaas A, Gadiot J, Morrow M, Stewart R, Schumacher T, Blank CU. Selective BRAF inhibition decreases tumor-resident lymphocyte frequencies in a mouse model of human melanoma. Oncoimmunology. 2012;1:609–17. doi: 10.4161/onci.20226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hu-Lieskovan S, Robert L, Homet Moreno B, Ribas A. Combining Targeted Therapy With Immunotherapy in BRAF-Mutant Melanoma: Promise and Challenges. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2014;32:2248–54. doi: 10.1200/JCO.2013.52.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. The New England journal of medicine. 2013;368:1365–6. doi: 10.1056/NEJMc1302338. [DOI] [PubMed] [Google Scholar]
- 57.Ackerman A, Klein O, McDermott DF, Wang W, Ibrahim N, Lawrence DP, et al. Outcomes of patients with metastatic melanoma treated with immunotherapy prior to or after BRAF inhibitors. Cancer. 2014 doi: 10.1002/cncr.28620. [DOI] [PubMed] [Google Scholar]
- 58.Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494:251–5. doi: 10.1038/nature11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cooper ZA, Frederick DT, Juneja VR, Sullivan RJ, Lawrence DP, Piris A, et al. BRAF inhibition is associated with increased clonality in tumor-infiltrating lymphocytes. Oncoimmunology. 2013;2:e26615. doi: 10.4161/onci.26615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nature medicine. 2013;19:747–52. doi: 10.1038/nm.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31:e439–42. doi: 10.1200/JCO.2012.47.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sullivan RJ, Lawrence DP, Wargo JA, Oh KS, Gonzalez RG, Piris A. Case records of the Massachusetts General Hospital. Case 21-2013. A 68-year-old man with metastatic melanoma. The New England journal of medicine. 2013;369:173–83. doi: 10.1056/NEJMcpc1302332. [DOI] [PubMed] [Google Scholar]
- 63.Barker CA, Postow MA. Combinations of Radiation Therapy and Immunotherapy for Melanoma: A Review of Clinical Outcomes. International journal of radiation oncology, biology, physics. 2014;88:986–97. doi: 10.1016/j.ijrobp.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Postow MA, Callahan MK, Barker CA, Yamada Y, Yuan J, Kitano S, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. The New England journal of medicine. 2012;366:925–31. doi: 10.1056/NEJMoa1112824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Golden EB, Demaria S, Schiff PB, Chachoua A, Formenti SC. An Abscopal Response to Radiation and Ipilimumab in a Patient with Metastatic Non-Small Cell Lung Cancer. Cancer immunology research. 2013;1:365–72. doi: 10.1158/2326-6066.CIR-13-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Begley J, Ribas A. Targeted therapies to improve tumor immunotherapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14:4385–91. doi: 10.1158/1078-0432.CCR-07-4804. [DOI] [PubMed] [Google Scholar]
- 67.Blanke CD, Rankin C, Demetri GD, Ryan CW, von Mehren M, Benjamin RS, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008;26:626–32. doi: 10.1200/JCO.2007.13.4452. [DOI] [PubMed] [Google Scholar]
- 68.Druker BJ, Guilhot F, O'Brien SG, Gathmann I, Kantarjian H, Gattermann N, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. The New England journal of medicine. 2006;355:2408–17. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 69.Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nature medicine. 2007;13:84–8. doi: 10.1038/nm1517. [DOI] [PubMed] [Google Scholar]
- 70.Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer discovery. 2013;3:1355–63. doi: 10.1158/2159-8290.CD-13-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]