

Abstract
Aims/hypothesis
T cell protein tyrosine phosphatase (TCPTP, encoded by PTPN2) regulates cytokine-induced pancreatic beta cell apoptosis and may contribute to the pathogenesis of type 1 diabetes. However, the role of TCPTP in pancreatic endocrine function and insulin secretion remains largely unknown.
Methods
To investigate the endocrine role of pancreatic TCPTP we generated mice with pancreas Ptpn2/TCPTP deletion (panc-TCPTP KO).
Results
When fed regular chow, panc-TCPTP KO and control mice exhibited comparable glucose tolerance. However, when challenged with prolonged high fat feeding panc-TCPTP KO mice exhibited impaired glucose tolerance and attenuated glucose-stimulated insulin secretion (GSIS). The defect in GSIS was recapitulated in primary islets ex vivo and after TCPTP pharmacological inhibition or lentiviral-mediated TCPTP knockdown in the glucose-responsive MIN6 beta cells, consistent with this being cell autonomous. Reconstitution of TCPTP in knockdown cells reversed the defect in GSIS demonstrating that the defect was a direct consequence of TCPTP deficiency. The reduced insulin secretion in TCPTP knockdown MIN6 beta cells was associated with decreased insulin content and glucose sensing. Furthermore, TCPTP deficiency led to enhanced tyrosyl phosphorylation of signal transducer and activator of transcription 1 and 3 (STAT 1/3), and substrate trapping studies in MIN6 beta cells identified STAT 1/3 as TCPTP substrates. STAT3 pharmacological inhibition and small interfering RNA-mediated STAT3 knockdown in TCPTP deficient cells restored GSIS to control levels, indicating that the effects of TCPTP deficiency were mediated, at least in part, through enhanced STAT3 phosphorylation and signalling.
Conclusions/interpretation
These studies identify a novel role for TCPTP in insulin secretion and uncover STAT3 as a physiologically relevant target for TCPTP in the endocrine pancreas.
Keywords: Insulin secretion, Ptpn2, STAT1, STAT3, Tcell protein–tyrosine phosphatase, Type 2 diabetes
Introduction
Type 2 diabetes is one of the most prevalent metabolic diseases and is characterised by hyperinsulinaemia, insulin resistance and pancreatic beta cell dysfunction [1]. Beta cells dynamically respond to fluctuations in blood glucose levels by the regulated secretion of insulin to modulate glucose homeostasis [2]. Tyrosyl phosphorylation is an important regulator of insulin secretion and action, and is tightly controlled by the opposing actions of protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs). Pancreas-specific deletion of receptor PTKs (RTKs) implicated the insulin receptor (IR) [3] and insulin growth factor 1 receptor [4] in beta cell function in mice. However, the role of PTPs in beta cell function remains largely unexplored.
T cell protein tyrosine phosphatase (TCPTP; encoded by PTPN2) is a ubiquitously expressed PTP [5]. TCPTP has two splice variants: a 48 kDa variant, which is anchored to the endoplasmic reticulum (ER), and a 45 kDa variant that has access to cytosolic and nuclear substrates [6]. TCPTP has several substrates that include: (1) RTKs such as the IR [7] and epidermal growth factor receptor [6]; (2) non-receptor PTKs such as c-Src [8] and Janus family kinases 1/3 [9]; (3) substrates of RTKs such as signal transducer and activator of transcription (STAT) 1, 3, 5 and 6 [10–13]. Accordingly, TCPTP has the capacity to modulate diverse signalling pathways.
Insights into the physiological functions of TCPTP have emerged from studies in mice with global TCPTP deficiency. TCPTP whole-body knockout (KO) mice exhibit haematopoietic defects and progressive systemic inflammatory disease, and die after birth thus hampering detailed assessment of their metabolic phenotype [14]. More recently, tissue-specific TCPTP deletion has helped define the cell type and tissue-specific functions of this phosphatase. T cell-specific TCPTP deficiency establishes a role for this phosphatase in attenuating T cell signalling [15]. While muscle-specific TCPTP deficiency does not alter insulin signalling and glucose homeostasis [16], brain-specific TCPTP deficiency leads to enhanced leptin sensitivity and resistance to high fat diet (HFD)-induced weight gain [17]. Moreover, liver-specific TCPTP deficiency promotes hepatosteatosis, obesity and insulin resistance [18]. The function of TCPTP in the pancreas remains largely unresolved, but a growing body of evidence suggests a role for this phosphatase in beta cell function. Genome-wide association studies identified PTPN2 as a susceptibility gene in the pathogenesis of type 1 diabetes [19]. In addition, TCPTP regulates cytokine-induced beta cell apoptosis [19, 20]. Notably, progressive erosion of beta cell function is observed prior to the full manifestation of type 1 diabetes [21], and TCPTP could modulate insulin secretion during pathogenesis of the disease. Previously, we demonstrated that TCPTP modulates ER stress in MIN6 beta cells and that alterations in pancreatic TCPTP expression may serve to mitigate chronic ER stress [22]. More recently, we reported that pancreatic TCPTP deficiency mitigates cerulein-induced acute pancreatitis in mice [23]. In the current study, we investigated the endocrine role of pancreatic TCPTP and its underlying molecular mechanism.
Methods
Mouse studies
Details of mouse studies are provided in the electronic supplementary material (ESM) Methods. Briefly, metabolic variables, insulin tolerance tests (ITTs), glucose tolerance tests (GTTs) and glucose-stimulated insulin secretion (GSIS) tests were performed as previously described [24]. All mouse studies were conducted according to federal guidelines and were approved by the Institutional Animal Care and Use Committee at University of California, Davis, CA, USA.
Cellular and biochemical studies
MIN6 beta cell culture, knockdown and biochemical studies, pancreas immunostaining and morphometric analysis of islets are described in detail in the ESM Methods. Pharmacological inhibition of TCPTP and STAT3 was achieved by treating MIN6 beta cells with TCPTP inhibitor compound 8 (provided by Z-Y Zhang; 50 nmol/l) [25] and STAT3 inhibitor BP-1-102 (10 μmol/l; Millipore, Temecula, CA, USA) [26], respectively. TCPTP enzymatic activity was determined as described in the ESM Methods. SYBR Green (BioRad, Hercules, CA, USA) quantitative real time PCR was used to determine mRNA of CaCna1d, Gk, Glut2 (also known as Slc2a2), Ins1, Ins2, Kir6.2 (also known as Kcnj11), Pdx1 and Ptpn2 using appropriate primers (ESM Table 1).
