

Abstract
mTOR is a central nutrient sensor that signals a cell to grow and proliferate. Through distinct protein complexes it regulates different levels of available cellular energy substrates required for cell growth. One of the important functions of the complex is to maintain available amino acid pool by regulating protein translation. Dysregulation of mTOR pathway leads to aberrant protein translation which manifests into various pathological states. Our review focuses on the role mTOR signaling plays in protein translation and its physiological role. It also throws some light on available data that show translation dysregulation as a cause of pathological complexities like cancer and the available drugs that target the pathway for cancer treatment.
1. Overview of Translation Initiation
The regulation of translation is crucial for controlling cell growth and proliferation while translation dysregulation results in aberrant growth and tumorigenicity [1]. Translational control is mediated by the 7-methyl-GTP cap structure present at the 5′ termini of all eukaryotic mRNAs where multiprotein complexes are formed during translation initiation. The eukaryotic initiation factor 4G (eIF4G) acts as a scaffold protein for eukaryotic initiation factor 4E (eIF4E) and eukaryotic initiation factor 4A (eIF4A) to form a protein complex eIF4F, which binds to the cap structure and positions the ribosome near the 5′ terminus of mRNA [2]. Because of its low availability, the cap binding protein eIF4E is the rate limiting factor and inhibitory proteins, namely, eIF4E binding proteins (4E-BPs), regulate this process by binding to eIF4E which prevents its association with eIF4G, thus inhibiting protein translation [3]. Upon mitogenic stimulation 4E-BP1 is phosphorylated which is believed to cause its dissociation from eIF4E leading to the subsequent formation of the eIF4F complex, thus resulting in stimulation of translation initiation. Overall translation levels are therefore lowered when 4E-BP1 is active and this activity is thought to be regulated by mTOR dependent phosphorylation [4]. The mTOR activity itself is regulated by growth factors and amino acid availability as well as the energy status of the cell [4]. When mTOR activity is low, 4E-BP1 is hypophosphorylated which allows it to bind efficiently to eIF4E and block translation initiation whereas when mTOR activity is high, 4E-BP1 is phosphorylated causing it to release eIF4E, thus allowing cap dependent translation to begin [5].
2. mTOR
TOR is the target of rapamycin, a highly conserved serine/threonine kinase that plays a significant role in controlling cell growth and metabolism [6]. Rapamycin is an antifungal compound produced by the bacteria Streptomyces hygroscopicus that was isolated from a soil sample of Rapa Nui islands in the 1970s [7]. It is an anticancer compound that inhibits cell growth and proliferation [8] as well as a potent immunosuppressant that effectively prevents allograft rejection [9]. In 1990s, the isolation of yeast mutants that were resistant to growth inhibition by rapamycin led to the discovery of TOR which was later followed by the identification of the mammalian TOR (mTOR) as the physical target of rapamycin [10].
mTOR belongs to the phosphatidylinositol 3-kinase (PI3K) kinase-related kinase (PIKK) superfamily as the catalytic domain of PI3K has strong homology with the C-terminus of mTOR [11]. It consists of 2549 amino acids and several conserved domain structures. Tandemly repeated HEAT (for huntingtin, elongation factor 3 (EF3), a subunit of PP 2A, and TOR) motifs comprise its first 1200 amino acids [12]. These tandem HEAT repeats create a superhelical structure with large interfaces that facilitates protein-protein interaction. A FAT (FRAP, ATM, and TRRAP) domain lies downstream the HEAT repeat region which is followed by an FKPB12-rapamycin binding (FRB) domain. Rapamycin binds to FK506 binding protein 12 (FKBP12), thereby inhibiting its enzymatic activity as prolylisomerase, and this rapamycin-FKBP12 complex then binds to the FRB domain of mTOR and inhibits its activity [10]. The FRB domain is followed by a catalytic kinase domain (KD), an autoinhibitory or repressor domain (RD domain), and a FAT carboxy-terminal (FATC) domain (Figure 1). The FATC domain is crucial for the kinase activity of mTOR since deletion of even a single amino acid from this domain inhibits mTOR kinase activity. The FAT domain interacts with the FATC region and this interaction between the two domains might expose the catalytic domain, thus regulating the kinase activity of mTOR [13].
Figure 1.
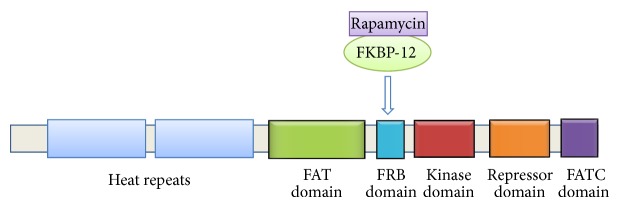
Schematic representation of mTOR domain structure.
The mTOR pathway regulates cell growth and proliferation in response to mitogen, nutrient, and energy status within the cell and is often dysregulated in various diseases, such as cancer and diabetes [14]. Recent findings have indicated a key role of mTOR signaling in tumorigenesis and activation of the mTOR pathway has been reported in several human cancers [15]. mTOR interacts with different proteins and forms two structurally and functionally distinct multiprotein complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) [16]. These complexes have important differences in their protein composition, rapamycin sensitivities, upstream signals, and substrates [17].
mTORC1. mTORC1 is composed of mTOR, raptor (regulatory associated protein of mTOR), mLST8 (also called G-protein β-subunit-like protein, GβL), PRAS40 (proline rich AKT/PKB substrate 40 kDA), and deptor (death domain containing mTOR interacting protein) (Figure 2) [18]. Rapamycin inhibits the mTORC1 activity since rapamycin bound to FKBP12 interacts with either free mTOR or mTOR in the mTORC1 complex causing conformational changes in mTORC1 which are responsible for rapamycin's effect on mTORC1 targets [19]. mTORC1 regulates translation by phosphorylating its various downstream effectors, with S6K1 and 4E-BP1 being the most imperative targets.
