

Abstract
Background
Massive computational power is needed to analyze the genomic data produced by next-generation sequencing, but extensive computational experience and specific knowledge of algorithms should not be necessary to run genomic analyses or interpret their results.
Findings
We present BamBam, a package of tools for genome sequence analysis. BamBam contains tools that facilitate summarizing data from BAM alignment files and identifying features such as SNPs, indels, and haplotypes represented in those alignments.
Conclusions
BamBam provides a powerful and convenient framework to analyze genome sequence data contained in BAM files.
Electronic supplementary material
The online version of this article (doi:10.1186/1756-0500-7-829) contains supplementary material, which is available to authorized users.
Keywords: SAM, BAM, Haplotype, SNP, Indel, Consensus, Methylation, Expression
Findings
Massive amounts of data are involved in genome sequence research, requiring researchers to use supercomputing clusters and complex algorithms to analyze their sequence data. Genomic analyses frequently include next-generation sequencing to produce millions of short reads, followed by aligning of reads to a reference genome sequence with software like GSNAP and Bowtie 2 [1, 2]. These programs generate SAM files, the accepted standard for storing short read alignment data, which are subsequently compressed to BAM format via SAMtools [3]. The BAM files must then be analyzed and compared to produce meaningful results. Here we expand on the body of tools for analyzing and comparing BAM files.
We present BamBam, a package of bioinformatics tools to carry out a variety of genomic analyses on BAM files (Table 1). The included tools perform such tasks as counting the number of reads mapped to each gene in a genome (as for gene expression analyses), identifying SNPs (Single Nucleotide Polymorphisms) and CNVs (Copy Number Variants), and extracting consensus sequences. The purpose of BamBam is to provide a consistent framework to perform common tasks, without requiring extensive knowledge of computation or algorithms to select or interpret appropriate parameters.
Table 1.
The core independent tools of BamBam
| Section | Tool | Purpose |
|---|---|---|
| Single nucleotide Polymorphisms | InterSnp | Call SNPs between two or more samples |
| Pebbles | Impute genotypes in output from InterSnp | |
| HapHunt | Phase haplotypes with K-means | |
| Copy number Variants | GapFall | Identify deletions between two samples |
| Elfen | Identify covered regions | |
| HMMph | Call copy number variants with HMM | |
| Bisulfite-sequence Analysis | MetHead | Summarize base pair methylation in bisulfite-sequence data |
| GeneVisitor | Bam2Consensus | Generate consensus sequences from one or more samples |
| Bam2Fastq | Extract mapped and unmapped reads from BAM files | |
| Counter | Summarize read coverage of sequences or regions | |
| SubBam | Extract subset of mapped reads | |
| Allopolyploid analysis | PolyCat | Categorize reads by genome based on similarity to parents |
| Scripts | Various | Various |
The BamBam package includes several independent programs, briefly described below. Brief tests were carried out to compare InterSnp, GapFall, and HapHunt with similar tools (Additional file 1). The latest version of PolyCat is also included [4]. The README in the download package provides example commands for various common analyses, including phylogeny inference, molecular evolution estimation, methylation analysis, and differential expression analysis. A usage guide (see Additional file 2) provides a more detailed walkthrough of some workflows.
Single nucleotide polymorphisms
InterSnp calls SNPs between samples, represented by separate BAM files. InterSnp examines each position in the genome, assigning consensus alleles to each site for each sample. A SNP is called whenever two samples differ at the same position, producing a table with the genotypes of all samples at all polymorphic sites. The output is a table with the sequence name, position, and genotype for each sample at that site on each row, which can be readily processed by common command-line programs or scripts to calculate statistics or produce marker data for other programs.
Pebbles imputes genotypes using the K-nearest neighbor algorithm [5, 6]. For each unknown genotype, Pebbles finds the samples that are most similar at nearby loci. Then it assigns a genotype to the unknown locus based on the weighted contributions of those neighbors. Pebbles operates on InterSnp output—a table of genotypes—and produces a file of the same form.