Statistical analyses
Data are expressed as means±SEM. Repeated measures ANOVA was used to analyse multiple comparisons. Single data point comparisons were performed using Tukey–Kramer honest significant difference analyses; both analyses were performed using JMP program (SAS Institute, Cary, NC, USA). Differences were considered significant at p<0.05 and highly significant at p<0.01.
Results
Generation of pancreatic TCPTP KO mice
To determine pancreatic TCPTP endocrine function we generated mice with pancreas Ptpn2 deletion by crossing Ptpn2fl/fl (TCPTPfl/fl) mice to transgenic mice expressing Cre recombinase under the control of pancreatic and duodenal homeobox 1 (Pdx1) promoter. Pancreatic Ptpn2/TCPTP KO (panc-TCPTP KO) mice survived to adulthood and did not display gross defects in the pancreas. Immunoblot analysis of primary islets lysates revealed loss of TCPTP protein expression in panc-TCPTP KO compared with control mice (Fig. 1a). Similarly, immunoblotting of total pancreas lysates demonstrated significant reduction in TCPTP protein in panc-TCPTP KO compared with control mice (Fig. 1a), consistent with TCPTP deletion occurring in islet and non-islet cells. In addition, expression of protein tyrosine phosphatase 1B (PTP1B) and Src homology phosphatase 2 (SHP2) was not altered upon TCPTP deletion. Importantly, TCPTP expression was not changed in liver, muscle, adipose tissue or hypothalamus (Fig. 1b). Immunostaining of pancreas sections from control mice revealed abundant TCPTP expression in the pancreas, which was predominantly nuclear, suggesting that the major murine isoform is the 45 kDa variant (Fig. 1c). Consistent with immunoblotting data, TCPTP staining was reduced in panc-TCPTP KO islets confirming its ablation in these mice. Together, these data demonstrate efficient TCPTP deletion in pancreatic islets enabling the determination of its metabolic function in the endocrine pancreas.
Fig. 1.
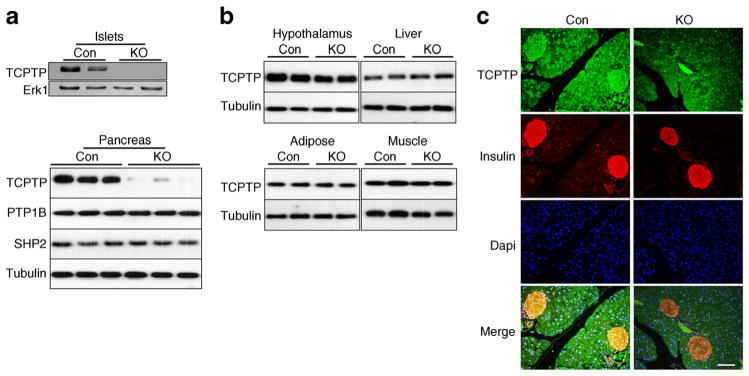
Pancreatic Ptpn2 (TCPTP) deletion. (a) Islets lysates from TCPTPfl/fl (Con) and panc-TCPTP KO mice immunoblotted for TCPTP and Erk1. Representative immunoblots are shown, n=3. Whole pancreas lysates from Con and KO mice immunoblotted for TCPTP, PTP1B, SHP2 and tubulin. (b) Representative immunoblots of TCPTP and tubulin in lysates of hypothalamus, liver, adipose and muscle of Con and KO mice, n =4. (c) Pancreases of Con and KO female mice fed HFD for 36 weeks were sectioned and immunostained for TCPTP (green), insulin (red) and DAPI (blue). Scale bar 100 μm
Prolonged high fat feeding impairs glucose tolerance in panc-TCPTP KO mice
To investigate the endocrine effects of pancreatic TCPTP deletion we assessed metabolic alterations in control and panc-TCPTP KO mice fed a regular chow diet and HFD. Immunoblotting of pancreas lysates revealed decreased TCPTP expression in high fat-fed mice (ESM Fig. 1), consistent with previous studies [22]. Body weights were determined and, as expected, male mice fed HFD gained significantly more weight than their chow-fed counterparts, with control and panc-TCPTP KO mice exhibiting comparable body weights on both diets (Fig. 2a). Fed and fasted glucose concentrations were comparable between panc-TCPTP KO and control mice on either diet (ESM Fig. 2a). In addition, fed glucagon levels were comparable between groups, while fasted concentrations were lower in HFD-fed panc-TCPTP KO mice compared with chow-fed mice (ESM Fig. 2b). To directly assess the effect of pancreatic TCPTP deletion on glucose homeostasis, we determined the ability of mice to dispose of a glucose load by conducting GTTs at different ages. Control and panc-TCPTP KO mice exhibited comparable glucose tolerance when fed regular chow (Fig. 2b, c). However, when fed HFD panc-TCPTP KO mice had a reduced ability to dispose of glucose load compared with controls. This was more pronounced when mice were older and fed HFD for prolonged periods. To determine whether or not differences in peripheral insulin sensitivity contribute to the glucose intolerance of panc-TCPTP KO mice, we performed ITTs. The two groups exhibited comparable insulin sensitivity when fed regular chow or HFD (Fig. 2d). Similar results were obtained in an independent cohort of mice (ESM Fig. 3). Together, these findings indicate that pancreatic TCPTP deficiency impairs glucose tolerance in mice upon prolonged high fat feeding.
Fig. 2.