Figure 2.

mTORC1 and mTORC2 complexes, different interaction partners, and cellular functions.
Raptor is a 150 kDa mTORC1 binding protein, having a highly conserved N-terminal domain followed by three HEAT repeats and seven WD40 repeats in the C-terminal half [20, 21]. It binds strongly with mTOR via its N-terminal domain containing the HEAT repeats [20] and functions as a vital scaffold protein by linking the mTOR kinase with the mTORC1 substrates 4E-BP1 and S6K1, thus regulating mTORC1 activity in response to various mitogenic signals [22]. The phosphorylation status of raptor governs mTORC1 activity since phosphorylation of raptor on S722/792 by 5′ AMP-activated kinase (AMPK) inhibits mTORC1 whereas mTOR mediated phosphorylation of raptor on S863 is crucial for the activation of mTORC1 in response to mitogen stimulation [23, 24]. PRAS40, another subunit of mTORC1, is a negative regulator of mTORC1 function that was initially identified as an AKT substrate because of its direct phosphorylation at T246 by AKT when activated by insulin [25]. However, later studies showed that PRAS40 inhibits mTORC1 activity and is phosphorylated by mTORC1 on S183 when associated with mTORC1 via raptor and this binding is abolished by mutation of the S183 to aspartate [26, 27]. The role of mLST8 in mTORC1 function is unclear since loss of this protein does not affect mTORC1 activity in vivo [28].
mTORC1 activity is regulated by multiple growth factor signals such as insulin and nutrients. mTORC1 is activated by the PI3K/AKT pathway whereas TSC1-TSC2 (tuberous sclerosis complex) inhibits it [14]. Insulin binds to insulin receptor that recruits IRS (insulin receptor substrate) and activates phosphoinositide 3-kinase (PI3K) which induces cell proliferation and cell survival [29]. Besides other proteins, the downstream targets of PI3K include S6K1 and the serine/threonine kinase AKT [30]. Upon activation by growth factors, PI3K phosphorylates the D3 position of phosphatidylinositols producing the second messenger PtdIns(3,4,5)P3 that binds to the pleckstrin homology (PH) domain of AKT and translocates this kinase to the plasma membrane where it gets activated by upstream kinases [31]. PDK1 phosphorylates AKT at T308 and mTORC2 phosphorylates it at S473, both phosphorylations being necessary for the full activation of its kinase activity [32, 33]. Phosphatase and tensin homolog (PTEN) acts as a negative regulator of AKT activation by converting PtdIns(3,4,5)P3 into PtdIns(4,5)P2 which leads to a reduced recruitment of AKT to the cell membrane [34]. The negative regulators of mTORC1, TSC2, and PRAS40 are two of the several downstream substrates of activated AKT that are phosphorylated and inhibited by AKT [35]. Rheb (Ras homolog enriched in brain) GTPase directly binds to the catalytic domain of mTOR and acts as a positive regulator of mTORC1 kinase activity [26]. TSC1 and TSC2 proteins form a complex in vivo which negatively regulates mTORC1 by acting as Rheb GAP (GTPase activation protein) that converts Rheb into an inactive GDP bound form [36, 37]. In response to growth factors, AKT directly phosphorylates TSC2 on several distinct residues [35] that prevents the formation of TSC1-TSC2 complex, thus allowing GTPase Rheb to convert back into the GTP-bound active state [38] which leads to mTORC1 activation [39, 40]. Thus PI3K/AKT signalling pathway regulates mTORC1 by phosphorylation and inhibition of TSC2 which impairs its ability to inhibit Rheb, thereby resulting in the subsequent activation of Rheb and mTORC1.
mTORC2. mTORC2 is composed of RICTOR (rapamycin-insensitive companion of mTOR), mSin1 (mammalian stress-activated protein kinase (SAPK) interacting protein 1), Protor (protein observed with RICTOR), mLST8, deptor, and PRAS40 [16]. The mTORC2 complex was initially thought to be insensitive to acute rapamycin treatment since this RICTOR containing mTOR complex is not bound by FKBP12-rapamycin [41] but later studies demonstrated that prolonged treatment with rapamycin can inhibit the assembly and function of mTORC2 as well [42]. Deptor and mLST8 are components of both mTOR complexes and whereas deptor negatively regulates mTORC1 as well as mTORC2, mLST8 is essential only for mTORC2 function [28, 43]. mSin1 is another important subunit of mTORC2 that is indispensable for mTORC2 function and integrity because the interaction between RICTOR and mTOR is impaired in its absence [44, 45]. Protor interacts with RICTOR but it is not required for the assembly of other subunits of the mTORC2 complex [46].
The best-characterized function of mTORC2 is the activation of AKT by phosphorylating it on S473 which is essential for the regulation of various important cellular processes such as cell growth, proliferation, glucose metabolism, and apoptosis by activated AKT [33]. The phosphorylation of another conserved motif on AKT is also mediated by mTORC2 [47] and thus mTOR lies both upstream (mTORC2) and downstream (mTORC1) of AKT. The other important substrates of mTORC2 are SGK (serum and glucocorticoid-inducible kinase) and PKCα (protein kinase Cα) [48]. mTORC2 functions also include regulation of PKC maturation and stability [47] in addition to organization of actin cytoskeleton [41].
