

Abstract
Clinical and basic science research suggests that stress and/or changes in central stress signaling intermediates may be involved in Alzheimer’s disease (AD) pathogenesis. Although the links between stress and AD remain unsettled, data from our group and others have established that stress exposure in rodents may confer susceptibility to AD pathology by inducing hippocampal tau phosphorylation (tau-P). Work in our lab has shown that stress-induced tau-P requires activation of the type-1 corticotropin-releasing factor receptor (CRFR1). CRF overexpressing (CRF-OE) mice are a model of chronic stress that display cognitive impairment at 9–10 month of age. In this study we used 6–7 month old CRF-OE mice to examine whether sustained exposure to CRF and stress steroids would impact hippocampal tau-P and kinase activity in the presence or absence of the CRFR1-specific antagonist, R121919, given daily for 30 days. CRF-OE mice had significantly elevated tau-P compared to wild type (WT) mice at the AT8 (S202/T204), PHF-1 (S396/404), S262, and S422 sites. Treating CRF-OE mice with R121919 blocked phosphorylation at the AT8 (S202/T204) and PHF-1 (S396/404) sites, but not at the S262 and S422 sites and reduced phosphorylation of c-Jun N Terminal Kinase (JNK). Examination of hippocampal extracts from CRF-OE mice at the ultrastructural level revealed negatively stained round/globular aggregates that were positively labeled by PHF-1. These data suggest critical roles for CRF and CRFR1 in tau-P and aggregation and may have implications for the development of AD cognitive decline.
Keywords: Alzheimer’s disease, corticotropin-releasing, corticotropin-releasing factor receptor (CRFR), electron microscopy, hippocampus, immunohistochemistry, stress, tau phosphorylation (tau-P), western blot
INTRODUCTION
Alzheimer’s disease (AD) is characterized pathologically by extensive cell loss, the accumulation of senile plaques composed of amyloid-β (Aβ) and neurofibrillary tangles (NFT) consisting of hyperphosphorylated tau protein. The incidence of NFT is positively correlated with cognitive deficits and neuronal loss in AD [1, 2]. Tau is a soluble phospho-protein that stabilizes microtubules, plays a critical role in the establishment and maintenance of neuronal structure, polarity, transport, and exists in multiple isoforms (six in humans, four in mice) produced from a single gene [3]. Whereas tau is phosphorylated at 2–3 moles/mole of protein under basal conditions, tau from the AD brain is hyperphosphorylated at a 7–10 molar ratio [4, 5] and at least 25 distinct sites [6, 7]. Although information is known about the general role of tau in AD, the precise consequences of tau phosphorylation (tau-P) have been difficult to determine. A leading hypothesis in the field postulates that phosphorylation reduces the ability of tau to bind and stabilize microtubules and leads to tau aggregation and formation of paired helical filaments, which comprise NFT [8–10]. Several studies demonstrate that exposure to acute physiological and emotional stress paradigms induces reversible hippocampal tau-P in rodents (for review, [11]), a process that, at least in the case of emotional stress, is reliant on signaling through the type-1 corticotropin-releasing factor receptor (CRFR1) [12, 13]. Our previous data demonstrate that exposure to chronic/repeated emotional stress leads to cumulative effects on tau-P and its sequestration to detergent-soluble cellular fractions of mice hippocampus [12, 14].
Constitutive overexpression of CRF (CRF-OE) in mice is considered a valid animal model of chronic stress [15, 16]. CRF-OE mice display significant brain atrophy at 3–6 months of age [17] and show hippocampal-dependent learning and memory deficits when tested at 9 months of age [18]. Furthermore, studies using CRF-OE mice have found increased levels of hyperphosphorylated tau compared with wild-type littermates [19]. Although published data with stress and/or CRF-OE suggest that stress steroids are not required for stress-induced tau-P [12, 19, 20], the circuitry involved is unknown and whether sustained levels of CRF can induce tau-P via CRFR1 in the absence of environmental stress is uncertain. In the present study, we used CRF-OE mice to examine the impact of sustained exposure to CRF on hippocampal tau-P at several AD-relevant sites (S262, S422, S202/T205 (AT8), and S396/404 (PHF-1). To identify the potential role of CRFR1 in facilitating tau-P, we compared groups of CRF-OE mice treated for 30 days with vehicle or the CRFR1 antagonist, R121919 (NBI 30775) [21].
MATERIALS AND METHODS
CRF-OE transgenic mice
As described previously, CRF-OE mice were originally generated with a chimeric CRF transgene composed of the rat genomic CRF gene replacing the 5′ regulatory region by a mouse metallothionein-1 promoter that was microinjected into the male pronucleus of fertilized eggs (C57/B6 × SJL) [15, 22]; CRF over-expression has been confirmed through administration of CRF antagonists [16]. CRF-OE mice were obtained from Jackson Laboratories. Experiments were conducted in adult (6–7 months old) female CRF-OE mice (n = 20) and age-matched wild type (WT) female littermates (n = 10). Mice were housed (2–4/cage) with ad libitum access to standard laboratory rodent diet and water in a temperature controlled room (22°C) with a 12 h light-dark cycle. The University of California, San Diego’s Institutional Animal Care and Use Committee approved all experimental protocols.
In vivo pharmacology
CRF-OE mice received daily subcutaneous (sc) injections either the CRFR1 antagonist, R121919 at 20 mg/kg (gift of Dr. K. Rice, NIH) (n = 10), or drug vehicle (n = 10) for 30 days leading up to euthanasia. The dose of 20 mg/kg/d was based on our previous studies in mice with R121919 and stress-induced tauP [12, 14]. R121919 was dissolved in 0.3% tartaric acid and 5% v/v polyethoxylated castor oil. Both R121919 and vehicle solution were vortexed and sonicated immediately before use. The volume of daily sc injection was 0.1 mL.