Impaired glucose tolerance in HFD-fed panc-TCPTP KO mice. (a) Body weights of control (Con; white symbols) and panc-TCPTP KO (black symbols) male mice fed regular chow (full lines) or HFD (dotted lines). GTTs in male mice fed regular chow or HFD at (b) 9 weeks of age and (c) 45 weeks of age. (d) ITT in the same cohort at 51 weeks of age. Data are presented as mean±SEM. *p<0.05 control vs panc-TCPTP KO. Numbers of mice were as follows: HFD KO (a) 11, (b) 7, (c) 11, (d) 11; HFD Con (a) 7, (b) 6, (c) 5, (d) 6; chow KO (a) 9, (b) 7, (c) 7, (d) 8; chow Con (a) 4, (b) 4, (c) 3, (d) 4
Pancreatic TCPTP deficiency attenuates GSIS
Impaired glucose tolerance in HFD-fed panc-TCPTP KO mice suggests potential defects in insulin secretion. To test this possibility, we determined concentrations of circulating insulin under basal and glucose-stimulated conditions in control and panc-TCPTP KO mice. Chow-fed panc-TCPTP KO exhibited comparable circulating insulin levels to controls (Fig. 3a). However, HFD-fed control mice exhibited a robust increase in insulin after glucose injection, whereas panc-TCPTP KO mice exhibited lower circulating insulin levels (Fig. 3a, b). To test whether the observed defect in insulin secretion in panc-TCPTP KO mice fed HFD was due to TCPTP deficiency in islets and not in other pancreatic cell types and/or secondary to neural effects, we determined insulin secretion in isolated primary islets ex vivo (Fig. 3c). A glucose dose-dependent increase in insulin secretion was observed in primary islets from control mice. On the other hand, primary islets from panc-TCPTP KO mice exhibited attenuated GSIS compared with controls. Of note, glucagon-like peptide 1 can partially mitigate the attenuated insulin secretion in panc-TCPTP KO islets. To examine whether or not decreased circulating insulin concentrations in panc-TCPTP KO mice was due to decreased islet mass we immunostained pancreas sections for insulin and quantified islet surface area and number as detailed in the ESM Methods. Islet/pancreas surface area and islet number were comparable between HFD-fed control and panc-TCPTP KO mice (Fig. 3d, e). In addition, islet number/range of islet size was determined in mice fed regular chow and HFD (Fig. 3f). As expected, HFD-fed control and panc-TCPTP KO mice exhibited increased islet number/range of sizes compared with those fed regular chow, but islet number was comparable between HFD-fed control and panc-TCPTP KO mice.
Fig. 3.

TCPTP deficiency attenuates GSIS in HFD-fed mice and in islets. Circulating insulin concentrations in control (Con; white symbols) and panc-TCPTP KO (black symbols) male mice fed regular chow (full lines) or HFD (dotted lines) at (a) 52 weeks of age and (b) 58 weeks of age. (c) Primary islets from HFD-fed female control (white bars) and panc-TCPTP KO mice (black bars; 35 weeks old) were treated with glucose with or without glucagon-like peptide 1 (GLP-1). Data are presented as mean±SEM, n=3. *p<0.05 control vs panc-TCPTP KO. (d) Per cent islet surface area/pancreas surface area, (e) islet number and (f) islet number/range of sizes from HFD control (white bars) and panc-TCPTP KO (black bars) and chow-fed control (light grey bars) and panc-TCPTP KO (dark grey bars) male mice at 60 weeks of age. Data are presented as mean±SEM. *p<0.05; **p<0.01 chow vs HFD and †p<0.05 control vs panc-TCPTP KO. Numbers of mice were as follows: HFD KO (a) 7, (b) 5, (c) 3, (d) 6, (e) 6, (f) 6; HFD Con (a) 5, (b) 5, (c) 3, (d) 4, (e) 4, (f) 4; chow KO (a) 8, (f) 4; chow Con (a) 3, (f) 3
To determine whether or not the defect in insulin secretion was directly caused by TCPTP deficiency, we performed lentiviral small hairpin (shRNA)-mediated TCPTP knockdown in MIN6 beta cells. TCPTP knockdown (~74%) was achieved without compensatory changes in PTP1B and SHP2 expression (Fig. 4a). In addition, knockdown cells were reconstituted with shRNA-resistant TCPTP. As a complementary approach, pharmacological inhibition of TCPTP was achieved using the selective inhibitor compound 8 [25]. TCPTP activity was significantly reduced (~80%) in compound 8-treated control cells (Fig. 4b). Knockdown cells exhibited approximately 60% reduction in TCPTP activity and this was restored to control levels upon TCPTP reconstitution. In line with ex vivo islet studies, TCPTP knockdown cells exhibited reduced GSIS at 15, 30, 60 and 120 min (Fig. 4c). Importantly, reconstitution of knockdown cells with TCPTP restored insulin secretion to levels comparable with controls. Further, pharmacological inhibition of TCPTP in control cells led to attenuated GSIS that was comparable with knockdown cells (Fig. 4c). This finding was associated with decreased insulin content and glucose sensing in knockdown cells. Indeed, mRNA level of genes involved in beta cell function and differentiation, including Glut 2 [27], Pdx1 [28], Gk [29], Ins1 [30], Ins2 [31], Kir6.2 [32] and CaCna1d [33], was reduced in knockdown cells compared with controls (Fig. 4d). Importantly, expression of these genes was restored to control level upon reconstitution of TCPTP in knockdown cells. Moreover, protein expression of GLUT2, PDX1, GK and KIR6.2 was decreased in TCPTP knockdown cells and restored to control level upon reconstitution (Fig. 4e, f). Collectively, these findings demonstrate that glucose intolerance in HFD-fed panc-TCPTP KO mice was likely to be due to a cell autonomous defect in GSIS that was directly caused by TCPTP deletion.
Fig. 4.