Feedback Regulation. Multiple negative feedback loops regulate the mTOR pathway and various studies have shown that mTORC1 negatively affects the insulin-PI3K-AKT pathway. The insulin receptor substrate 1 (IRS1) is directly phosphorylated by S6K1 at multiple sites which impairs its function and leads to inhibitory effects on the insulin-PI3K-AKT pathway. Growth factor receptor-bound protein 10 (GRB10) is a recently discovered substrate of mTOR that is activated by mTOR phosphorylation and negatively controls insulin-PI3K-AKT signaling pathway by inhibiting the insulin receptor in its active form [49]. The TSC/Rheb axis also regulates mTOR pathway via another feedback loop. Rheb activates mTORC1 but inhibits mTORC2 while TSC1/2 inhibits Rheb/mTORC1 but activates mTORC2 most likely by overcoming the negative feedback loop [50]. TSC2 also interacts with mTORC2 via RICTOR and activates the mTORC2 complex independently of its effects on mTORC1. mTORC2 is inhibited by mTORC1 through a negative feedback loop that involves S6K1, since RICTOR is inhibited by its S6K1 mediated phosphorylation at T1135 [51]. The PI3K-AKT-mTOR pathway is connected with the mitogen activated protein kinase (MAPK) pathway as AKT is activated by treatment with MEK inhibitors [52] and also by another feedback loop since the inhibition of mTORC1 results in the activation of the PI3K pathway as well as the MAPK pathway [53].
3. Downstream the mTOR Pathway
The best-characterized downstream effectors of mTORC1 which are phosphorylated by activated mTOR kinase are S6K1 and 4E-BP1.
3.1. S6K1
The ribosomal protein S6 kinase (S6K), a serine/threonine kinase that belongs to the AGC kinase family, is an important regulator of cell growth and cell size. The S6K family consists of two genes (S6K1 and S6K2) which share an overall 70% sequence homology [54]. S6Ks have five domains, the N-terminal regulatory domain, catalytic domain, linker domain, an autoinhibitory domain, and the C-terminal domain. S6K1 activity is regulated by sequential phosphorylations at multiple serine/threonine sites. The phosphorylation of four serine/threonine-proline sites in the autoinhibitory domain opens the kinase domain and relieves autoinhibition, which then allows subsequent phosphorylations of critical sites in the kinase domain [55]. mTOR kinase phosphorylates S6K1 at T389 which is the hydrophobic motif (HM) phosphorylation site [56, 57] followed by the PDK1 mediated phosphorylation at T229 present in the activation loop (AL) which leads to full activation of S6K1 [58]. The N-terminus of S6K1 has a conserved TOR signaling (TOS) motif that interacts with raptor and enables mTORC1 mediated phosphorylation of S6K1 [59]. The C-terminal region of S6K1 has an RSPRR motif which is important for the inhibitory role of this region, because a negative regulator of S6K1 binds to this motif [60] and a deletion mutant of S6K1 lacking the C-terminal region is phosphorylated by mTORC2 [61]. Although PDK1 is a rapamycin resistant kinase, T229 is also lost along with T389 upon rapamycin treatment therefore suggesting that T229 phosphorylation is dependent on T389 phosphorylation. On the other hand it has been reported that the T389 phosphorylation is not required for T229 phosphorylation [62]. More recently, we have reported that these phosphorylations might be coordinate instead of being sequential and the loss of these HM and AL phosphorylations is consequential and not because of inhibition of S6K1 by rapamycin [63]. Earlier we had also shown that the activity and rapamycin sensitivity of the exogenously expressed S6K1 are independent of any TOR dependent phosphorylations [64].
The ribosomal protein S6 (rpS6) was the first identified substrate of S6K. It is phosphorylated on five C-terminal serine sites by S6Ks in the following sequential order: S236 > S235 > S240 > S244 > S247 [65]. The study of rpS6P/− knock-in mice (in which alanine residues replaced the rpS6 phosphorylation sites) established that rpS6 phosphorylation is crucial for cell size and proliferation since cells isolated from rpS6P−/− displayed defective cell growth [66, 67]. S6K1 is very essential for regulating cell and body size as S6K1 null mice are much smaller at birth because of a decrease in the size of all organs [66] and a majority of dS6K null Drosophila show embryonic lethality [68]. The study of rpS6P−/− mice has also confirmed that the translation of mRNAs having a 5′ terminus oligopyrimidine tract (5′TOP mRNAs), a process that was earlier considered to be regulated by rpS6 phosphorylation, is not dependent on this event [69].
S6K1 targets a number of proteins that control protein translation (Figure 3). S6K1 regulates translation initiation by phosphorylating the cap binding complex component eIF4B at S422 [70]. It also controls initiation of translation by phosphorylating PDCD4, a tumor suppressor that is a negative regulator of eIF4A [71], and targets it for degradation by the ubiquitin ligase, βTRCP [72]. S6K1 inactivates eukaryotic elongation factor-2 kinase (eEF2K) which is a negative regulator of eukaryotic elongation factor 2 (eEF2), by phosphorylating it at S366, and thus regulates the elongation step of translation [73]. Eukaryotic translation initiation factor 4B (eIF4B) is also phosphorylated by S6K1 and this phosphorylation promotes the recruitment of eIF4B to eukaryotic initiation factor 4A (eIF4A) at the translation initiation complex where it functions as a cofactor of eIF4A and increases its processivity [74]. S6K1 controls transcription by phosphorylating the cAMP response element binding protein (CREB) isoform, CREMt, and also regulates ribosome biogenesis by phosphorylating the transcription factor UBF-1 which results in the activation of RNA pol1 mediated transcription of genes that encode rRNA [75, 76]. S6K1 directly phosphorylates estrogen receptor (ERα) to stimulate its transcriptional activity in breast cancer cell lines [77]. S6K1 also phosphorylates the p53 ubiquitin ligase, Mdm2, on S166 [78] and proapoptotic protein BAD, thus regulating cell survival [79]. S6K1 regulates mRNA processing since it phosphorylates SKAR (S6K1 Aly/REF-like target) which is involved in mRNA splicing [80].