Antibodies
Several well-characterized antibodies were used to detect phosphorylated forms of tau and major tau kinases. For western blots, S202/T205 (1 : 500; AT8; Pierce Biotechnology), S262, S422 (1 : 1000; Biosource, Camarillo, CA), and S396/404 (1 : 1000; PHF-1; gift from Dr. P. Davies, Albert Einstein, NY), were selected based on their ability to reveal target bands at the appropriate molecular weight for phosphorylated tau (~50–75 kDa). Antibodies against S199, T212, T231, and S409 sites were tested but were found either to target only high-molecular weight proteins (>100 kDa) (S199, T231) or have no signal (T212, S409) and were therefore excluded from the analysis. For assessment of tau kinases, antibodies to phosphorylation sites or activator proteins were also used: activated GSK-3β (pY216; 1 : 1000; BD Transduction), inactive GSK-3β (pS9; 1 : 1000; Cell Signaling Technology, Danvers, MA), cyclin-dependent kinase 5 (cdk5; 1 : 1000; EMD Biosciences), cdk5 activator proteins, p25 and p35 (1 : 1000; Santa Cruz Biotechnology, Santa Cruz, CA), phosphorylated c-Jun-N-terminal kinase (JNK; 1 : 1000; Cell Signaling Technology), mitogen-activated protein kinases, extracellular signal-regulated kinases 1 and 2 (ERK1/2, 1 : 500; Cell Signaling Technology). Total protein levels of GSK-3 (GSK-3β; 1 : 2500; BD Biosciences, San Diego, CA), JNK, CDK-5, and ERKS (Cell Signaling Technology) were measured. Beta-actin (Sigma-Aldrich) was used as a control for protein loading for each blot.
Immunohistochemistry
Due to the propensity of anesthesia to impact tau phosphorylation (e.g., [23]), mice (n = 4 for each group) were euthanized by cervical dislocation 24 h after the last injection of R121919, then perfused through the ascending aorta with 0.9% saline and brains were removed. Left side hemibrains were drop fixed in 4% paraformaldehyde for 4 h and then cryopotected overnight in 20% sucrose-phosphate buffered saline (PBS). The next morning, 30 μm-thick sections were cut on a freezing-sliding microtome and stored at −20°C in cryoprotectant solution, containing 20% glycerol and 30% ethylene glycol in 0.1 M phosphate buffer. Before immunostaining, endogenous peroxidase was quenched with 0.3% H2O2. After incubating in PHF-1 primary antibody for 48 h at 4°C, sections were incubated in biotinylated anti-mouse antibody followed by avidin-biotin complex. Reaction product was developed using a nickel-enhanced DAB reaction [24]. Antibody specificity for phosphorylated mouse tau was confirmed previously by pretreating sections from mice exposed to 30 min restraint stress with alkaline phosphatase (40 mg/ml), which eliminated immunoreactivity [12].
Western analysis
The hippocampus from each animal was rapidly dissected from the right hemisphere and immediately homogenized in detergent-containing radioimmunoprecipitation assay (RIPA) buffer containing protease and phosphatase inhibitor cocktail EDTA-free (Thermo Scientific), as previously described [12–14, 25]. Protein concentrations were determined using a bicinchoninic acid (BCA) Protein Assay kit (Pierce Biotechnology). 6 μg of protein was loaded and electrophoretically separated on a bis-tris polyacrylamide gel. Proteins were transferred to 0.2 μm nitrocellulose membrane and incubated in primary antibody followed by HRP-linked secondary antibodies and developed with a chemiluminescence western blot detection kit (Pierce Supersignal). All blots were then stripped with stripping buffer (Thermo Scientific) and probed for actin (1 : 1000, Sigma), which served as a loading control. Background subtraction was performed and quantitative band intensity readings were obtained using NIH ImageJ software. All data were then normalized to WT controls, with all western blot data presented as a percent change in intensity relative to WT controls.
Immunoelectron microscopy
RIPA fractions were also examined by immuno-gold electron microscopy to assess whether structural changes in hippocampal tau occur as a function of CRF-OE in the presence or absence of R121919. RIPA extracts were postfixed in 1% glutaraldehyde, treated with osmium tetraoxide, embedded in epon araldite and sectioned with an ultramicrotome (Leica, Germany). Grids were analyzed with a Zeiss OM 10 electron microscope as previously described [26]. For immunogold labeling, sections were mounted on nickel grids, etched and incubated with PHF-1 antibody followed by gold-tagged antibodies against mouse IgG1 (10 nm Aurion ImmunoGold particles) (1 : 50, Electron Microscopy Sciences, Fort Washington, PA) and silver enhancement.
Statistical analyses
Optical density readings from western blots were analyzed using either one- or two-way (genotype x treatment) ANOVA using Prism 6 software (GraphPad, San Diego, CA). Data were plotted on histograms, and expressed as mean ± SEM percentage of control values. For the epitopes that had no signal in WT animals, OD readings are presented.
RESULTS
CRF overexpressing mice have increased hippocampal tau-P
Vehicle-treated CRF-OE mice had a widespread profile of tau-P protein expression in the hippocampus (Fig. 1A), with significantly increased tau-P at all sites tested compared to WT controls (all p < 0.05). In particular, AT8, PHF-1, and S422 were 350%, 350%, and 170% (respectively) greater in CRF-OE mice than their WT counterparts (Fig. 1B, C). Of interest, tauP at the S262 site, which was not observed in WT, was observed in CRF-OE although at a modest level (OD = 1.25) in comparison to other epitopes (Fig. 1C). This suggests that tau-P at S262 may be directly due to the overexpression of CRF in this model. Treatment for 30 days with R121919 (CRF-OE + antag) completely prevented the rise in tau-P at the AT8 and PHF-1 sites in CRF-OE mice (p < 0.05 each) as shown by the levels of phosphorylation at these two sites not significantly different between R121919 treated CRF-OE mice and WT counterparts (AT8, p = 0.17; PHF-1, p = 0.25, Fig. 1B). By contrast, levels at the S262 and S422 sites were unchanged by R121919 pretreatment (p > 0.05, Fig. 1C). These results demonstrate that overexpression of CRF can induce tau-P in the hippocampus, a process that requires CRFR1 for specific sites but not others.
Fig. 1.