HapHunt uses K-means clustering to solve the haplotype-phasing problem, which consists of identifying all haplotypes in a sampled individual or population. Many programs have attempted to solve haplotype phasing and the closely related haplotype assembly problems using a variety of strategies, including Max-Cut, hidden Markov models, and dynamic programming [7–9]. The K-means clustering algorithm (Figure 1) is an unsupervised machine learning algorithm, and is mathematically equivalent to Principle Component Analysis [10, 11].
Figure 1.
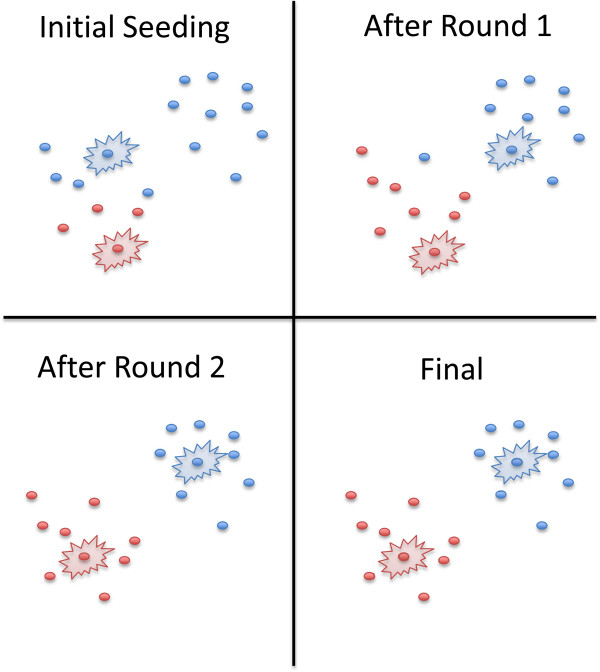
K-means clustering algorithm. An example 2-cluster run is shown, with the clusters distinguished by color and the current cluster seeds marked by a starburst. In the first round, each point is assigned to its closest seed, and a new seed is chosen for each cluster based on the average of all points in that cluster. As a result, the blue cluster seed moves to the right side. In the second round, both cluster seeds drift to their correct locations, resulting in a proper division. Note that, after two rounds, the clusters have reached a steady-state, and would not change further through an infinite number of iterations.
HapHunt first selects K reads distant from one another to serve as haplotype seeds. It assigns each other read to the haplotype with the closest consensus sequence. Then it recalculates the consensus sequences based on the reads in each haplotype and repeats the process of assigning each read to the haplotype with the closest consensus sequence. It repeats this process a given number of times, calculating a score at the end of each round based on the difference of the smallest interhaplotype distance and greatest intrahaplotype distance. This score favors clusterings in which haplotypes are individually compact and most distinct from one another. This score can optionally be scaled by the average size ratio for each pair of haplotypes, favoring clusterings that are more evenly divided. The consensus sequences of the final haplotypes are printed as an aligned FASTA file for each sequence in the original reference.
Copy number variants
Gapfall identifies large deletions between samples based on read coverage. It searches the genome for extended regions that have high coverage in one sample but no coverage in the other. A large region with no coverage could indicate a physical deletion (for genomic samples) or a deactivated gene (for RNA-seq). These putative deletions are reported as an annotation file that can be visualized with a genome browser such as IGV [12].
Eflen identifies and extracts regions in a BAM file that are covered by at least a user-specified number of reads and outputs those regions as a GFF file. Provided with multiple BAMs, Eflen will identify regions that are covered in at least a user-specified fraction of those BAMs. This tool can be especially useful for analyzing GBS or RNA-seq data.
HMMph identifies CNVs between samples based on read coverage. BAM files must be provided for a control and for the sample of interest. The coverage ratio between those two BAM files is normalized by the total read coverage. Then the copy number of each locus in a sliding window is modeled based on a Poisson distribution in an untrained Hidden Markov Model [13, 14].
