

Abstract
Quantum dots were used as fluorescent probes to investigate nanoparticle penetration into biofilms. The particle penetration behavior was found to be controlled by surface chemical properties.
Bacterial biofilm formation plays an important role in many persistent diseases1 and medical device-associated infections.2 These infections are particular challenging, as biofilm bacteria are both embedded in and protected by the sticky and strong framework generated by extracellular polymeric substances (EPS).3 The EPS matrix possesses a complex composition, architecture, and dynamic function, all of which are believed to result in high resistance to antibiotics. 4 Creating an efficient treatment to biofilm-associated infections requires a fundamental understanding of how materials penetrate the complex milieu presented by biofilms.
Much of the protection provided by EPS comes from barrier characteristics, which can profoundly impede the penetration of antibiotics.5 Moreover, the EPS matrix is capable of deactivating antibiotics in the surface layers more rapidly than they diffuse, causing limited penetration.6 The failure of antibiotics to penetrate the full depth of biofilms is one mechanism behind the biofilm resistance.7 In recent studies, the surface functionality of NPs has been used to control their interactions with biomolecules and cells.8 Engineered nanoparticles (NPs) possess the ability to permeate into cells, 9 tumors, 10 and even the blood-brain barrier,11 presenting a potentially powerful vehicle to infiltrate the biofilm EPS barrier.12
We report here the use of quantum dots (QDs) to determine the role of particle surface properties in dictating biofilm penetration. QDs functionalized with different charges and head functional groups were used to systematically investigate the role of surface chemistry in QD penetration and distribution inside biofilms using fluorescence microscopy. The results show that neutral and anionic QDs cannot penetrate or accumulate efficiently into biofilms. In contrast, cationic particles readily penetrate fully into biofilms. With these cationic QDs we observed that hydrophobic particles are more homogeneously distributed through the biofilm than hydrophilic analogs. Our studies demonstrate that control of surface functionality on the NP surface provides an effective approach to predict the NP behavior in biofilms (Figure 1).
Fig. 1.
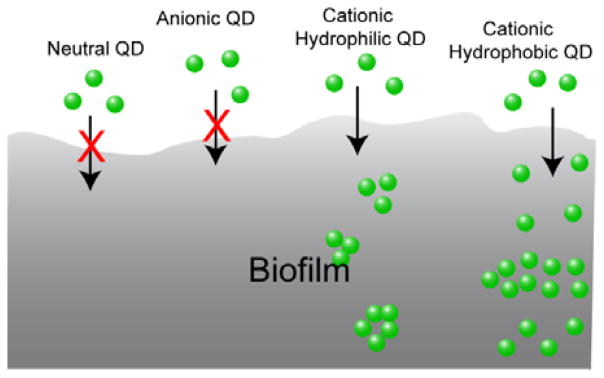
Schematic illustration of surface functionality-controlled QD penetration into biofilms.
Green fluorescent CdSe/ZnS core-shell QDs (em 535 nm) were used to prepare water soluble QDs through a two-step ligand exchange process (See ESI for QD preparation).13,14 Dithiolate ligands15 presenting different functional head groups were synthesized for QD surface functionalization (Fig. 2a). The ligand design features a dihydrolipoic acid (DHLA) bidentate anchor, an oligo(ethylene glycol) (OEG) spacer and a tunable functional head group.16 The terminal functionalities of the ligands were designed with different surface charges (i.e., neutral, anionic, and cationic). Additionally, two types of cationic ligands were synthesized to impart differing hydrophobicity to the cationic head groups. As shown in Figure 2, the neutral QDs (PEG-QDs) were prepared by using the poly(ethelene glycol)-appended DHLA ligands, and the charged QDs were synthesized with different functionalities including carboxyl terminus (COOH-QD), trimethyl ammonium terminus (TTMA-QD), and dimethylhexyl ammonium terminus (Hexyl-QD). The absorption peak positions of these QDs were very similar while the emission peaks showed modest differences, as is commonly observed after surface modification.17,18,19 Dynamic light scattering data indicated that QDs had comparable sizes, ranging from 7.5 to 11.7 nm, with PEG-QD having slightly larger hydrodynamic size of 24 nm, presumably due to modest level of aggregation (see ESI for QD physicochemical properties).