TCPTP deficiency and pharmacological inhibition attenuate GSIS in MIN6 beta cells. (a) Representative immunoblots of TCPTP, PTP1B, SHP2 and tubulin in lysates of MIN6 beta cells (Con), TCPTP knockdown (KD) and reconstituted (KDR) cells, n=4. (b) TCPTP activity in Con, KD, KDR and TCPTP inhibitor (C8)-treated cells, n=4. **p<0.01 C8-treated vs non-treated cells; ††p<0.01 KD vs Con; and ‡p<0.05, ‡‡p<0.01 KDR vs KD. (c) Basal insulin and GSIS in Con (white bars), KD (black bars), KDR (light grey bars) and C8-treated control (dark grey bars) cells, n=5. *p<0.05, **p<0.01 each cell type at the indicated time vs basal; †p<0.05, ††p<0.01 each cell type vs Con at indicated time; ‡‡p<0.01 KDR vs KD at indicated time. (d) Glut2, Pdx1, Gk, Ins1, Ins2, Kir6.2 and CaCna1d mRNA in Con (white bars), KD (black bars) and KDR (light grey bars) cells, n=3. **p<0.01 KD vs Con; †p<0.05, ††p<0.01 KDR vs KD. Representative immunoblots (e) and corresponding quantitative bar graph (f) of GLUT2, PDX1, GK, KIR6.2, CaCna1d, TCPTP and tubulin in lysates of Con (white bars), KD (black bars) and KDR (light grey bars) cells, n=3. Bar graph represents normalised data for the indicated protein/tubulin as means±SEM. **p<0.01 KD vs Con; and †p<0.05, ††p<0.01 KDR vs KD
TCPTP regulates STAT1/3 tyrosyl phosphorylation in beta cells
To investigate the molecular basis for decreased GSIS we evaluated tyrosyl phosphorylation of STAT3, a bona fide TCPTP substrate [34]. STAT3 is activated by phosphorylation at Tyr705 leading to relocation to the nucleus to promote gene expression [35]. Primary islets from HFD-fed control and panc-TCPTP KO mice were isolated and stimulated with glucose. Immunoblots revealed elevated STAT3 Tyr705 phosphorylation in panc-TCPTP KO islets compared with controls (Fig. 5a). We used MIN6 beta cells to determine whether or not the enhanced STAT3 tyrosyl phosphorylation in KO islets was cell autonomous and caused by TCPTP deficiency. Glucose stimulation enhanced STAT3 Tyr705 phosphorylation in control cells and this was further enhanced in TCPTP knockdown cells (Fig. 5b). Reconstitution of knockdown cells with TCPTP restored STAT3 phosphorylation to control levels. Similarly, STAT3 immunoprecipitation demonstrated that STAT3 total tyrosyl phosphorylation was enhanced in knockdown cells compared with controls and restored to control levels upon TCPTP reconstitution (Fig. 5c). The elevated STAT3 tyrosine phosphorylation in TCPTP knockdown cells prompted us to test whether or not STAT3 is a direct TCPTP substrate in beta cells. To that end, we used the substrate trapping mutant (wherein the catalytic essential aspartate-182 is mutated to alanine; D/A) [6]. TCPTP D/A was transiently expressed in knockdown cells and TCPTP co-association with STAT3 was determined (Fig. 5d). Modest co-immunoprecipitation was observed between TCPTP wild-type and STAT3 upon glucose stimulation and this was disrupted upon lysis in the stringent buffer RIPA (R; Fig. 5d). On the other hand, co-association of TCPTP D/A and STAT3 was detected under basal conditions and was significantly enhanced upon glucose stimulation. Importantly, treatment with pervanadate (V), an inhibitor of PTPs, disrupted TCPTP D/A–STAT3 interactions (Fig. 5d).
Fig. 5.

TCPTP regulates STAT1/3 tyrosyl phosphorylation. (a) Representative immunoblots of pSTAT3 (Tyr705), STAT3 and TCPTP in islet lysates from HFD-fed male mice (59 weeks old) under basal and glucose-stimulated (25 mmol/l for 10 min) conditions. Bar graph represents normalised data for pSTAT3 (Tyr705)/STAT3 as means±SEM, n=3. *p<0.05 glucose-treated vs non-treated islets; ††p<0.01 KO (black bars) vs control (Con; white bars). (b) Immunoblots of pSTAT3 (Tyr705), STAT3, TCPTP and tubulin in lysates of Con (white bars), KD (black bars) and reconstituted (KDR; grey bars) cells under basal (0) and glucose-stimulated conditions (25 mmol/l for 15, 30 or 60 min), n=3. (c) STAT3 and e STAT1 immunoprecipitates from Con (white bars), KD (black bars) and KDR (grey bars) cells were immunoblotted for phosphotyrosine (PY99), STAT3 and STAT1 under basal (0) and glucose-stimulated conditions (25 mmol/l for 15, 30 or 60 min). TCPTP immunoprecipitates from Con and TCPTP D/A-transfected cells were immunoblotted for (d) STAT3, (f) STAT1 and TCPTP under basal (0) and glucose-stimulated conditions (25 mmol/l for 15 min). (R) indicates RIPA lysis and (V) vanadate treatment. In (b), (c) and (e), representative immunoblots are shown from independent repeats and bar graphs represent data for pSTAT3 (Tyr705)/STAT3, pSTAT3/STAT3, and pSTAT1/ STAT1 as means±SEM (n=4), respectively. For (b), (c) and (e) *p<0.05, **p<0.01 for each cell type at indicated times vs basal; †p<0.05, ††p<0.01 for each cell type vs Con at indicated times; ‡p<0.05, ‡‡p<0.01 KDR vs KD
Additional members of the STAT family are likely to be regulated by TCPTP in the pancreas. Indeed, STAT1 immunoprecipitation revealed that STAT1 total tyrosyl phosphorylation was enhanced in knockdown cells compared with controls and restored to control levels upon TCPTP reconstitution (Fig. 5e). Moreover, STAT1 co-associated with TCPTP D/A in response to glucose was consistent with STAT1 serving as a TCPTP substrate (Fig. 5f).
STAT3 is a mediator of TCPTP endocrine function
To investigate whether or not the effects of TCPTP deficiency on insulin secretion were mediated through STAT3 we determined the effects of the STAT3 pharmacological inhibitor BP-1-102 on insulin secretion. We reasoned that STAT3 inhibition should mitigate the effects of TCPTP deficiency on GSIS. Indeed, STAT3 inhibitor-treated TCPTP knockdown exhibited significantly improved GSIS and insulin levels were comparable with controls (Fig. 6a). Of note, BP-1-102 treatment inhibited phosphorylation of STAT3 and STAT1 (Fig. 6b). Accordingly, siRNA-mediated knockdown was used to further assess the contributions of STAT3 vs STAT1 to insulin secretion (Fig. 6c, d). Indeed, STAT3 knockdown phenocopied STAT3 pharmacological inhibition. STAT3 knockdown significantly enhanced GSIS at 60 min in control cells, whereas in TCPTP knockdown cells STAT3 deficiency restored GSIS to control levels. By contrast, siRNA-mediated STAT1 knockdown did not significantly alter GSIS in control cells but enhanced GSIS in TCPTP knockdown cells, albeit to a lower extent than that observed for STAT3 knockdown (Fig. 6c). Together, these data demonstrate that TCPTP regulates insulin secretion, at least in part, through STAT3 and that it is a physiologically relevant target of TCPTP in the endocrine pancreas.