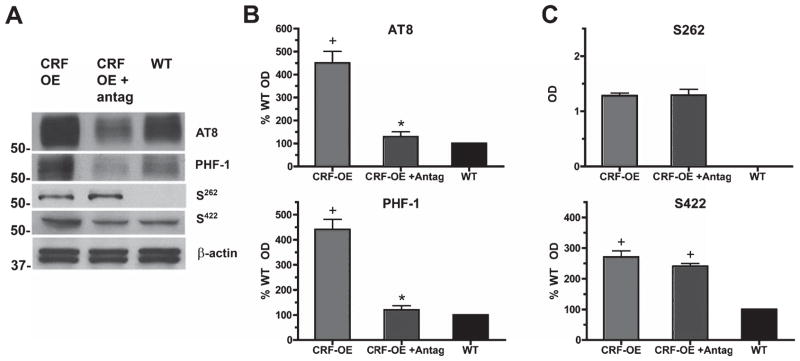
Increased tau-P in CRF-OE mice and involvement of CRFR1. A) CRF-OE mice had significantly elevated basal levels of tau-P in the hippocampus compared to WT cohorts at all phosphoepitopes examined. B) Treatment with the selective CRFR1 antagonist, R121919, significantly reduced tau-P at the AT8 and PHF-1 sites to at or below WT levels. C) R121919 treatment did not impact phosphorylation of the S262 or S422 sites. With the exception of S262 that had no WT baseline signal for comparison all data are presented as Mean ± SEM percentage of WT values, n = 10/group (*p < 0.05 Vehicle versus R121919; +p < 0.05 CRF-OE versus WT control).
To investigate the localization of tau-P responses, we used immunohistochemical methods to examine PHF-1 reactivity in the hippocampus of CRF-OE mice and its CRFR1 dependence. The detection of immunolabeling was consistent with biochemical data in showing a prominent increase in tau-P staining in CRF-OE mice that was blocked by treatment with R121919 (Fig. 2). Hippocampal PHF-1 labeling in CRF-OE was restricted to Ammon’s horn, with stained perikarya scattered throughout the stratum oriens and pyramidale of CA1 and CA2 subregions. Relatively fewer PHF-1 positive cells were observed in stratum radiatum shown in Fig. 2A. Other prevailing features included bands of axonal staining in stratum laconosum-moleculare. CRF-OE mice treated with drug (CRF-OE + R121919) had no cellular labeling in any region of the hippocampus shown in Fig. 2B. We observed low levels of discernible cell or fiber labeling in CA3, hilus, or dentate gyrus as shown in Fig. 2C. Similar to that seen in CRF-OE mice, CRF-OE treated with R121919 had no discrete labeling in CA3, hilus, or dentate gyrus (Fig. 2D). We did, however, observe persistence in fiber immunoreactivity within white matter tracts of the alveus, which appeared weaker in intensity compared to vehicle treated CRF-OE mice. PHF-1 immunoreactivity in the white matter of mice has been seen previously (R. A. Rissman and P.E Sawchenko, unpublished observations), though whether this labeling is specific or represents an artifact due to fixation is uncertain. White matter labeling was observed to varying degrees in both WT and CRF-OE mice with a localization primarily restricted to the alveus and dorsal portions of corpus callosum. No immunoreactivity was observed in the lateral or ventral aspects or splenium of the corpus callosum or in motor nuclei or tracts and no obvious preference was seen in immunoreactivity with proximity to ventricular or pial surfaces (data not shown).
Fig. 2.

Cellular localization of PHF-1 site tau-P in vehicle or CRF-antagonist treated CRF-OE mice. A) Bands of phospho-tau positive axons in stratum lacunosum-moleculare. B) Treatment with R121919 blocked −1 labeling in cells in CRF-OE mice (CRF-OE + R121919), leaving unlabeled neuronal profiles starkly visible against the neuropil. C) CRF-OE mice had widespread tau-P (PHF-1) labeling in the dentate gyrus and CA3 areas, and included perikarya scattered throughout the pyramidal and proximal dendritic zones. D) No PHF-1 immunoreactivity was observed in the dentate/hilar areas in CRF-OE mice regardless of drug treatment. Scale bar = 50 μm.
CRFR1 regulation of tau kinases
Our data demonstrate that CRFR1 blockade reduces hippocampal tau-P to WT levels in CRF-OE at select phosphorylation sites, N-terminal to the microtubule binding domain (AT8, S202/T205) and at the C-terminus (PHF-1, S396/404). Based on these results and that reported by others with CRF-OE mice [19], we hypothesized that differential modulation of kinases by CRFR1 existed in this context. We therefore used antibodies directed to active and inactive states of kinases implicated in tau-P at the epitopes examined to determine which kinases were altered in the hippocampus of CRF-OE treated with vehicle or R121919. Compared to WT cohorts, CRF-OE mice displayed significantly increased phosphorylation levels of GSK-3β, MapK P38 and ERK1/2 kinases (all p < 0.05 compared to WT), but not JNK (p = 0.12) (Fig. 3). Levels of CDK5 activator protein, p35, were robustly upregulated in CRF-OE but the p35-truncated protein, p25 was not observed. Levels of total GSK-3, ERK1/2, and CDK5 were unchanged in CRF-OE mice regardless of treatment with R121919 (all p > 0.05, data not shown). Although several of the kinases examined are implicated in tau-P at the AT8 and PHF-1 sites shown in Fig. 3A, we found only levels of activated JNK to be sensitive R121919 pretreatment (p < 0.05) (Fig. 3A, C). These findings suggest that induction of tau-P via JNK activation is central to tau-P at the AT8 and PHF-1 sites in CRF-OE mice.
Fig. 3.

Modulation of tau kinases in vehicle or CRFR1 antagonist treated CRF-OE mice. A) In the hippocampus, CRF-OE had significantly elevated levels of both inhibited and activated forms of GSK-3 compared to WT animals. B) GSK-3β Y216 phosphorylation levels were unaffected by drug treatment. C) Phosphorylation levels of JNK in CRF-OE were not significantly different from WT, but were significantly reduced by R121919 treatment. This is in line with our tau-P data showing dramatically reduced AT8 and PHF-1 reactivities in the hippocampus. No significant modulations of ERK1/2, CDK5, or MAPK p38 kinases were seen in the hippocampus of CRF-OE mice regardless of treatment. All data are expressed as mean ± SEM percentage of WT values, n = 10/group (*p < 0.05 Vehicle versus R121919).