Fig. 2.

Representative 3D projection of image z-stacks showing distribution of bacterial cells (red) in E. coli biofilms and QDs (green): a), PEG-QD; b), COOH-QD; c), TTMA-QD; d), Hexyl-QD. Upper panels are projections at 247° angle turning along Y axis and lower panels are at 270° angle turning along Y axis. Scale bar is 20 μm. e) and f) are plot profiles of the three linear selections (yellow lines) in c) and d), illustrating horizontal distribution of e) TTMA-QD and f) Hexyl-QD.
We chose E. coli strain DH53 that expresses E2-crimson, a bright far-red fluorescent protein (emission peak at 646 nm), as a model strain for our study. The biofilms were cultured onto coverslips in a 1/10 strength LB broth supplemented with 100 μM IPTG (isopropyl 3-D-1-thiogalactopyranoside) according to a reported protocol20 (see ESI for culture conditions) and were 7–8 mm in thickness. The penetration experiments were performed with 1 h incubation of QDs with three-day old biofilms followed by washing with PBS. The cover slips were then mounted on glass slides using 4% anti-fade mounting solution and examined by confocal laser scanning microscopy (CLSM).
The CLSM images were obtained using two different fluorescent channels to simultaneously detect bacterial cells (red fluorescence) and QDs (green fluorescence). Figure 2 shows the 3D projection of images from a single z-stack to illustrate the overall contribution of QDs to the biofilms. In the case of PEG-QD and COOH-QD, there was no green fluorescence observed either on the surface or inside the biofilms (Figure 2a,b), indicating no QD adhesion or penetration during the 1 h incubation (see Figure S1 and S2 for all the images). The absence of neutral and negative QDs on the surface of biofilms was likely due to weak adsorption of QDs that was disrupted during the washing step. In contrast, strong green fluorescence was observed throughout the film from the cationic QDs TTMA-QD and Hexyl-QD (Figure 2c,d). This finding is surprising, since it is reasonable to expect the cationic QDs interact with and stick strongly to the negatively charged biofilm EPS, and remaining in the top layers instead of penetrating. This behavior is, however, in agreement with recent reports where cationic NPs were shown to have a better penetration into negative matrix than anionic NPs.21,22,23 The different penetration patterns of these two cationic QDs are indicated by the projected sample images at 270° angle (turning along the Y axis, Figure 2c–d, lower panel). These images show that TTMA-QD accumulated preferentially near the bottom of the biofilms while Hexyl-QD was more concentrated in the middle of the film.
To quantify the penetration profiles of the two cationic QDs, three stacks of CLSM images taken at random locations were analyzed using ImageJ (see Figure S3–S6 for all stacks of images). For each slice in one image stack, the green and red channels were separated and then analyzed for integrated intensity within a fixed 512×512 frame. The integrated intensity from the green and red channel hence represented the intensity of QDs and bacteria in the biofilms, respectively. The biofilm 3D architecture is inherently heterogeneous, causing the biofilm mass distribution to vary among the three image stacks. The integrated intensity from both channels was therefore plotted versus the biofilm depth. Both TTMA-QD and Hexyl-QD were present at 7.2 μm biofilm depth, corresponding to the bottom (coverslip) surface of the film (Figure 3). The distribution of the two particles, however, was quite different. TTMA-QD was relatively evenly dispersed along the vertical direction of biofilms at both 1 and 3h, with the highest intensity at 7.2 μm. Hexyl-QD, exhibited a different pattern in which the intensity reached a peak value around 4.4 μm and then decreased. This same pattern was observed after 3h as well, suggesting that a steady state had been reached for this QD as well.
Fig. 3.