Fig. 6.

STAT3 pharmacological inhibition and deficiency restore attenuated GSIS in TCPTP knockdown cells. (a) Basal insulin and GSIS in control (Con) and KD cells without (white bars and black bars, respectively) and with (light grey and dark grey bars, respectively) STAT3 inhibitor (BP-1-102) treatment, n=4. *p<0.05, **p<0.01 Con vs Con+ BP-1-102 at indicated times; †p<0.05, ††p<0.01 KD vs KD+BP-1-102 at indicated times. (b) pSTAT3 (Tyr705), STAT3, pSTAT1 (Tyr701) and STAT1 immunoblots of lysates from KD cells under basal and glucose-stimulated conditions (25 mmol/l for 15, 30 or 60 min), without (black bars) and with (grey bars) BP-1-102 treatment. Bar graph represents normalised data for pSTAT3 (Tyr705)/STAT3 and pSTAT1 (Tyr701)/STAT1 as means±SEM, n=4. **p<0.01 glucose-treated vs non-treated cells at indicated times; †p<0.05, ††p<0.01 KD vs KD+BP-1-102 at indicated times. (c) Basal insulin and GSIS in Con and KD cells with and without Stat1 and Stat3 silencing using siRNA, n=3. **p<0.01 each cell type at indicated time vs basal; †p<0.05, ††p<0.01 each cell type vs Con at indicated times; ‡p<0.05, ‡‡p<0.01 cells with the indicated siRNA vs the same cell transfected with a non-silencing siRNA. (d) Representative immunoblots from three independent repeats of STAT1, STAT3 and tubulin in control and knockdown cells treated with non-silencing (SCR), Stat1 (1) or Stat3 (3) siRNA
Discussion
Deciphering the molecular mechanisms that modulate insulin secretion is a prerequisite for repairing beta cell dysfunction in type 2 diabetes. The role of TCPTP in beta cell function and insulin secretion has heretofore remained largely unexplored. The current study demonstrated that pancreatic TCPTP deficiency impaired glucose tolerance and attenuated GSIS in mice with prolonged high fat feeding. At the molecular level, we identified STAT3 as a TCPTP substrate in the endocrine pancreas and demonstrated that TCPTP regulated GSIS, at least in part, through STAT3. Together, these findings provide new insights into the endocrine function of pancreatic TCPTP.
In this study we used Pdx1-Cre mice to investigate the role of TCPTP in the endocrine pancreas. Some reports suggest that Pdx1-Cre transgenic lines exhibit Cre expression in select regions of the brain, including the hypothalamus [36]. This issue was not a factor in our studies since we observed selective TCPTP deletion in the pancreas. Although immunoblotting of hypothalamic lysates is not adequate to detect deletion in select hypothalamic neurons, the comparable gains in body weight of panc-TCPTP KO and controls on chow and HFD suggest this is not a confounding factor in these studies. This is particularly relevant given that TCPTP deletion in the brain has overt effects on body weight and adiposity [17], which was not observed in these studies. When challenged with prolonged high fat feeding panc-TCPTP KO mice exhibited glucose intolerance that was independent of altered insulin sensitivity in the periphery and was due, in large part, to attenuated GSIS. The defect in insulin secretion was cell autonomous since it was recapitulated in primary islets and in MIN6 beta cells after TCPTP knockdown or pharmacological inhibition. However, we cannot completely rule out secondary effects in vivo and contributions from non-beta cells. Indeed, TCPTP inhibition and knockdown in MIN6 beta cells reduced GSIS but this was only observed in vivo after prolonged high fat feeding, implying that non-beta cell autonomous mechanisms modulate the panc-TCPTP KO mice phenotype. The reason for this finding is not clear but may be due to redundancies with other potential STAT3 regulatory phosphatases in chow-fed mice, such as PTP1B [24, 37] or SHP2 [38]. Additionally, we cannot rule out the potential influence of the genetic background. We used the C57Bl/6J strain, which has been reported to have a deletion in Nnt, and consequent reduced insulin secretion [39]. Of note, panc-TCPTP KO mice exhibit a phenotype that is comparable with obese glutathione peroxidase 1-deficient mice which display defective insulin secretion associated with oxidation of PTPs (including TCPTP) and decreased PDX1 and insulin content [40]. Further, we recently demonstrated beneficial effects of pancreatic TCPTP deficiency in mitigating cerulein-induced acute pancreatitis [23], highlighting the diverse functions of TCPTP in the exocrine and endocrine pancreas. Finally, the finding that the metabolic effects of pancreatic TCPTP deficiency are influenced by diet are relevant to human type 2 diabetes, which is known to correlate with old age and obesity. We also observed that the phenotypes in male panc-TCPTP KO mice were more pronounced compared with females. Such a sexual dichotomy in metabolic studies at the whole-animal level has been reported [41, 42], but the underlying reasons are not fully understood.
The current studies identified STAT3 as a TCPTP substrate in the endocrine pancreas. Islets and MIN6 beta cells lacking TCPTP exhibited increased STAT3 phosphorylation, and substrate trapping established a direct association between STAT3 and TCPTP D/A that is consistent with an enzyme-substrate interaction. Pharmacological inhibition of STAT3 in TCPTP knockdown cells mitigated the defect in GSIS demonstrating that the effects of the phosphatase deficiency are mediated through STAT3. Knockdown of STAT3 phenocopied the effects of STAT3 pharmacological inhibition consistent with the enhanced STAT3 signalling associated with TCPTP deficiency being important in the attenuated GSIS. The precise mechanism is not clear but it is worth noting that we observed increased GLUT2, PDX1 and GK protein expression in TCPTP knockdown cells with STAT3 deficiency (data not shown), consistent with the normalisation of insulin secretion in these cells. Conflicting reports suggest a role for STAT3 in insulin secretion. Pancreatic STAT3 deletion using rat insulin promoter (RIP-Cre) leads to glucose intolerance, defects in early phase insulin secretion and mild obesity [43, 44]. However, the RIP-Cre transgene has been reported to be expressed in the hypothalamus [45] and STAT3 was deleted in leptin-receptor positive neurons. Other studies aimed at determining the pancreas-specific function of STAT3 using Pdx1-Cre have also yielded conflicting data. While STAT3 appeared dispensable for pancreatic beta cell function in one study [46], another demonstrated that the KO mice exhibited impaired insulin secretion [47]. It is important to note that TCPTP can conceivably modulate pancreatic endocrine function through numerous effectors and signalling pathways. Indeed, TCPTP deficiency altered STAT1 phosphorylation, which has been shown to regulate beta cell function, differentiation and apoptosis [48, 49]. In addition, TCPTP is a regulator of insulin [7], leptin [17], c-Src [50] and ER stress [22] signalling pathways all of which play important roles in beta cell function. Previously, we demonstrated that TCPTP deficiency in MIN6 cells mitigated chemically induced ER stress signalling that coincided with increased STAT3 activation [22]. However, our current findings suggest that pancreatic TCPTP deletion coupled with high fat feeding has detrimental effects on insulin secretion. Although we cannot exclude other pathways contributing to the attenuated insulin secretion in HFD-fed panc-TCPTP KO mice, we propose that elevated STAT3 signalling is an important contributing factor.