Bisulfite-sequence analysis
Bisulfite treatment converts unmethylated cytosines to thymines. MetHead summarizes methylation at all cytosine positions in the genome, based on BAM files of mapped bisulfite-treated reads. It totals the number of mapped cytosines and thymines at each position (indicating methylated and unmethylated states, respectively), then performs a one-tailed binomial test for the methylation of that site.Different protocols are used for bisulfite treatment. If PCR is not performed after bisulfite treatment but before sequencing, then only 2 possibilities exist: conversions on the forward and reverse strand. But if PCR is performed, 4 possibilities exist (Figure 2). To properly count the number of cytosines and thymines in the 4-possibility protocol, the origin of the pre-PCR DNA fragment must be inferred. MetHead determines this—if necessary—by counting the number of C- > T conversions and G- > A conversions (indicative of a conversion on the reverse strand). It generates a BAM file with the orientation of each read matching its origin strand. That BAM can then be analyzed as if it were data produced by the 2-possibility protocol. Note that, in the produced BAM, the orientation of reads is not based on the direction in which the read was sequenced. Instead, the orientation of the read indicates the type of conversion caused by bisulfite treatment: C- > G or A- > T.
Figure 2.
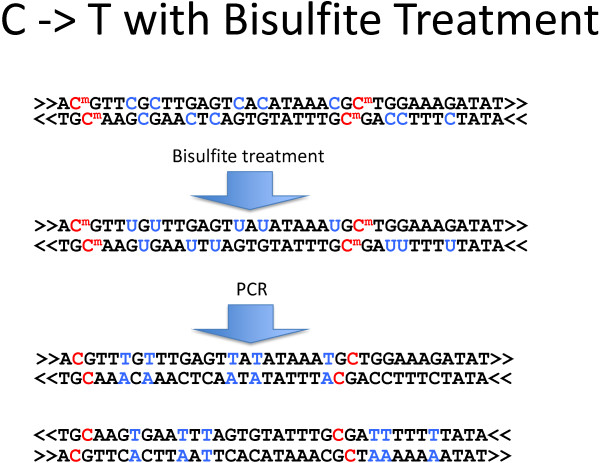
Bisulfite treatment. The effects of bisulfite treatment on DNA are shown. An “m” superscript indicates a methylated cytosine. The orientation of each strand is indicated by “<<” and “>>”. Bisulfite treatment converts unmethylated cytosines into uracils and, ultimately, thymines. After PCR, however, a given fragment may have C- > T conversions or G- > A conversions, depending on its orientation relative to its origin fragment.
GeneVisitor
It is often useful to be able to compute on specific genomic intervals, such as genes. GeneVisitor provides a quick and easy way to do this, using an annotation file (GFF or BED format) to call a function on each indicated region of the genome. This class can be used by C++ programmers to run custom functions. In addition, pre-built tools utilize GeneVisitor without the need for programming.
Bam2Consensus converts one or more BAM files into a series of FASTA-formatted consensus sequences. If desired, multiple sequences—essentially unphased haplotypes—can be produced per BAM file, facilitating analyses of heterozygosity, nucleotide diversity, and molecular evolution. Suppose you have several BAM files representing different accessions of a species, all mapped to a common genome reference sequence. With a single command, Bam2Consensus can produce an aligned FASTA file for each gene, each containing the consensus sequences for each accession.
Bam2Fastq extracts mapped or unmapped reads from a BAM file, or from select regions of the BAM file.
Counter summarizes the number of reads mapped to each annotated region in one or more BAM files. RPKM (Reads Per Kilobase per Million mapped reads) normalization can be applied if desired. The output of Counter is a table of features and read counts, ready to be imported into EdgeR for differential expression analysis [15].
SubBam extracts a subset of a BAM file. It can optionally modify the BAM file, changing the coordinates of mapped reads to match a new reference that is a subset of the original reference. Suppose you have WGS reads mapped to a reference sequence and are interested in several loci. SubBam can produce BAMs that only contain the loci of interest, with a coordinate system corresponding to the position in the locus, rather than in the genome as a whole.