CRF-OE increases formation of globular tau aggregates
To examine the functional consequences of increased tau-P and its regulation by R121919 in CRF-OE mice, we used immunogold electron microscopy techniques. We used detergent-soluble hippocampal extracts generated for biochemical studies to examine whether structural changes occur as a consequence of chronically elevated tau-P. We found negatively stained round/globular aggregates (~50 nm diameter) (Fig. 4A, left panel) with a density as high as 10/μm2. These aggregates were not present in mice that received R121919 (Fig. 4A, right panel). Extracts from CRF-OE mice had aggregates that were positive for PHF-1 immunogold labeling (Fig. 4B, both panels). An average of 3–6 gold particles were seen per aggregate. CRF-OE mice treated with R121919 had no immunogold signal (data not shown). Although requiring confirmation and extension, our findings may represent evidence of pre-pathologic hippocampal tau aggregates, in vivo.
Fig. 4.

Immunoelectron microscopy of tau-P aggregates in the hippocampal extracts from CRF-OE mice. A) CRF-OE mice had negatively stained round/globular aggregates (~50 nm diameter) which were not present with R121919 treatment. B) Aggregates in CRF-OE mice could be decorated with PHF-1 immunogold labeling. C) Negative controls included trials without primary or secondary antibodies. Scale bar = 20 nm.
DISCUSSION
In the present study, we examined the status of hippocampal tau-P in the presence or absence of CRFR1 antagonism in CRF-OE mice aged 6–7 month, a time point demonstrated to be prior to the onset of reported hippocampal memory deficits (9 months) [18]. Our data provide evidence of increased hippocampal levels of active tau kinases, increased tau-P and detectable aggregates of tau in detergent-soluble cellular fractions. We also demonstrate that administration of CRFR1 antagonist results in a reduction of tau-P to levels seen in WT controls and prevents the formation of observed globular tau aggregates. The aggregates we identified in CRF-OE mice are very similar in size and shape to that which we have seen in mice exposed to repeated stress (cf, [14]), and are similar to that seen in in vitro studies with truncated tau species (e.g., [27]). In addition to the possibility of CRF overexpression inducing tau truncation, given the granular appearance of our aggregates, these inclusions may be tau oligomers which have been reported in AD mice and in humans with AD [28–32]. Because our previously published [14] and current data appear to be the first to report aggregated structures of tau with purely WT murine tau, additional studies are needed to determine the precise nature of these inclusions. Our findings also suggest that although increased tau-P is present at several AD-relevant tau epitopes in CRF-OE mice, phosphorylation at S202/T205 and S396/404 sites via increased GSK 3β activity is regulated directly through CRF-CRFR1 signaling in the mouse hippocampus.
The role of stress and CRF in AD
Stress exposure and/or exposure to stress peptides can impact learning, memory, and hippocampal morphology and function in animals models [33–36]. Although CRF is best known as the hypothalamic neuropeptide that governs the endocrine stress response [37], its distribution and actions in the CNS suggest a broad involvement in integrating hormonal, autonomic, and behavioral adaptations [38–41]. Supporting the hypothesis of broader central involvement, studies have found prominent changes in CRF in AD, particularly in brain regions vulnerable to AD neuropathology [11, 42, 43]. In addition to CRF itself, considerable attention has been focused on effectors of the stress cascade, such as glucocorticoids, as mediators of neuronal vulnerability in AD. Increased circulating levels of these hormones in aging have been linked to brain atrophy and a range of adverse effects in the hippocampus [36, 44–46]. Stress disorders, such as depression, are very common in AD patients, which are thought to mechanistically involve alterations in the hypothalamic-pituitary-adrenal axis (reviewed in [47]). Whether stress and/or depression can be considered preclinical or prodromal risk factors for AD is still a matter of debate.
Relationship between tau-P in CRF-OE mice and stress-induced tau-P
The stress response can be initiated by the two primary categories of physical or emotional stressors. They are recognized and distinguished by their differential sensory modalities that register the challenges and patterns of neuronal activity they induce, and thereby mediate adaptive responses and the extent to which they invoke affective responses [48–50]. Regardless of the salience or strength of the stressors, they induce consistent increased tau-P expression in the hippocampus [11]. In terms of epitopes examined, our analysis of the published literature revealed no stark difference in the induction of tau-P between physical and emotional stressors. Acute challenges generally yielded readily reversible increases in tauP (range, minutes-hours) [12, 23, 51–55], while more chronic paradigms lead to longer lasting effects (days) [12, 56–59]. In particular, there is evidence of relatively widespread phosphorylation at various sites, with robust changes seen consistently at the S202/T205 (AT8) and S396/404 (PHF-1) phosphorylation sites [11, 12, 14, 60]. In the studies that examined them, few, if any, changes have been seen in the phosphorylatable amino acids between the PHF-1 site (S396/404) and S422 [11, 14, 61], except at S413 in the starvation model [58]. Likewise, with the exception of glucoprivation [57], small or no changes have been seen in the S262 site with stress exposure [11, 14] which is consistent with our findings in CRF-OE mice. In terms of kinases, nearly all studies implicate GSK-3β in the induction of tau-P (reviewed in [11]). We also observed increased phosphorylation of tyrosine 216 of GSK-3β in CRF-OE mice, however only JNK activity was sensitive to CRFR1 antagonism and resulted in ablation of phosphorylation increases at the AT8 and PHF-1 sites (Fig. 3). Interestingly, we observed relationships between JNK phosphorylation and tau phosphorylation at AT8 and PHF-1 in WT mice. R121919 treatment diminished tau-P responses at both AT8 and PHF-1 sites, presumably via JNK but yet under WT conditions, elevated JNK only resulted in observed increased basal levels of AT8. JNK phosphorylation and tau-P at AT8 sites may be due to acute stress exposure occurring during handling/sacrifice or be involved in tau-P seen in the white matter that we have observed. Inconsistent relationships between tau-P and upstream kinases have been reported previously [51], and the mediating entities and mechanisms underlying this selective regulation are unresolved and require further investigation.