Figure 3.

Major substrates and functions of ribosomal protein S6K.
3.2. 4E-BP1
4E-BP1, which belongs to a family of three small (10–12 kda) proteins that act as inhibitors of translation initiation by binding and inactivating eIF4E, is the second well-characterized mTORC1 target. In eukaryotic cells, the mRNAs transcribed by RNA pol II have a cap structure (m7Gppp) at their 5′ end. This cap structure has several functions such as pre-mRNA processing, mRNA stability, and its export and translation. As the mRNA is exported into the cytoplasm, its cap structure interacts with the eIF4F complex, whose main function is to facilitate the recruitment of ribosome to the 5′ end of mRNA and the consequent initiation of translation. The eIF4 complex is composed of three polypeptides: eIF4E (the cap binding protein), eIF4A (an RNA helicase), and eIF4G (scaffolding protein). eIF4G interacts simultaneously with both eIF4E and eIF4A along with eIF3 which is a multiprotein complex that is associated with the 43S ribosomal particle. Briefly, the eIF4F complex facilitates the association of mRNA with ribosome by interacting with mRNA 5′ cap via eIF4E, unwinds the mRNA secondary structures via eIF4A, and recruits the ribosome through eIF4G-eIF3 interaction, thus acting as a cap dependent translation initiation factor [81].
eIF4E is regulated by phosphorylation as well as its sequestration by eIF4E binding proteins (4E-BPs). These eIF4E inhibitory proteins as well as eIF4G possess a conserved amino acid motif (YxxxxLΦ) which is the eIF4E binding motif [82]. Thus 4E-BPs compete with eIF4G for binding to the same site on eIF4E, thereby preventing the assembly of eIF4F complex and inhibiting initiation of cap dependent translation [83]. eIF4E is phosphorylated at S209 by mitogen activated protein kinase (MAPK) interacting protein kinase Mnk1/2 which uses a docking site in the carboxy-terminus of eIF4G to phosphorylate eIF4E, thus making it certain that eIF4E is phosphorylated only after the assembly of the eIF4F complex [84].
mTORC1 phosphorylates 4E-BP1 at several residues which promotes the dissociation of eIF4E from 4E-BP1 consequently mitigating the inhibitory effect of 4E-BP1 on eIF4E dependent translation initiation whereas the inhibition of mTOR by rapamycin is believed to cause 4E-BP1 dephosphorylation, which results in inhibition of protein translation (Figure 4) [85]. The main phosphorylation sites that have been identified in 4E-BP1 are T37, T46, S65, T70, S83, S101, and S112 [86] but the ability of 4E-BP1 to bind and inhibit eIF4E is mainly regulated by the phosphorylation of four residues: T37, T46, S65, and T70. In HEK293 cells, T37 and T46 were earlier shown to be phosphorylated significantly even in the absence of serum and the phosphorylation at these residues increased slightly with serum stimulation [87]. The phosphorylation of T37 and T46 was reported to be required for the phosphorylation of some unknown serum sensitive sites since the mutation of T37 and T46 to alanine residues prevented the phosphorylation of these serum sensitive sites while their substitution by glutamic acid residues restored the phosphorylation of the same serum sensitive sites to some extent [87]. Subsequently S65 and T70 were identified as the serum sensitive phosphorylation sites of 4E-BP1 and the phosphorylation of 4E-BP1 in HEK 293 was reported to occur in a hierarchical order. The phosphorylation of T36/T47 acts as the priming step which is followed by the phosphorylation of T70 and finally S65 [5]. However during ischemic stress in brain tissues, a new hierarchical phosphorylation for 4E-BP1 has been proposed in which T70 phosphorylation is the priming event for subsequent phosphorylation of T36/T47 [88].
Figure 4.
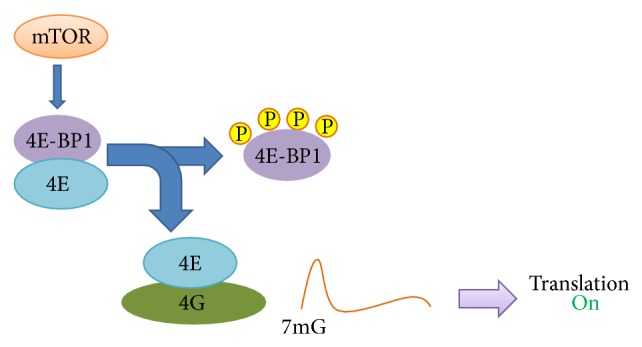
Regulation of cap dependent translation.
S101 is necessary for phosphorylation of S65 and S112 has been shown to affect binding to eIF4E although it does not affect phosphorylation at other sites. The kinase responsible for the phosphorylation of these two serine sites has not been identified. S83 is conserved in all three 4E-BPs but does not seem to control translation initiation. The exact function and regulation of each phosphorylation site and which phosphorylations are dependent on one another are still not very well understood [89].