Allopolyploid analysis
The latest version of PolyCat is included in BamBam. PolyCat uses an index of known homoeo-SNPs (polymorphisms that distinguish the genomes of an allopolyploid) to identify the source genome for each read in a library, which cannot be distinguished through typical next-generation sequencing protocols [4].The MultiIndex class is used by PolyCat and MetHead, and can be used to make novel tools in C++. The MultiIndex is appropriate for random access to hundreds of millions of individual base positions in a genome sequence. It provides quick random access to base positions scattered across a genome sequence. Each sequence in the reference is indexed with a linked-list, with an index of landmark nodes spaced along the sequence at a resolution specified by the user (Figure 3).
Figure 3.
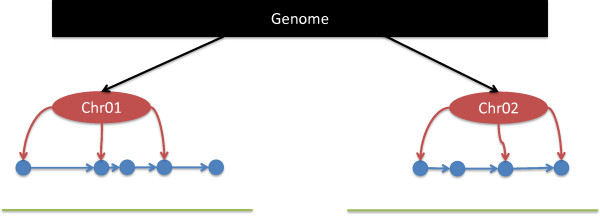
Multi-level index. The multi-level index provides random access to large numbers of individual base positions across a genome. Each sequence (green) is indexed by a linked-list (blue), and that index is indexed by a set of landmark nodes (red) to provide rapid access to any location.
Scripts
In addition to the core tools mentioned above, BamBam includes many Perl scripts, many of which use BioPerl modules [16]. Script functions include calculation of nucleotide diversity (π) and molecular evolution rates (Ka and Ks), paralog identification, differential expression with EdgeR [15], summarization of results from MetHead, and summarization of genotype tables produced by InterSnp and Pebbles.
Conclusions
The BamBam tools form a simple interface between the researching biologist and the wealth of data contained in next-generation sequence alignments. They provide a means to efficiently identify interesting genomic features and summarize data, facilitating many next-generation sequence analysis experiments.
BamBam is freely available under the MIT license at http://sourceforge.net/projects/bambam/. It depends on both SAMtools and BAMtools [3, 17].
Availability and requirements
Project Name: BamBam
Project Home Page:http://sourceforge.net/projects/bambam/
Operating System: Unix
Dependencies: SamTools, BamTools, BioPerl
Programming Language: C++ and Perl
License: MIT
Authors’ information
JP has a B.S. in Computer Science and is currently a graduate student in Biology, focusing on developing tools for polyploid genome analysis. ZL is an undergraduate student in the Udall lab. MH is a graduate student in the Udall lab. JU is an Associate professor at Brigham Young University and is the academic advisor of JP, ZL, and MH.
Electronic supplementary material
Additional file 1:Supplementary Material. Figure S1 Read alignment of cotton A-genome and d-genome reads to a common reference, rendered in IGV. Highlights indicate differences compared to the reference, so highlights in the upper sequence (A-genome) and the lack of those highlights in the lower sequence (D-genome) indicate SNPs between the two genomes. In this region, InterSnp identified 17 SNPs but SAMtools failed to identify any. Figure S2 Haplotypes identified by SAMtools and HapHunt, compared to the known haplotype. Figure S3 Phylogenetic tree. This neighbor-joining tree was built by neighbor based on SNPs identified by InterSnp. Then Geneious was used to render the actual tree. Table S1 The number of deletions identified in each accession (row), along with the percentage of those deletions that were shared with other members of the same species and with the entire group of samples. (DOCX 207 KB)
Acknowledgements
The authors would like to thank NSF Plant Genome Research Program, Cotton, Inc., and BYU Mentoring Grants for funding. They would also like to thank the Fulton Supercomputing Lab for their invaluable computational resources and technical support.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JP designed, wrote, and tested the programs and scripts and prepared the final manuscript. ZL and MH performed additional testing of the tools, pipelines, usage guides, and portability. JU provided direction, resources, and data, designed the larger research projects that produced various tools, and edited and approved the final manuscript. All authors read and approved the final manuscript.