Possible sites of CRF-CRFR1 interaction and the role of corticosterone
We previously reported that restraint stress-induced tau-P was dependent on signaling through CRFR1 [12]. To bypass the neuronal circuitries activated by various stressors, we examined whether chronic elevation in CRF occurring without stress exposure could result in tau-P and its dependence on CRFRs. Several aspects of our analysis warrant further consideration. First, CRF-OE mice treated with R121919, a selective CRFR1 antagonist that crosses the blood-brain barrier [19], displayed dramatic reduction in tau-P at two of four sites and in activating phosphorylation of one kinase. The role of CRFR1 as the primary signaling pathway involved in regulating hippocampal tau-P is further supported by the expression of CRFR1 as the dominant receptor subtype expressed in the mouse hippocampus [62] and by our previous findings showing that increased hippocampal tau-p induced by an acute stress is mediated by CRFR1 [12, 14]. CRFR2 actions, if relevant here, may occur outside the hippocampus because CRFR2 is expressed at low level in the mice hippocampus [55]. In the mouse hippocampus there is paucity of CRFR2 ligands in hippocampal afferents. However, extrinsic CRF inputs to hippocampus have not been described, leaving local interneurons and the CSF as possible sources of CRF to hippocampal CRFR1 [63, 64]. Second, we cannot fully parse out the role of chronically elevated corticosterone in this CRF-OE expressing mice [15]. CRFR1 activation is associated with initiation of the endocrine stress response and CRF-OE mice display high circulating levels of corticosterone [37, 38]. However as we allude to above, it is our hypothesis that stress-induced tau-P is a phenomenon associated with local CRF circuitry in the hippocampus. Our contention is supported by data from our lab and others demonstrating that stress-associated increases in glucocorticoids are not required for either physical or psychological stress-induced tau-P [12, 20]. Our preliminary studies exploring the impact of prolonged corticosterone also support these findings. We treated groups of WT mice subcutaneously with corticosterone (5–25 mg) for 21-days and observed no obvious increase in tau-P in the hippocampus compared to placebo-treated mice (R.A. Rissman, unpublished observations). Although requiring verification and extension, data thus far suggest that glucocorticoids may not be integral for tau-P responses.
Implications
Consistent reports indicate that exposure to a range of stressors induce reversible tau-P at many AD-relevant sites (reviewed in [11]). Some studies propose that stress-induced tau-P is a functional component of neuroplasticity, conferring adaptation to stressful stimuli [56, 59], while our group and others suggest that the relationship between stress and tau-P may also define a potential means by which chronic stress exposure may translate into neuropathology [12, 51, 54, 55, 57]. Our present and previous experimental studies demonstrating chronic elevations in tau-P with altered tau solubility under conditions of chronic CRF expression and repeated stress are supportive of this hypothesis, as well as clinical evidence with AD patients demonstrating high levels of distress proneness [65–67]. Collectively it therefore seems plausible that stress-induced tau-P is an essential process required for stress adaptation that becomes dysfunctional with advancing age and/or with chronic overstimulation. Although the phenomenology is certainly distinct, a similar case can be made for toxicity associated with Aβ. Aβ may be present throughout life, but with advancing age the ability to process the fragments may become reduced or lost, leading to accumulation. Although the mechanisms involved remain to be determined, our data suggest a possible underpinning for evidence relating chronic stress to memory impairment and tau pathology, which may facilitate the development of new therapeutic strategies for AD.
Acknowledgments
This work was supported by grants to RAR from NIA AG032755, the Alzheimer’s Art Quilt Initiative (AAQI), the Alzheimer’s Association and the Shiley-Marcos Alzheimer’s Disease Research Center at UCSD (AG005131). The authors thank Drs. P.E. Sawchenko (Salk Inst) and L. Wang (UCLA) for helpful discussions. We also thank Dr. P. Davies for antibody PHF-1. A portion of this work was also supported by the Intramural Research Programs of the National Institute on Drug Abuse and the National Institute on Alcohol Abuse and Alcoholism. YT is in receipt of the senior Career Scientist Award from Veteran Administration.
Footnotes
Authors’ disclosures available online (http://www.j-alz.com/disclosures/view.php?id=2440).