Besides the eIF4E binding motif, there are two other key regulatory motifs in 4E-BP1, a RAIP motif, named after its sequence Arg-Ala-Ile-Pro at the N-terminus, and an mTOR signaling (TOS) motif at the C-terminus (Figure 5) [90, 91]. The TOS motif is also present in S6K1 and PRAS40, the other substrates of mTORC1 [27], and this motif is required for the interaction of 4E-BP1 with raptor since the mutation of specific residues within this motif abrogates the binding of raptor to 4E-BP1 [91, 92]. The RAIP motif has been found to be unable to bind with raptor [91] although another study reported that raptor does not bind to a 4E-BP variant with disrupted RAIP motif [93]. This motif is necessary for phosphorylation of residues present in both the N-terminus and the C-terminus of 4E-BP1. On the other hand, the TOS motif primarily affects the phosphorylation of S65 and T70, while the phosphorylation of the N-terminal T36/T47 residues is not affected by inactivation of the TOS motif to a great extent [91]. Also the phosphorylation of these sites is rather insensitive to rapamycin which could suggest that these phosphorylations are possibly mTOR independent however since these phosphorylations are inhibited by amino acids starvation of cells and by mTOR kinase inhibitors like wortmannin, activated by Rheb (an mTOR activator), and are decreased in mTOR knockdown cells which suggests that the phosphorylation of these N-terminal residues is mediated by mTOR but through a raptor independent mechanism [94]. Recently it has been shown that, in addition to the YxxxxLΦ motif, other conserved regions in the N- and C-termini of 4E-BP1 might also be involved in its binding to eIF4E [95, 96].
Figure 5.
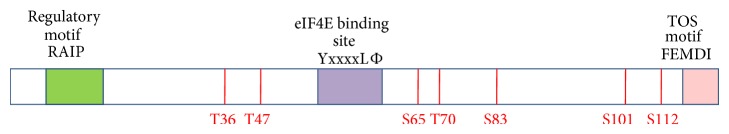
Important domains and phosphorylation sites of 4E-BP1.
Although mTORC1 has been implicated in the regulation of 4E-BP1 phosphorylation there are various conflicting reports. For instance, rapamycin treatment leads to loss of phosphorylation at S65 and T70 on 4E-BP1 while mTORC1 has a modest effect on the phosphorylation of these sites in vitro [12]. On the other hand T37 and T46 are phosphorylated in vitro by mTORC1 but these sites are considered rapamycin insensitive in cells [12]. It is possible that rapamycin does not inhibit mTORC1 dependent phosphorylation of 4E-BP1 completely. Also it is not clear whether the rapamycin sensitive sites of 4E-BP1 are directly phosphorylated by mTOR in vivo; moreover some studies suggest that another unidentified 4E-BP1 kinase might exist [94]. In different cell types originating from solid or hematological tumors where 4E-BP1 phosphorylation becomes resistant to rapamycin, the Pim-2 serine/threonine kinase has been found to phosphorylate 4E-BP1 at S65 in a raptor-independent and rapamycin-insensitive way [97]. Whether this Pim-2 kinase dependent and rapamycin insensitive phosphorylation of 4E-BP1 is carried out by a rapamycin insensitive mTORC1 complex or somehow by the mTORC2 complex is not known [98].
There are some major discrepancies regarding the identity of phosphorylation sites required for the release of 4E-BP1 from eIF4E. T37 and T46 phosphorylations have been reported to have either little effect on eIF4E binding [85, 87] or to cause a major reduction in eIF4E binding affinity [99] while the effect of S65 phosphorylation on 4E-BP1 binding to eIF4E also remains in question [86, 100]. Moreover a 4E-BP1 variant that mimics hyperphosphorylation of the four main phosphorylation sites does not release eIF4E from 4E-BP1 in sea urchin, therefore suggesting that other mechanisms in addition to the phosphorylation at these four sites might play a role in 4E-BP1 binding to eIF4E [101].
Rapamycin resistant phosphorylation of 4E-BP1 has been reported in regenerating rat livers where rapamycin inhibits activation of S6K1 in response to partial hepatectomy, but 4E-BP1 phosphorylation remains uninhibited, suggesting that the effect of rapamycin on 4E-BP1 function in vivo can be significantly different from its effect in cultured cells [102]. Rapamycin has also been reported to differentially regulate S6K1 in comparison with 4E-BP1 in various cell lines including HeLa, MEFs, and HEK293, where rapamycin initially decreased 4E-BP1 phosphorylation but this decrease recovered within 6 h while S6K1 phosphorylation continued to be inhibited and by 12 h after treatment since 4E-BP1 was mostly hyperphosphorylated, it dissociated from eIF4E leading to a recovery in cap dependent translation even though S6K1 inhibition by rapamycin continued [103]. Several mechanisms such as association/dissociation of mTOR associated proteins or posttranslational modifications on mTORC1 have been put forward to explain this phenomenon of differential phosphorylation [104]. Torin is an ATP-competitive inhibitor that inhibits 4E-BP1 phosphorylation more strongly than rapamycin in various human cell lines. It inhibits phosphorylation of T37/T46 as well as S65 but fails to inhibit T70 phosphorylation in MEFs. These studies with Torin suggest that rapamycin resistance of mTORC1 might be a general feature of a good number of mammalian systems [105]. The activation of the tumour suppressor protein p53 induces a proteasome mediated specific cleavage of 4E-BP1 that gives rise to an N-terminally truncated, completely unphosphorylated and more stable form of 4E-BP1 that interacts with eIF4E in preference to full-length 4E-BP1, which results in long-term unavailability of eIF4E, thereby contributing to the growth-inhibitory and proapoptotic effects of p53 [106, 107]. 4E-BP1 activity is regulated in apoptosis since treatment of cells with DNA damaging drugs such as etoposide and staurosporine results in dephosphorylation of 4E-BP1 due to inhibition of mTOR signaling which impairs cap dependent protein translation and drives the IRES-mediated cap independent protein synthesis [108]. One mechanism through which DNA damage could result in the inhibition of mTOR involves p53 [109] while another different link between DNA damage and mTOR signaling is the tyrosine kinase c-Abl, which once activated by DNA damage can inactivate mTOR [110].