Contributor Information
Justin T Page, Email: jtpage68@gmail.com.
Zachary S Liechty, Email: zliechty@gmail.com.
Mark D Huynh, Email: markdhuynh@gmail.com.
Joshua A Udall, Email: jaudall@byu.edu.
References
- 1.Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873–881. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Meth. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J. The sequence alignment/map format and SAMtools. Oxford, UK: Bioinformatics; 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Page JT, Gingle AR, Udall JA. PolyCat: a resource for genome categorization of sequencing reads from allopolyploid organisms. G3 (Bethesda) 2013;3:517–525. doi: 10.1534/g3.112.005298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rutkoski JE, Poland J, Jannink J-L, Sorrells ME. Imputation of unordered markers and the impact on genomic selection accuracy. G3 (Bethesda) 2013;3:427–439. doi: 10.1534/g3.112.005363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Troyanskaya O, Cantor M, Sherlock G, Brown P, Hastie T, Tibshirani R, Botstein D, Altman RB. Missing value estimation methods for DNA microarrays. Bioinformatics. 2001;17:520–525. doi: 10.1093/bioinformatics/17.6.520. [DOI] [PubMed] [Google Scholar]
- 7.Bansal V, Bafna V. HapCUT: an efficient and accurate algorithm for the haplotype assembly problem. 2008. [DOI] [PubMed] [Google Scholar]
- 8.Browning BL, Browning SR. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet. 2009;84:210–223. doi: 10.1016/j.ajhg.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He D, Eskin E. Hap-seqX: expedite algorithm for haplotype phasing with imputation using sequence data. Gene. 2012;518:2–6. doi: 10.1016/j.gene.2012.11.093. [DOI] [PubMed] [Google Scholar]
- 10.Lloyd S. Least squares quantization in PCM. IEEE Trans Inf Theory. 1982;28:129–137. doi: 10.1109/TIT.1982.1056489. [DOI] [Google Scholar]
- 11.Ding C, He X. K-Means Clustering via Principal Component Analysis. New York, NY, USA: ACM Press; 2004. p. 29. [Google Scholar]
- 12.Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao M, Wang Q, Wang Q, Jia P, Zhao Z. Computational tools for copy number variation (CNV) detection using next-generation sequencing data: features and perspectives. BMC Bioinformatics. 2013;14:S1. doi: 10.1186/1471-2105-14-S11-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rabiner LR. A tutorial on hidden Markov models and selected applications in speech recognition. Proc IEEE. 1989;77:1–30. doi: 10.1109/5.18626. [DOI] [Google Scholar]
- 15.Robinson MD, Robinson MD, McCarthy DJ, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2009;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stajich JE, Block D, Boulez K, Brenner SE. The bioperl toolkit: Perl modules for the life sciences. Genome. 2002;12:1611–1618. doi: 10.1101/gr.361602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barnett DW, Garrison EK, Quinlan AR. BamTools: A C++ API and Toolkit for Analyzing and Managing BAM Files. Oxford, UK: Bioinformatics; 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drummond AJ, Ashton B, Buxton S, Cheung M. Drummond: Geneious v5. 4 - Google Scholar. Aukland, New Zealand: Biomatters Ltd; 2011. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1:Supplementary Material. Figure S1 Read alignment of cotton A-genome and d-genome reads to a common reference, rendered in IGV. Highlights indicate differences compared to the reference, so highlights in the upper sequence (A-genome) and the lack of those highlights in the lower sequence (D-genome) indicate SNPs between the two genomes. In this region, InterSnp identified 17 SNPs but SAMtools failed to identify any. Figure S2 Haplotypes identified by SAMtools and HapHunt, compared to the known haplotype. Figure S3 Phylogenetic tree. This neighbor-joining tree was built by neighbor based on SNPs identified by InterSnp. Then Geneious was used to render the actual tree. Table S1 The number of deletions identified in each accession (row), along with the percentage of those deletions that were shared with other members of the same species and with the entire group of samples. (DOCX 207 KB)