References
- 1.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 2.Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- 3.Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 4.Kenessey A, Yen SH. The extent of phosphorylation of fetal tau is comparable to that of PHF-tau from Alzheimer paired helical filaments. Brain Res. 1993;629:40–46. doi: 10.1016/0006-8993(93)90478-6. [DOI] [PubMed] [Google Scholar]
- 5.Kopke E, Tung YC, Shaikh S, Alonso AC, Iqbal K, Grundke-Iqbal I. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J Biol Chem. 1993;268:24374–24384. [PubMed] [Google Scholar]
- 6.Anderton BH, Betts J, Blackstock WP, Brion JP, Chapman S, Connell J, Dayanandan R, Gallo JM, Gibb G, Hanger DP, Hutton M, Kardalinou E, Leroy K, Lovestone S, Mack T, Reynolds CH, Van Slegtenhorst M. Sites of phosphorylation in tau and factors affecting their regulation. Biochem Soc Symp. 2001:73–80. doi: 10.1042/bss0670073. [DOI] [PubMed] [Google Scholar]
- 7.Derkinderen P, Scales TM, Hanger DP, Leung KY, Byers HL, Ward MA, Lenz C, Price C, Bird IN, Perera T, Kellie S, Williamson R, Noble W, Van Etten RA, Leroy K, Brion JP, Reynolds CH, Anderton BH. Tyrosine 394 is phosphorylated in Alzheimer’s paired helical filament tau and in fetal tau with c-Abl as the candidate tyrosine kinase. J Neurosci. 2005;25:6584–6593. doi: 10.1523/JNEUROSCI.1487-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gustke N, Steiner B, Mandelkow EM, Biernat J, Meyer HE, Goedert M, Mandelkow E. The Alzheimer-like phosphorylation of tau protein reduces microtubule binding and involves Ser-Pro and Thr-Pro motifs. FEBS Lett. 1992;307:199–205. doi: 10.1016/0014-5793(92)80767-b. [DOI] [PubMed] [Google Scholar]
- 9.Bramblett GT, Goedert M, Jakes R, Merrick SE, Trojanowski JQ, Lee VM. Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron. 1993;10:1089–1099. doi: 10.1016/0896-6273(93)90057-x. [DOI] [PubMed] [Google Scholar]
- 10.Alonso AC, Grundke-Iqbal I, Iqbal K. Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med. 1996;2:783–787. doi: 10.1038/nm0796-783. [DOI] [PubMed] [Google Scholar]
- 11.Rissman RA. Stress-induced tau phosphorylation: Functional neuroplasticity or neuronal vulnerability? J Alzheimers Dis. 2009;18:453–457. doi: 10.3233/JAD-2009-1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rissman RA, Lee KF, Vale W, Sawchenko PE. Corticotropin-releasing factor receptors differentially regulate stress-induced tau phosphorylation. J Neurosci. 2007;27:6552–6562. doi: 10.1523/JNEUROSCI.5173-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roe AD, Staup MA, Serrats J, Sawchenko PE, Rissman RA. Lipopolysaccharide-induced tau phosphorylation and kinase activity–modulation, but not mediation, by corticotropin-releasing factor receptors. Eur J Neurosci. 2011;34:448–456. doi: 10.1111/j.1460-9568.2011.07764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rissman RA, Staup MA, Lee AR, Justice NJ, Rice KC, Vale W, Sawchenko PE. Corticotropin-releasing factor receptor-dependent effects of repeated stress on tau phosphorylation, solubility, and aggregation. Proc Natl Acad Sci U S A. 2012;109:6277–6282. doi: 10.1073/pnas.1203140109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stenzel-Poore MP, Cameron VA, Vaughan J, Sawchenko PE, Vale W. Development of Cushing’s syndrome in corticotropin-releasing factor transgenic mice. Endocrinology. 1992;130:3378–3386. doi: 10.1210/endo.130.6.1597149. [DOI] [PubMed] [Google Scholar]
- 16.Stenzel-Poore MP, Heinrichs SC, Rivest S, Koob GF, Vale WW. Overproduction of corticotropin-releasing factor in transgenic mice: A genetic model of anxiogenic behavior. J Neurosci. 1994;14:2579–2584. doi: 10.1523/JNEUROSCI.14-05-02579.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goebel M, Fleming SM, Million M, Stengel A, Tache Y, Wang L. Mice overexpressing corticotropin-releasing factor show brain atrophy and motor dysfunctions. Neurosci Lett. 2010;473:11–15. doi: 10.1016/j.neulet.2010.01.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heinrichs SC, Stenzel-Poore MP, Gold LH, Battenberg E, Bloom FE, Koob GF, Vale WW, Pich EM. Learning impairment in transgenic mice with central overexpression of corticotropin-releasing factor. Neuroscience. 1996;74:303–311. doi: 10.1016/0306-4522(96)00140-6. [DOI] [PubMed] [Google Scholar]
- 19.Carroll JC, Iba M, Bangasser DA, Valentino RJ, James MJ, Brunden KR, Lee VM, Trojanowski JQ. Chronic stress exacerbates tau pathology, neurodegeneration, and cognitive performance through a corticotropin-releasing factor receptor-dependent mechanism in a transgenic mouse model of tauopathy. J Neurosci. 2011;31:14436–14449. doi: 10.1523/JNEUROSCI.3836-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korneyev A, Binder L, Bernardis J. Rapid reversible phosphorylation of rat brain tau proteins in response to cold water stress. Neurosci Lett. 1995;191:19–22. doi: 10.1016/0304-3940(95)11546-3. [DOI] [PubMed] [Google Scholar]
- 21.Chen C, Wilcoxen KM, Huang CQ, Xie YF, McCarthy JR, Webb TR, Zhu YF, Saunders J, Liu XJ, Chen TK, Bozigian H, Grigoriadis DE. Design of 2,5-dimethyl-3-(6-dim-ethyl-4-methylpyridin-3-yl)-7-dipropylaminopyrazolo[1,5-a ]py rimidine (NBI 30775/R121919) and structure–activity relationships of a series of potent and orally active corticotropin-releasing factor receptor antagonists. J Med Chem. 2004;47:4787–4798. doi: 10.1021/jm040058e. [DOI] [PubMed] [Google Scholar]
- 22.Stenzel-Poore MP, Heldwein KA, Stenzel P, Lee S, Vale WW. Characterization of the genomic corticotropin-releasing factor (CRF) gene from Zenopus laevis: Two members of the CRF family exist in amphibians. Mol Endocrinol. 1992;6:1716–1724. doi: 10.1210/mend.6.10.1448118. [DOI] [PubMed] [Google Scholar]