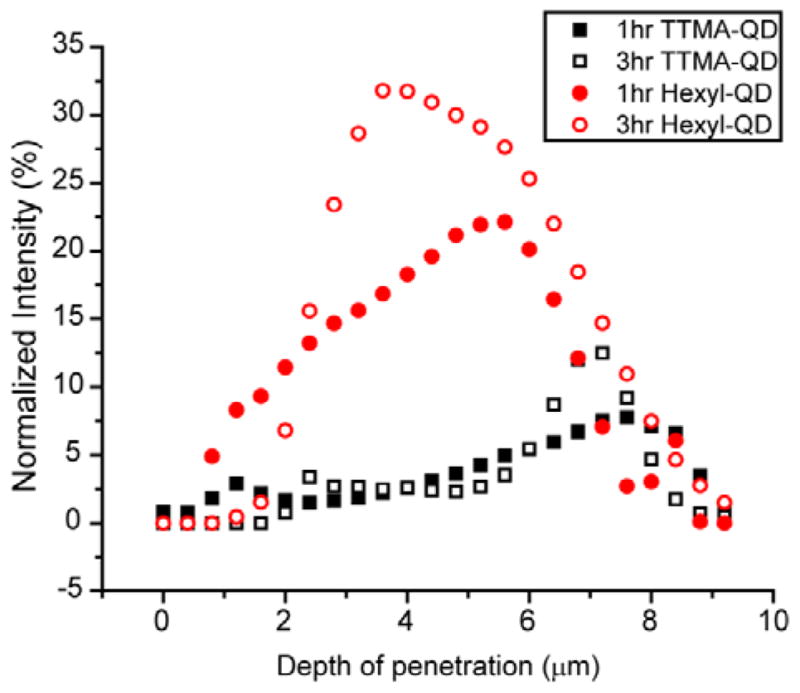
Integrated intensity of TTMA-QD and Hexyl-QD and biofilm after 1h and 3h hr incubation. The y-axis, normalized intensity, is the integrated QD intensity normalized by the integrated biofilm intensity. The x-axis is the depth of penetration of biofilms, where 0 μm represents the top and ~7.2 μm represents the bottom. The data are average of three image stacks.
In addition to regulating penetration depth, hydrophobicity markedly affects the localization of the cationic QDs. Overlay studies (Figure 4) show that TTMA-QD were not co-localized with the bacteria in the biofilms (Figure 4a), and were instead found exclusively in the EPS. In contrast, Hexyl-QDs co-localized with the red fluorescence from the bacterial cells (Figure 4b). The different distributions of the two QDs can be explained thermodynamically and kinetically. First, different partitioning affinities of the QD head groups to biofilm components, e.g. selective binding TTMA-QD to EPS results in extracellular localization. Dynamically, changes in NP surface functionality could lead to differing uptake rates, with Hexyl-QD more efficiently taken up by the bacteria.10 This observation suggests that rational design of NP surface can facilitate selective targeting of biofilm components, for example, targeting the EPS matrix for delivery of enzymes to induce dispersion of biofilms; or delivery into bacterial cells for delivery of antibiotics.
Fig. 4.

CLSM images of biofilms after 1hr incubation with QDs, showing association sites of (a) TTMA-QD (extracellular) and (b) Hexyl-QD (intracellular).
We next investigated the cytotoxicity profiles of these engineered QDs against human airway smooth muscle (ASM) cells (see ESI for detailed method). At the concentrations (300 nM) used for biofilm penetration studies, both TTMA-QD and Hexyl-QD showed minimal cytotoxicity on three different ASM cell lines after 24-hour incubation (Figure S7, S8, and S9). These results revealed the low cytotoxicity properties of these engineered QDs and their potential as a biofilm penetrating agent for in vivo applications.