Various growth-inhibitory conditions and physiological stresses also lead to shutdown of mTOR signaling and 4E-BP1 dephosphorylation, which indicates that this may be a common response of cells to unfavorable conditions [111].
4. Other Cellular Processes Downstream of mTORC1
mTORC1 upregulates protein synthesis by various other mechanisms. Maf1 which is a Pol III repressor is inhibited by mTORC1 phosphorylation and thus induces 5S rRNA and tRNA transcription [112, 113]. The regulatory element tripartite motif containing protein-24 (TIF-1A) is activated by mTORC1, which enhances the expression of ribosomal RNA (rRNA) by promoting TIF-1A interaction with RNA Pol I [114].
Proliferating cells also require lipids in addition to protein to synthesize plasma membranes and other macromolecules and mTORC1 controls this synthesis of lipids [115] via the transcription factors SREBP1/2 (sterol regulatory element binding protein 1/2) that regulate the expression of genes involved in fatty acid biosynthesis. The inhibition of mTORC1 reduces SREBP1/2 levels which results in the downregulation of lipogenic genes [116–118]. Lipin-1 lowers SREBP1/2 levels inside the nucleus and mTORC1 mediated phosphorylation of Lipin-1 prevents it from entering the nucleus, thus suppressing this inhibition [119]. The peroxisome proliferator-activated receptor γ (PPAR-γ) is the main regulator of adipogenesis which is also activated by mTORC1 [120, 121]. mTORC1 positively regulates cellular metabolism and ATP production by activating the transcription and translation of hypoxia inducible factor 1α (HIF1α) which is a positive regulator of many glycolytic genes [116, 122].
mTORC1 is also an important negative regulator of autophagy, a eukaryotic homeostatic process in which various cytoplasmic components such as damaged organelles and intracellular pathogens are degraded inside lysosomes [123]. mTOR induces autophagy in response to reduced growth factor signalling, starvation, and other metabolic and genotoxic stresses [124] which leads to the formation of phagophores, inside which the lysosomal hydrolases degrade the targeted substrates [125]. During physiological conditions, the phagophore formation is inhibited by mTORC1, since it directly interacts with and phosphorylates the Ulk1 kinase complex (Ulk1-Atg13-FIP200-Atg101) which is required for the initiation of autophagy [126, 127]. mTORC1 regulates WIPI2 (mammalian orthologue of Atg18), which is also essential for phagophore formation [128], as well as DAP1 (death associated protein 1), an inhibitor of autophagy [129]. During unfavourable conditions, such as cell starvation or rapamycin mediated inhibition of mTOR kinase, the Ulk1 complex is released from mTOR, thereby allowing it to associate with the membranes from which phagophores are formed [130].
5. The mTOR Pathway and Cancer
The mTOR pathway is related to tumorigenesis because of its vital role in cell growth, proliferation, and metabolism. The aberrant activation of mTOR pathway either by loss of tumor suppressors or activation of oncogenes promotes tumor growth in various malignant cell lines. The upstream and downstream elements of the mTOR pathway are dysregulated in different human cancers. The overexpression of different growth factor receptors like IGFR (insulin like growth factor receptor) and HER2 (human epidermal growth factor receptor 2) and mutations in the PI3K can lead to activation of AKT and mTOR pathways [131]. This mTOR activation causes an increase in ribosome biogenesis that promotes cell proliferation by providing the machinery which is required by cells to maintain high levels of growth [132].
The downstream effectors of mTORC1, 4E-BP1, eIF4E, and S6K1 are also associated with various malignancies. eIF4E is an oncogene as it is overexpressed in many human cancers with poor prognosis and its overexpression results in transformation of cells in vivo [133, 134]. eIF4E promotes the translation of specific mRNAs that code for prooncogenic proteins which promote cell survival and cell-cycle progression, energy metabolism, and metastasis, thus affecting cell proliferation and tumorigenesis [135]. Dysregulated expression as well as increased phosphorylation of 4E-BPs in cancer also results in poor patient prognosis and the loss of 4E-BP1 with the resulting activation of cap dependent translation promotes cell-cycle progression and cell proliferation in culture [136, 137] whereas overexpression of constitutively active 4E-BP1 suppresses tumor growth in vivo [138, 139]. The increase in cell proliferation by downregulation of 4E-BP1 might be due to the removal of the inhibition of translation of mRNAs that encode proteins such as vimentin, Y-box protein, and CD44, which promote cell growth, proliferation, and metastasis [140]. S6K1 is also overexpressed in lung and ovary cancer [141] and its gene expression has been found to be upregulated in brain tumors [142].
mTORC2 also plays a role in cancer since the mTORC2 subunit RICTOR is also overexpressed in multiple cancer types [143] and its overexpression increases mTORC2 activity which causes the cancer cells to become more proliferative and invasive [143, 144]. Dysregulation of several other elements of the mTOR pathway also results in tumorigenesis. The loss of the tumor suppressor PTEN decreases its expression in many human cancers [145] and in mice results in the development of prostate cancer [146]. PROTOR 1 is downregulated in human breast tumors and cell lines [147] whereas deptor is overexpressed in various tumors such as myelomas and hepatocellular carcinomas [43, 148] and RHEB is overexpressed in some human lymphomas [149, 150].