- 23.Planel E, Richter KE, Nolan CE, Finley JE, Liu L, Wen Y, Krishnamurthy P, Herman M, Wang L, Schachter JB, Nelson RB, Lau LF, Duff KE. Anesthesia leads to tau hyperphosphorylation through inhibition of phosphatase activity by hypothermia. J Neurosci. 2007;27:3090–3097. doi: 10.1523/JNEUROSCI.4854-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shu S, Ju G, Fan L. The glucose oxidase-DAB-nickel method in peroxidase histochemistry of the nervous system. Neurosci Lett. 1988;85:169–171. doi: 10.1016/0304-3940(88)90346-1. [DOI] [PubMed] [Google Scholar]
- 25.Zhang C, Rodriguez C, Spaulding J, Aw TY, Feng J. Age-dependent and tissue-related glutathione redox status in a mouse model of Alzheimer’s disease. J Alzheimers Dis. 2012;28:655–666. doi: 10.3233/JAD-2011-111244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rockenstein E, Mallory M, Mante M, Sisk A, Masliaha E. Early formation of mature amyloid-beta protein deposits in a mutant APP transgenic model depends on levels of Abeta(1-42) J Neurosci Res. 2001;66:573–582. doi: 10.1002/jnr.1247. [DOI] [PubMed] [Google Scholar]
- 27.King ME, Gamblin TC, Kuret J, Binder LI. Differential assembly of human tau isoforms in the presence of arachidonic acid. J Neurochem. 2000;74:1749–1757. doi: 10.1046/j.1471-4159.2000.0741749.x. [DOI] [PubMed] [Google Scholar]
- 28.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 29.Maeda S, Sahara N, Saito Y, Murayama M, Yoshiike Y, Kim H, Miyasaka T, Murayama S, Ikai A, Takashima A. Granular tau oligomers as intermediates of tau filaments. Biochemistry. 2007;46:3856–3861. doi: 10.1021/bi061359o. [DOI] [PubMed] [Google Scholar]
- 30.Maeda S, Sahara N, Saito Y, Murayama S, Ikai A, Takashima A. Increased levels of granular tau oligomers: An early sign of brain aging and Alzheimer’s disease. Neurosci Res. 2006;54:197–201. doi: 10.1016/j.neures.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 31.Sahara N, Maeda S, Murayama M, Suzuki T, Dohmae N, Yen SH, Takashima A. Assembly of two distinct dimers and higher-order oligomers from full-length tau. Eur J Neurosci. 2007;25:3020–3029. doi: 10.1111/j.1460-9568.2007.05555.x. [DOI] [PubMed] [Google Scholar]
- 32.Sahara N, Maeda S, Yoshiike Y, Mizoroki T, Yamashita S, Murayama M, Park JM, Saito Y, Murayama S, Takashima A. Molecular chaperone-mediated tau protein metabolism counteracts the formation of granular tau oligomers in human brain. J Neurosci Res. 2007;85:3098–3108. doi: 10.1002/jnr.21417. [DOI] [PubMed] [Google Scholar]
- 33.Sapolsky RM. Why stress is bad for your brain. Science. 1996;273:749–750. doi: 10.1126/science.273.5276.749. [DOI] [PubMed] [Google Scholar]
- 34.Sapolsky RM. Stress, glucocorticoids, and damage to the nervous system: The current state of confusion. Stress. 1996;1:1–19. doi: 10.3109/10253899609001092. [DOI] [PubMed] [Google Scholar]
- 35.McEwen BS, Sapolsky RM. Stress and cognitive function. Curr Opin Neurobiol. 1995;5:205–216. doi: 10.1016/0959-4388(95)80028-x. [DOI] [PubMed] [Google Scholar]
- 36.McEwen BS. Possible mechanisms for atrophy of the human hippocampus. Mol Psychiatry. 1997;2:255–262. doi: 10.1038/sj.mp.4000254. [DOI] [PubMed] [Google Scholar]
- 37.Vale W, Spiess J, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science. 1981;213:1394–1397. doi: 10.1126/science.6267699. [DOI] [PubMed] [Google Scholar]
- 38.Chadwick D, Marsh J, Ackrill K. Corticotropin-releasing factor. Wiley; Chichester, New York: 1993. [Google Scholar]
- 39.Turnbull AV, Rivier C. Corticotropin-releasing factor (CRF) and endocrine responses to stress: CRF receptors, binding protein, and related peptides. Proc Soc Exp Biol Med. 1997;215:1–10. doi: 10.3181/00379727-215-44108. [DOI] [PubMed] [Google Scholar]
- 40.Stengel A, Tache Y. Corticotropin-releasing factor signaling and visceral response to stress. Exp Biol Med (Maywood) 2010;235:1168–1178. doi: 10.1258/ebm.2010.009347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bale TL, Vale WW. CRF and CRF receptors: Role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol. 2004;44:525–557. doi: 10.1146/annurev.pharmtox.44.101802.121410. [DOI] [PubMed] [Google Scholar]
- 42.Rehman HU. Role of CRH in the pathogenesis of dementia of Alzheimer’s type and other dementias. Curr Opin Investig Drugs. 2002;3:1637–1642. [PubMed] [Google Scholar]
- 43.Davis KL, Mohs RC, Marin DB, Purohit DP, Perl DP, Lantz M, Austin G, Haroutunian V. Neuropeptide abnormalities in patients with early Alzheimer disease. Arch Gen Psychiatry. 1999;56:981–987. doi: 10.1001/archpsyc.56.11.981. [DOI] [PubMed] [Google Scholar]
- 44.Sapolsky RM, Krey LC, McEwen BS. Prolonged glucocorticoid exposure reduces hippocampal neuron number: Implications for aging. J Neurosci. 1985;5:1222–1227. doi: 10.1523/JNEUROSCI.05-05-01222.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sapolsky RM, Krey LC, McEwen BS. The neuroendocrinology of stress and aging: The glucocorticoid cascade hypothesis. Endocr Rev. 1986;7:284–301. doi: 10.1210/edrv-7-3-284. [DOI] [PubMed] [Google Scholar]
- 46.Bremner JD. Stress and brain atrophy. CNS Neurol Disord Drug Targets. 2006;5:503–512. doi: 10.2174/187152706778559309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chi S, Yu JT, Tan MS, Tan L. Depression in Alzheimer’s disease: Epidemiology, mechanisms, and management. J Alzheimers Dis. 2014 doi: 10.3233/JAD-140324. [DOI] [PubMed] [Google Scholar]