In summary, we have employed functionalized QDs as model nanoparticles to demonstrate that the engineering of surface properties can be used to direct the penetration and distribution of nanomaterials into biofilms. This strategy presents new opportunities for using nanomaterials with different core materials to fight biofilm-based infections through both delivery of therapeutics and by creation of self-therapeutic systems. Both routes offer a potential approach for a low-stress, surgery-free, and efficient treatment of biofilm-associated infections.
Supplementary Material
Acknowledgments
VR acknowledges the NIH (EB014277) and VR and YP acknowledge support from the HIPFA program at the Mayo Clinic.
Footnotes
Electronic Supplementary Information (ESI) available: [QD synthesis; Biofilm cultures; CLSM image stacks; Mammlican cell toxicity]. See DOI: 10.1039/b000000x/
Notes and references
- 1.(a) Hall-Stoodley L, Costerton JW, Stoodley P. Nat Rev Microbiol. 2004;2:95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]; (b) Donlan RM. Emerg Infect Dis. 2002;8:881–890. doi: 10.3201/eid0809.020063. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) James GA, Swogger E, Wolcott R, Pulcini ED, Secor P, Sestrich J, Costerton JW, Stewart PS. Wound Repair Regen. 2008;16:37–44. doi: 10.1111/j.1524-475X.2007.00321.x. [DOI] [PubMed] [Google Scholar]
- 2.(a) Costerton JW, Stewart PS, Greenberg EP. Science. 1999;284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]; (b) Donlan M. Emerg Infect Dis. 2001;7:277–281. doi: 10.3201/eid0702.010226. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) van de Belt H, Neut D, Schenk W, van Horn JR, van der Mei HC, Busscher HJ. Acta Orthop Scand. 2001;72:557–571. doi: 10.1080/000164701317268978. [DOI] [PubMed] [Google Scholar]
- 3.(a) Flemming H-C, Neu TR, Wozniak DJ. J Bacteriol. 2007;189:7945–7947. doi: 10.1128/JB.00858-07. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Flemming H-C, Wingender J. Nature Rev Microbiol. 2010;8:623–633. doi: 10.1038/nrmicro2415. [DOI] [PubMed] [Google Scholar]
- 4.(a) Romling U, Balsalobre C. J Intern Med. 2012;272:541–561. doi: 10.1111/joim.12004. [DOI] [PubMed] [Google Scholar]; (b) Oubekka SD, Briandet R, Fontaine-Aupart M-P, Steenkeste K. Antimicrob Agents Chemother. 2012;56:3349–3358. doi: 10.1128/AAC.00216-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Kumon H, Tomochika K, Matunaga T, Ogawa M, Ohmori H. Microbiol Immunol. 1994;38:615–619. doi: 10.1111/j.1348-0421.1994.tb01831.x. [DOI] [PubMed] [Google Scholar]; (b) Shigeta M, Tanaka G, Komatsuzawa H, Sugai M, Suginaka H, Usui T. Chemotherapy. 1997;43:340–345. doi: 10.1159/000239587. [DOI] [PubMed] [Google Scholar]
- 6.Anderl JN, Franklin MJ, Stewart PS. Antimicrob Agents Chemother. 2000;44:1818–1824. doi: 10.1128/aac.44.7.1818-1824.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stewart PS, Costerton JW. Lancet. 2001;358:135–138. doi: 10.1016/s0140-6736(01)05321-1. [DOI] [PubMed] [Google Scholar]