Lipid synthesis is increased in proliferating cancer cells and PI3K/AKT mediated high glycolytic rates produce ATP and the other building blocks required for lipid synthesis [151]. The mTORC2 substrate GSK3 (glycogen synthase kinase 3) connects AKT to lipid synthesis since GSK phosphorylates lipogenic transcription factor SREBP (sterol responsive element binding protein) and targets it for protein degradation which is opposed by AKT mediated phosphorylation and inactivation of GSK. Thus dysregulated AKT/mTOR pathway can promote SREBP expression and activity which in turn enhances cancer cell lipid biosynthesis [116].
Activated mTOR pathway is also related to various familial cancer syndromes. The loss of tumor suppressor LKB1 (liver kinase B1), which is a key kinase for activating AMPK [152], results in Pentz-Jeghers syndrome [153]. Mutations in TSC1 or TSC2 cause tuberosis sclerosis [154], another familial cancer syndrome, and PTEN mutations result in Cowden's syndrome [155]. These syndromes result in benign tumors which may progress to malignancies.
6. Targeting the mTOR Pathway in Cancer
mTOR inhibitors can be broadly grouped into two classes: the allosteric inhibitors of mTORC1 (rapamycin and rapalogs) and the mTOR kinase inhibitors.
6.1. Rapamycin and Rapalogs
Rapamycin was the first mTOR inhibitor but despite its antitumor activity in preclinical models it was not successful as an anticancer drug. Subsequently several analogs of rapamycin, now called rapalogs, with better solubility and pharmacokinetic properties were synthesized such as temsirolimus, everolimus, and deforolimus. Like rapamycin these rapalogs form a complex with intracellular receptor FKBP12 which binds to mTOR and inhibits mTORC1 downstream signaling. In phase II and III clinical trials, everolimus and temsirolimus were effective in treating RCC (renal cell carcinoma), neuroendocrine tumors, and Mantle cell lymphoma and both have been approved by FDA for treatment of RCC [156]. However, the success of rapalogs as anticancer monotherapies was limited because the FKBP12-rapamycin complex cannot bind to mTORC2 [41] and also due to the activation of alternative signaling pathways such as the MAPK pathway [157] and AKT signaling resulting from the loss of a negative-feedback mechanism. Another reason why rapamycin and its analogs have limited efficacy in cancer treatment is because these drugs only partially inhibit the phosphorylation of 4E-BP1 [105, 158–160]. In AML, for example, the phosphorylation of S6K1 on T389 is abrogated by RAD001 while phosphorylation of 4E-BP1 on S65 residue remains unaffected, thus failing to inhibit mRNA translation and the assembly of eIF4F complexes [97].
6.2. Catalytic mTOR Inhibitors
In order to overcome the limitations of rapalogs, ATP-competitive mTOR kinase inhibitors were developed that directly target the mTOR catalytic site and inhibit phosphorylation of mTORC1 substrates S6K1 and 4E-BP1 as well as phosphorylation of the mTORC2 substrate AKT and mTORC2 [161]. These inhibitors include Ku-006379, Torin, PP242, PP40, OSI027, AZD2014, and AZD8055 which show better antitumorigenic effects in comparison with rapalogs because they bind to the ATP binding site of mTOR, thus inhibiting the catalytic activity of mTORC1 and mTORC2 [105, 159, 162]. The anticancer activity of these inhibitors has been superior to rapamycin in preclinical trials due to effective blocking of cell proliferation, 4E-BP1 phosphorylation, and protein translation [159]. Two such inhibitors, PP242 and PP40 which inhibit the insulin stimulated S473 phosphorylation of AKT, also inhibit protein synthesis and cell proliferation [159]. PP242 and OSI-027 show superior anticancer effects in BCR-ABL expressing cell lines [163]. OSI-027 is more potent in blocking 4E-BP1 phosphorylation and mRNA translation in acute myeloid leukemia in comparison with rapamycin [164]. Torin 1 is a selective ATP competitive inhibitor that is also more effective in inhibiting cell growth and proliferation than rapamycin [105]. AZD8055 potently (IC < 1 nM) inhibits the rapamycin-resistant T37/46 phosphorylation sites on 4E-BP1, resulting in significant inhibition of cap dependent translation. In vitro, AZD8055 effectively inhibits cell proliferation and induces autophagy and, in vivo, AZD8055 inhibits tumor growth [158]. In addition, it combines well with the MEK1/2 inhibitor selumetinib in preclinical studies [165]. Significant phosphorylation of 4E-BP1 at T37/T46 and S65 is still observed in mTORC2 deficient Sin-1 knockout MEFs when treated with rapamycin, even though the hydrophobic motifs of both AKT and S6K1 are not phosphorylated due to absence of mTOR activity in both of its complexes. In contrast, exposure of Sin-1 knockout MEFs to Ku-0063794 dephosphorylates 4E-BP1 to a much greater extent than rapamycin [160]. Treatment of cells with Torin and PP242 inhibitors indicates that antiproliferative effects of these mTOR kinase inhibitors are primarily through disruption of mTORC1 functions which are resistant to rapamycin [118].