In summary, pancreatic TCPTP deficiency in mice with prolonged high fat feeding led to delayed glucose clearance and impaired GSIS in a cell autonomous manner. Further, we report that STAT3 is a physiologically relevant mediator of TCPTP actions in islets. Together, these findings provide novel insight into the metabolic actions of TCPTP in the endocrine pancreas.
Supplementary Material
Acknowledgments
We thank D. Melton (Harvard University, Boston, MA, USA) for the Pdx1-Cre transgenic mice.
Funding statement: This work was supported by a research grant from the Juvenile Diabetes Research Foundation (1-2009-337) and National Institutes of Health (NIH) grants R56 DK084317, R01 DK090492 and R01 DK095359 to FGH and K99 DK100736 to AB. Research in the Kulkarni laboratory is funded by NIH grants R01 DK67536 and R01 DK103215, Zhang laboratory by R01 CA69202 and the Tiganis laboratory by the National Health and Medical Research Council (NHMRC) of Australia (APP1047055). T. Tiganis is an NHMRC Principal Research Fellow (APP1002693).
Abbreviations
- ER
Endoplasmic reticulum
- GK
Glucokinase
- GLUT2
Glucose transporter 2
- GSIS
Glucose-stimulated insulin secretion
- GTT
Glucose tolerance test
- HFD
High fat diet
- IR
Insulin receptor
- ITT
Insulin tolerance test
- KO
Knockout
- Panc-TCPTP KO
Pancreatic Ptpn2/TCPTP knockout
- PDX1
Pancreatic and duodenal homeobox 1
- PTKs
Protein tyrosine kinases
- PTP1B
Protein tyrosine phosphatase 1B
- PTPs
Protein tyrosine phosphatases
- RIP
Rat insulin promoter
- RTKs
Receptor protein tyrosine kinases
- SHP2
Src homology phosphatase 2
- shRNA
Small hairpin RNA
- siRNA
Small interfering RNA
- STAT
Signal transducer and activator of transcription
- TCPTP
T cell protein tyrosine phosphatase
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00125-014-3413-7) contains peer-reviewed but unedited supplementary material, which is available to authorised users.
Duality of interest: The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement: YX, SL and AB designed and performed research, analysed data and revised the manuscript. KM, IM, EH, SC, FWand SZ performed research and revised the manuscript. ZYZ, RK and TT contributed reagents, analysed data, provided intellectual input and revised the manuscript. FH designed research, analysed data, provided intellectual input and wrote the manuscript. All authors approved the final version. FH is the guarantor of this work.
Contributor Information
Yannan Xi, Department of Nutrition, University of California Davis, 3135 Meyer Hall, Davis, CA 95616, USA.
Siming Liu, Department of Nutrition, University of California Davis, 3135 Meyer Hall, Davis, CA 95616, USA.
Ahmed Bettaieb, Department of Nutrition, University of California Davis, 3135 Meyer Hall, Davis, CA 95616, USA.
Kosuke Matsuo, Department of Nutrition, University of California Davis, 3135 Meyer Hall, Davis, CA 95616, USA.
Izumi Matsuo, Department of Nutrition, University of California Davis, 3135 Meyer Hall, Davis, CA 95616, USA.
Ellen Hosein, Department of Nutrition, University of California Davis, 3135 Meyer Hall, Davis, CA 95616, USA.
Samah Chahed, Department of Nutrition, University of California Davis, 3135 Meyer Hall, Davis, CA 95616, USA.
Florian Wiede, Department of Biochemistry and Molecular Biology, Monash University, Clayton, VIC, Australia.
Sheng Zhang, Department of Biochemistry and Molecular Biology, Indiana University, Indianapolis, IN, USA.
Zhong-Yin Zhang, Department of Biochemistry and Molecular Biology, Indiana University, Indianapolis, IN, USA.
Rohit N. Kulkarni, Islet Cell and Regenerative Biology, Research Division, Joslin Diabetes Center, Department of Medicine, Brigham and Women’s Hospital, Boston, MA, USA
Tony Tiganis, Department of Biochemistry and Molecular Biology, Monash University, Clayton, VIC, Australia.