- 48.Herman JP, Cullinan WE. Neurocircuitry of stress: Central control of the hypothalamo-pituitary-adrenocortical axis. Trends Neurosci. 1997;20:78–84. doi: 10.1016/s0166-2236(96)10069-2. [DOI] [PubMed] [Google Scholar]
- 49.Dayas CV, Buller KM, Crane JW, Xu Y, Day TA. Stressor categorization: Acute physical and psychological stressors elicit distinctive recruitment patterns in the amygdala and in medullary noradrenergic cell groups. Eur J Neurosci. 2001;14:1143–1152. doi: 10.1046/j.0953-816x.2001.01733.x. [DOI] [PubMed] [Google Scholar]
- 50.Sawchenko PE, Brown ER, Chan RK, Ericsson A, Li HY, Roland BL, Kovacs KJ. The paraventricular nucleus of the hypothalamus and the functional neuroanatomy of visceromotor responses to stress. Prog Brain Res. 1996;107:201–222. doi: 10.1016/s0079-6123(08)61866-x. [DOI] [PubMed] [Google Scholar]
- 51.Ikeda Y, Ishiguro K, Fujita SC. Ether stress-induced Alzheimer-like tau phosphorylation in the normal mouse brain. FEBS Lett. 2007;581:891–897. doi: 10.1016/j.febslet.2007.01.064. [DOI] [PubMed] [Google Scholar]
- 52.Yoshida S, Maeda M, Kaku S, Ikeya H, Yamada K, Nakaike S. Lithium inhibits stress-induced changes in tau phosphorylation in the mouse hippocampus. J Neural Transm. 2006;113:1803–1814. doi: 10.1007/s00702-006-0528-0. [DOI] [PubMed] [Google Scholar]
- 53.Planel E, Yasutake K, Fujita SC, Ishiguro K. Inhibition of protein phosphatase 2A overrides tau protein kinase I/glycogen synthase kinase 3 beta and cyclin-dependent kinase 5 inhibition and results in tau hyperphosphorylation in the hippocampus of starved mouse. J Biol Chem. 2001;276:34298–34306. doi: 10.1074/jbc.M102780200. [DOI] [PubMed] [Google Scholar]
- 54.Feng Q, Cheng B, Yang R, Sun FY, Zhu CQ. Dynamic changes of phosphorylated tau in mouse hippocampus after cold water stress. Neurosci Lett. 2005;388:13–16. doi: 10.1016/j.neulet.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 55.Okawa Y, Ishiguro K, Fujita SC. Stress-induced hyperphosphorylation of tau in the mouse brain. FEBS Lett. 2003;535:183–189. doi: 10.1016/s0014-5793(02)03883-8. [DOI] [PubMed] [Google Scholar]
- 56.Arendt T, Stieler J, Strijkstra AM, Hut RA, Rudiger J, Van der Zee EA, Harkany T, Holzer M, Hartig W. Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J Neurosci. 2003;23:6972–6981. doi: 10.1523/JNEUROSCI.23-18-06972.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Planel E, Miyasaka T, Launey T, Chui DH, Tanemura K, Sato S, Murayama O, Ishiguro K, Tatebayashi Y, Takashima A. Alterations in glucose metabolism induce hypothermia leading to tau hyperphosphorylation through differential inhibition of kinase and phosphatase activities: Implications for Alzheimer’s disease. J Neurosci. 2004;24:2401–2411. doi: 10.1523/JNEUROSCI.5561-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yanagisawa M, Planel E, Ishiguro K, Fujita SC. Starvation induces tau hyperphosphorylation in mouse brain: Implications for Alzheimer’s disease. FEBS Lett. 1999;461:329–333. doi: 10.1016/s0014-5793(99)01480-5. [DOI] [PubMed] [Google Scholar]
- 59.Hartig W, Stieler J, Boerema AS, Wolf J, Schmidt U, Weissfuss J, Bullmann T, Strijkstra AM, Arendt T. Hibernation model of tau phosphorylation in hamsters: Selective vulnerability of cholinergic basal forebrain neurons -implications for Alzheimer’s disease. Eur J Neurosci. 2007;25:69–80. doi: 10.1111/j.1460-9568.2006.05250.x. [DOI] [PubMed] [Google Scholar]
- 60.Clippinger AK, D’Alton S, Lin WL, Gendron TF, Howard J, Borchelt DR, Cannon A, Carlomagno Y, Chakrabarty P, Cook C, Golde TE, Levites Y, Ranum L, Schultheis PJ, Xu G, Petrucelli L, Sahara N, Dickson DW, Giasson B, Lewis J. Robust cytoplasmic accumulation of phosphorylated TDP-43 in transgenic models of tauopathy. Acta Neuropathol. 2013;126:39–50. doi: 10.1007/s00401-013-1123-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ko LW, DeTure M, Sahara N, Chihab R, Vega IE, Yen SH. Recent advances in experimental modeling of the assembly of tau filaments. Biochim Biophys Acta. 2005;1739:125–139. doi: 10.1016/j.bbadis.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 62.Van Pett K, Viau V, Bittencourt JC, Chan RKW, Li H-Y, Arias C, Prins GS, Perrin M, Vale W, Sawchenko PE. Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J Comp Neurol. 2000;428:191–212. doi: 10.1002/1096-9861(20001211)428:2<191::aid-cne1>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 63.Arborelius L, Owens MJ, Plotsky PM, Nemeroff CB. The role of corticotropin-releasing factor in depression and anxiety disorders. J Endocrinol. 1999;160:1–12. doi: 10.1677/joe.0.1600001. [DOI] [PubMed] [Google Scholar]
- 64.Chen Y, Brunson KL, Adelmann G, Bender RA, Frotscher M, Baram TZ. Hippocampal corticotropin releasing hormone: Pre- and postsynaptic location and release by stress. Neuroscience. 2004;126:533–540. doi: 10.1016/j.neuroscience.2004.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wilson RS, Schneider JA, Boyle PA, Arnold SE, Tang Y, Bennett DA. Chronic distress and incidence of mild cognitive impairment. Neurology. 2007;68:2085–2092. doi: 10.1212/01.wnl.0000264930.97061.82. [DOI] [PubMed] [Google Scholar]
- 66.Wilson RS, Bennett DA, Mendes de Leon CF, Bienias JL, Morris MC, Evans DA. Distress proneness and cognitive decline in a population of older persons. Psychoneuroendocrinology. 2005;30:11–17. doi: 10.1016/j.psyneuen.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 67.Wilson RS, Arnold SE, Schneider JA, Kelly JF, Tang Y, Bennett DA. Chronic psychological distress and risk of Alzheimer’s disease in old age. Neuroepidemiology. 2006;27:143–153. doi: 10.1159/000095761. [DOI] [PubMed] [Google Scholar]