- 8.(a) Verma A, Uzun O, Hu YH, Hu Y, Han HS, Watson N, Chen SL, Irvine DJ, Stellacci F. Nat Mater. 2008;7:588–595. doi: 10.1038/nmat2202. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Saha K, Bajaj A, Duncan B, Rotello VM. Small. 2011;7:1903–1918. doi: 10.1002/smll.201100478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slowing, Vivero-Escoto JL, Wu CW, Lin VSY. Adv Drug Delivery Rev. 2008;60:1278–1288. doi: 10.1016/j.addr.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 10.Kim B, Han G, Toley BJ, Kim CK, Rotello VM, Forbes NS. Nat Nanotech. 2010;5:465–472. doi: 10.1038/nnano.2010.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lockman PR, Mumper RJ, Khan MA, Allen DD. Drug Dev Ind Pharm. 2002;28:1–13. doi: 10.1081/ddc-120001481. [DOI] [PubMed] [Google Scholar]
- 12.(a) Morrow JB, Arango C, Holbrook RD. J Environ Qual. 2010;39:1934–1941. doi: 10.2134/jeq2009.0455. [DOI] [PubMed] [Google Scholar]; (b) Chalmers NI, Palmer RJ, Jr, Du-Thumm L, Sullivan R, Shi W, Kolenbrander PE. Appl Environ Microbiol. 2007;73:630–636. doi: 10.1128/AEM.02164-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Aldeek F, Mustin C, Balan L, Roques-Carmes T, Fontaine-Aupart M-P, Schneider R. Biomaterials. 2011;32:5459–5470. doi: 10.1016/j.biomaterials.2011.04.019. [DOI] [PubMed] [Google Scholar]; (d) Aldeek F, Balan L, Medjahdi G, Roques-Carmes T, Malval JP, Mustin C, Ghanbaja J, Schneider R. J Phys Chem C. 2009;113:19458–19467. [Google Scholar]; (e) Aldeek F, Mustin C, Balan L, Medjahdi G, Roques-Carmes T, Arnoux P, Schneider R. Eur J Inorg Chem. 2011;6:794–801. [Google Scholar]; (f) Aldeek F, Schneider R, Fontaine-Aupart M-P, Mustin C, Lécart S, Merlin C, Block J-C. Appl Environ Microbiol. 2013;79:1400–1402. doi: 10.1128/AEM.03054-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeh YC, Patra D, Yan B, Saha K, Miranda OR, Kim CK, Rotello VM. Chem Commun. 2011;47:3069–3071. doi: 10.1039/c0cc04975a. [DOI] [PubMed] [Google Scholar]
- 14.Susumu K, Mei BC, Mattoussi H. Nat Protocols. 2009;4:424–436. doi: 10.1038/nprot.2008.247. [DOI] [PubMed] [Google Scholar]
- 15.Susumu K, Uyeda HT, Medintz IL, Pons T, Delehanty JB, Mattoussi H. J Am Chem Soc. 2007;129:13987–13996. doi: 10.1021/ja0749744. [DOI] [PubMed] [Google Scholar]
- 16.Moyano DF, Rotello VM. Langmuir. 2011;27:10376–10385. doi: 10.1021/la2004535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thuy UTD, Liem NQ, Thanh DX, Protière M, Reiss P. Appl Phys Lett. 2008;91:241908–241911. [Google Scholar]
- 18.Leatherdale CA, Bawendi MG. Phys Rev B. 2001;63:165315. [Google Scholar]
- 19.Jin T, Fujii F, Yamada E, Nodasaka Y, Kinjo M. J Am Chem Soc. 2006;128:9288–9289. doi: 10.1021/ja062021k. [DOI] [PubMed] [Google Scholar]
- 20.Merritt JH, Kadouri DE, O’Toole GA. Curr Protoc Microbiol. 2005;1B:1.1. doi: 10.1002/9780471729259.mc01b01s00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin JQ, Zhang HW, Chen Z, Zheng YG. Acs Nano. 2010;4:5421–5429. doi: 10.1021/nn1010792. [DOI] [PubMed] [Google Scholar]
- 22.Ryman-Rasmussen JP, Riviere JE, Monteiro-Riviere NA. Toxicol Sci. 2006;91:159–165. doi: 10.1093/toxsci/kfj122. [DOI] [PubMed] [Google Scholar]
- 23.Briandet R, Lacroix-Gueu P, Renault M, Lecart S, Meylheuc T, Bidnenko E, Steenkeste K, Bellon-Fontaine M-N, Fontaine-Aupart M-P. Appl Environ Microbiol. 2008;74:2135–2143. doi: 10.1128/AEM.02304-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.