Fawaz G. Haj, Email: fghaj@ucdavis.edu, Department of Nutrition, University of California Davis, 3135 Meyer Hall, Davis, CA 95616, USA. Department of Internal Medicine, Division of Endocrinology, Diabetes and Metabolism, University of California Davis, Sacramento, CA, USA. Comprehensive Cancer Center, University of California Davis, Sacramento, CA, USA
References
- 1.Biddinger SB, Kahn CR. From mice to men: insights into the insulin resistance syndromes. Annu Rev Physiol. 2006;68:123–158. doi: 10.1146/annurev.physiol.68.040104.124723. [DOI] [PubMed] [Google Scholar]
- 2.Michael MD, Kulkarni RN, Postic C, et al. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell. 2000;6:87–97. [PubMed] [Google Scholar]
- 3.Kulkarni RN, Bruning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell. 1999;96:329–339. doi: 10.1016/s0092-8674(00)80546-2. [DOI] [PubMed] [Google Scholar]
- 4.Kulkarni RN, Holzenberger M, Shih DQ, et al. beta-cell-specific deletion of the Igf1 receptor leads to hyperinsulinemia and glucose intolerance but does not alter beta-cell mass. Nat Genet. 2002;31:111–115. doi: 10.1038/ng872. [DOI] [PubMed] [Google Scholar]
- 5.Tiganis T, Bennett AM. Protein tyrosine phosphatase function: the substrate perspective. Biochem J. 2007;402:1–15. doi: 10.1042/BJ20061548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tiganis T, Bennett AM, Ravichandran KS, Tonks NK. Epidermal growth factor receptor and the adaptor protein p52Shc are specific substrates of T cell protein tyrosine phosphatase. Mol Cell Biol. 1998;18:1622–1634. doi: 10.1128/mcb.18.3.1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galic S, Klingler-Hoffmann M, Fodero-Tavoletti MT, et al. Regulation of insulin receptor signaling by the protein tyrosine phosphatase TCPTP. Mol Cell Biol. 2003;23:2096–2108. doi: 10.1128/MCB.23.6.2096-2108.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Vliet C, Bukczynska PE, Puryer MA, et al. Selective regulation of tumor necrosis factor-induced Erk signaling by Src family kinases and the T cell protein tyrosine phosphatase. Nat Immunol. 2005;6:253–260. doi: 10.1038/ni1169. [DOI] [PubMed] [Google Scholar]
- 9.Simoncic PD, Lee-Loy A, Barber DL, Tremblay ML, McGlade CJ. The T cell protein tyrosine phosphatase is a negative regulator of janus family kinases 1 and 3. Curr Biol. 2002;12:446–453. doi: 10.1016/s0960-9822(02)00697-8. [DOI] [PubMed] [Google Scholar]
- 10.Fukushima A, Loh K, Galic S, et al. T cell protein tyrosine phosphatase attenuates STAT3 and insulin signaling in the liver to regulate gluconeogenesis. Diabetes. 59:1906–1914. doi: 10.2337/db09-1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.ten Hoeve J, de Jesus Ibarra-Sanchez M, Fu Y, et al. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol Cell Biol. 2002;22:5662–5668. doi: 10.1128/MCB.22.16.5662-5668.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aoki N, Matsuda T. A nuclear protein tyrosine phosphatase TC-PTP is a potential negative regulator of the PRL-mediated signaling pathway: dephosphorylation and deactivation of signal transducer and activator of transcription 5a and 5b by TC-PTP in nucleus. Mol Endocrinol. 2002;16:58–69. doi: 10.1210/mend.16.1.0761. [DOI] [PubMed] [Google Scholar]
- 13.Lu X, Chen J, Sasmono RT, et al. T cell protein tyrosine phosphatase, distinctively expressed in activated-B cell-like diffuse large B cell lymphomas, is the nuclear phosphatase of STAT6. Mol Cell Biol. 2007;27:2166–2179. doi: 10.1128/MCB.01234-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.You-Ten KE, Muise ES, Itie A, et al. Impaired bone marrow microenvironment and immune function in T cell protein tyrosine phosphatase-deficient mice. J Exp Med. 1997;186:683–693. doi: 10.1084/jem.186.5.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wiede F, Shields BJ, Chew SH, et al. T cell protein tyrosine phosphatase attenuates T cell signaling to maintain tolerance in mice. J Clin Investig. 2011;121:4758–4774. doi: 10.1172/JCI59492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loh K, Merry TL, Galic S, et al. T cell protein tyrosine phosphatase (TCPTP) deficiency in muscle does not alter insulin signalling and glucose homeostasis in mice. Diabetologia. 2012;55:468–478. doi: 10.1007/s00125-011-2386-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loh K, Fukushima A, Zhang X, et al. Elevated hypothalamic TCPTP in obesity contributes to cellular leptin resistance. Cell Metab. 2011;14:684–699. doi: 10.1016/j.cmet.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gurzov EN, Tran M, Fernandez-Rojo MA, et al. Hepatic oxidative stress promotes insulin-STAT-5 signaling and obesity by inactivating protein tyrosine phosphatase N2. Cell Metab. 2014;20:85–102. doi: 10.1016/j.cmet.2014.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moore F, Colli ML, Cnop M, et al. PTPN2, a candidate gene for type 1 diabetes, modulates interferon-gamma-induced pancreatic beta-cell apoptosis. Diabetes. 2009;58:1283–1291. doi: 10.2337/db08-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santin I, Moore F, Colli ML, et al. PTPN2, a candidate gene for type 1 diabetes, modulates pancreatic beta-cell apoptosis via regulation of the BH3-only protein Bim. Diabetes. 2011;60:3279–3288. doi: 10.2337/db11-0758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ize-Ludlow D, Lightfoot YL, Parker M, et al. Progressive erosion of beta-cell function precedes the onset of hyperglycemia in the NOD mouse model of type 1 diabetes. Diabetes. 2011;60:2086–2091. doi: 10.2337/db11-0373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bettaieb A, Liu S, Xi Y, et al. Differential regulation of endoplasmic reticulum stress by protein tyrosine phosphatase 1B and T cell protein tyrosine phosphatase. J Biol Chem. 2011;11:9225–9235. doi: 10.1074/jbc.M110.186148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bettaieb A, Xi Y, Hosein E, et al. Pancreatic T cell protein-tyrosine phosphatase deficiency ameliorates cerulein-induced acute pancreatitis. Cell Commun Signal. 2014;12:13. doi: 10.1186/1478-811X-12-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu S, Xi Y, Bettaieb A, et al. Disruption of protein-tyrosine phosphatase 1B expression in the pancreas affects beta-cell function. Endocrinology. 2014:en20132004. doi: 10.1210/en.2013-2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang S, Chen L, Luo Y, Gunawan A, Lawrence DS, Zhang ZY. Acquisition of a potent and selective TC-PTP inhibitor via a stepwise fluorophore-tagged combinatorial synthesis and screening strategy. J Am Chem Soc. 2009;131:13072–13079. doi: 10.1021/ja903733z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang X, Yue P, Page BD, et al. Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proc Natl Acad Sci U S A. 2012;109:9623–9628. doi: 10.1073/pnas.1121606109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guillam MT, Hummler E, Schaerer E, et al. Early diabetes and abnormal postnatal pancreatic islet development in mice lacking Glut-2. Nat Genet. 1997;17:327–330. doi: 10.1038/ng1197-327. [DOI] [PubMed] [Google Scholar]
- 28.Hani EH, Stoffers DA, Chevre JC, et al. Defective mutations in the insulin promoter factor-1 (IPF-1) gene in late-onset type 2 diabetes mellitus. J Clin Investig. 1999;104:R41–R48. doi: 10.1172/JCI7469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aizawa T, Asanuma N, Terauchi Y, et al. Analysis of the pancreatic beta cell in the mouse with targeted disruption of the pancreatic beta cell-specific glucokinase gene. Biochem Biophys Res Commun. 1996;229:460–465. doi: 10.1006/bbrc.1996.1826. [DOI] [PubMed] [Google Scholar]
- 30.Leroux L, Desbois P, Lamotte L, et al. Compensatory responses in mice carrying a null mutation for Ins1 or Ins2. Diabetes. 2001;50(Suppl 1):S150–S153. doi: 10.2337/diabetes.50.2007.s150. [DOI] [PubMed] [Google Scholar]
- 31.Wang J, Takeuchi T, Tanaka S, et al. A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Mody mouse. J Clin Investig. 1999;103:27–37. doi: 10.1172/JCI4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gupta RK, Vatamaniuk MZ, Lee CS, et al. The MODY1 gene HNF-4alpha regulates selected genes involved in insulin secretion. J Clin Investig. 2005;115:1006–1015. doi: 10.1172/JCI200522365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Namkung Y, Skrypnyk N, Jeong MJ, et al. Requirement for the L-type Ca(2+) channel alpha(1D) subunit in postnatal pancreatic beta cell generation. J Clin Investig. 2001;108:1015–1022. doi: 10.1172/JCI13310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fukushima A, Loh K, Galic S, et al. T cell protein tyrosine phosphatase attenuates STAT3 and insulin signaling in the liver to regulate gluconeogenesis. Diabetes. 2010;59:1906–1914. doi: 10.2337/db09-1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. 2007;282:20059–20063. doi: 10.1074/jbc.R700016200. [DOI] [PubMed] [Google Scholar]
- 36.Wicksteed B, Brissova M, Yan W, et al. Conditional gene targeting in mouse pancreatic ss-Cells: analysis of ectopic Cre trans-gene expression in the brain. Diabetes. 2010;59:3090–3098. doi: 10.2337/db10-0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kushner JA, Haj FG, Klaman LD, et al. Islet-sparing effects of protein tyrosine phosphatase-1b deficiency delays onset of diabetes in IRS2 knockout mice. Diabetes. 2004;53:61–66. doi: 10.2337/diabetes.53.1.61. [DOI] [PubMed] [Google Scholar]
- 38.Zhang SS, Hao E, Yu J, et al. Coordinated regulation by Shp2 tyrosine phosphatase of signaling events controlling insulin biosynthesis in pancreatic beta-cells. Proc Natl Acad Sci U S A. 2009;106:7531–7536. doi: 10.1073/pnas.0811715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mekada K, Abe K, Murakami A, et al. Genetic differences among C57BL/6 substrains. Exp Anim / Japan Assoc Lab Anim Sci. 2009;58:141–149. doi: 10.1538/expanim.58.141. [DOI] [PubMed] [Google Scholar]
- 40.Merry TL, Tran M, Stathopoulos M, et al. High-fat-fed obese glutathione peroxidase 1-deficient mice exhibit defective insulin secretion but protection from hepatic steatosis and liver damage. Antioxid Redox Signal. 2014;20:2114–2129. doi: 10.1089/ars.2013.5428. [DOI] [PubMed] [Google Scholar]
- 41.Kido Y, Burks DJ, Withers D, et al. Tissue-specific insulin resistance in mice with mutations in the insulin receptor, IRS-1, and IRS-2. J Clin Investig. 2000;105:199–205. doi: 10.1172/JCI7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yaekura K, Julyan R, Wicksteed BL, et al. Insulin secretory deficiency and glucose intolerance in Rab3A null mice. J Biol Chem. 2003;278:9715–9721. doi: 10.1074/jbc.M211352200. [DOI] [PubMed] [Google Scholar]
- 43.Gorogawa S, Fujitani Y, Kaneto H, et al. Insulin secretory defects and impaired islet architecture in pancreatic beta-cell-specific STAT3 knockout mice. Biochem Biophys Res Commun. 2004;319:1159–1170. doi: 10.1016/j.bbrc.2004.05.095. [DOI] [PubMed] [Google Scholar]
- 44.Cui Y, Huang L, Elefteriou F, et al. Essential role of STAT3 in body weight and glucose homeostasis. Mol Cell Biol. 2004;24:258–269. doi: 10.1128/MCB.24.1.258-269.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gannon M, Shiota C, Postic C, Wright CV, Magnuson M. Analysis of the Cre-mediated recombination driven by rat insulin promoter in embryonic and adult mouse pancreas. Genesis. 2000;26:139–142. doi: 10.1002/(sici)1526-968x(200002)26:2<139::aid-gene12>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 46.Lee JY, Hennighausen L. The transcription factor Stat3 is dispensable for pancreatic beta-cell development and function. Biochem Biophys Res Commun. 2005;334:764–768. doi: 10.1016/j.bbrc.2005.06.162. [DOI] [PubMed] [Google Scholar]
- 47.Kostromina E, Gustavsson N, Wang X, et al. Glucose intolerance and impaired insulin secretion in pancreas-specific signal transducer and activator of transcription-3 knockout mice are associated with microvascular alterations in the pancreas. Endocrinology. 2010;151:2050–2059. doi: 10.1210/en.2009-1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim S, Kim HS, Chung KW, et al. Essential role for signal transducer and activator of transcription-1 in pancreatic beta-cell death and autoimmune type 1 diabetes of nonobese diabetic mice. Diabetes. 2007;56:2561–2568. doi: 10.2337/db06-1372. [DOI] [PubMed] [Google Scholar]
- 49.Moore F, Naamane N, Colli ML, et al. STAT1 is a master regulator of pancreatic {beta}-cell apoptosis and islet inflammation. J Biol Chem. 2011;286:929–941. doi: 10.1074/jbc.M110.162131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shields BJ, Court NW, Hauser C, Bukczynska PE, Tiganis T. Cell cycle-dependent regulation of SFK, JAK1 and STAT3 signalling by the protein tyrosine phosphatase TCPTP. Cell Cycle. 2008;7:3405–3416. doi: 10.4161/cc.7.21.6950. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.