The drawback with mTOR kinase inhibitors as with rapalogs is that the mTORC1 feedback loop can be relieved which leads to activation of PI3K or MAP kinase signaling [166]. Thus dual specificity drugs that target both mTOR function and AKT activation can improve antitumor activity. Also rapalogs as well as catalytic inhibitors can lead to induction of autophagy which can be anti- as well as protumorigenic depending upon stimulus [167]. Autophagy has been recently shown to enable survival to mTOR inhibition [168]. Therefore combination treatment of cancers with rapalogs or catalytic inhibitors along with autophagy inhibitor may be a better strategy [18].
Another mechanism that explains the resistance to mTOR kinase inhibitors is that cancer cells downregulate the expression of 4E-BPs which leads to an increase in the eIF4E/4E-BP1 ratio, and it is this change in eIF4E/4E-BP1 stoichiometry that limits the sensitivity of cancer cells to catalytic site TOR inhibitors suggesting that the eIF4E/4E-BP1 ratio might act as a predictive marker for treatments using catalytic TOR inhibitors [169].
7. Dual PI3K/mTOR Inhibitors
Dual PI3K/mTOR inhibitors were developed because of the above concerns over mTOR inhibitors. This was made possible because of the high homology that is shared by the kinase domains of PI3K and mTOR [16]. These molecules inhibit mTORC1, mTORC2, and PI3K, thus inhibiting the phosphorylation of AKT, S6K1, and 4E-BP1, and are therefore attractive drugs for targeting cancers driven by PI3K activation [170]. These inhibitors include XL-765, PI-103, and NVP-BEZ235 which are undergoing phase I/II clinical trials [171, 172]. PI-103 and NVP-BEZ235 have been found to suppress AKT as well as S6K1 in breast tumors and leukemia cells [173], although some studies suggest that such broad inhibition of cellular signalling may also impair growth of normal cells [174].
8. Concluding Remarks
mTOR pathway plays a key role in nutrient homeostasis that regulates cellular growth and proliferation. mTOR regulates protein translation through effector molecules S6K1 and 4E-BP1. Dysregulation of the pathway is complicated by cross-talk between mTOR and other signalling pathways like AKT and PI3 kinase. Though mTOR pathway dysregulation manifests into various pathological states, it is not the only candidate responsible for the effect. Further downstream signalling is very complex that is understood by the fact that therapeutic regimens that target only mTOR are not very effective to treat cancer. Dual inhibitors that target both mTOR and PI3 kinase have shown promise in combating the disease.
Acknowledgments
Infrastructural grants in favour of Khurshid I. Andrabi under FIST program and Special Assistance grant under SAP program, from DST, India, and UGC, India, are gratefully acknowledged. Individual fellowships in favour of Mehvish Showkat (20-6/2009) from University Grants Commission (UGC) and Mushtaq A. Beigh (SB/FT/LS-362/2012) from SERB, DST, are duly acknowledged.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors’ Contribution
Mehvish Showkat and Mushtaq A. Beigh contributed equally.
References
- 1.Sonenberg N. Translation factors as effectors of cell growth and tumorigenesis. Current Opinion in Cell Biology. 1993;5(6):955–960. doi: 10.1016/0955-0674(93)90076-3. [DOI] [PubMed] [Google Scholar]
- 2.Pain V. M. Initiation of protein synthesis in eukaryotic cells. European Journal of Biochemistry. 1996;236(3):747–771. doi: 10.1111/j.1432-1033.1996.00747.x. [DOI] [PubMed] [Google Scholar]
- 3.Pause A., Belsham G. J., Gingras A.-C., Donzé O., Lin T.-A., Lawrence J. C., Jr., Sonenberg N. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature. 1994;371(6500):762–767. doi: 10.1038/371762a0. [DOI] [PubMed] [Google Scholar]
- 4.Hay N., Sonenberg N. Upstream and downstream of mTOR. Genes and Development. 2004;18(16):1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 5.Gingras A.-C., Raught B., Gygi S. P., Niedzwiecka A., Miron M., Burley S. K., Polakiewicz R. D., Wyslouch-Cieszynska A., Aebersold R., Sonenberg N. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes and Development. 2001;15(21):2852–2864. doi: 10.1101/gad.912401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Betz C., Hall M. N. Where is mTOR and what is it doing there? The Journal of Cell Biology. 2013;203(4):563–574. doi: 10.1083/jcb.201306041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vezina C., Kudelski A., Sehgal S. N. Rapamycin (AY 22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. Journal of Antibiotics. 1975;28(10):721–726. doi: 10.7164/antibiotics.28.721. [DOI] [PubMed] [Google Scholar]
- 8.Dazert E., Hall M. N. MTOR signaling in disease. Current Opinion in Cell Biology. 2011;23(6):744–755. doi: 10.1016/j.ceb.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 9.Abraham R. T., Wiederrecht G. J. Immunopharmacology of rapamycin. Annual Review of Immunology. 1996;14:483–510. doi: 10.1146/annurev.immunol.14.1.483. [DOI] [PubMed] [Google Scholar]
- 10.Brown E. J., Albers M. W., Ichikawa K., Keith C. T., Lane W. S., Schreiber S. L. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369(6483):756–758. doi: 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- 11.Kunz J., Henriquez R., Schneider U., Deuter-Reinhard M., Movva N. R., Hall M. N. Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell. 1993;73(3):585–596. doi: 10.1016/0092-8674(93)90144-F. [DOI] [PubMed] [Google Scholar]
- 12.Gingras A.-C., Raught B., Sonenberg N. Regulation of translation initiation by FRAP/mTOR. Genes and Development. 2001;15(7):807–826. doi: 10.1101/gad.887201. [DOI] [PubMed] [Google Scholar]
- 13.Perry J., Kleckner N. The ATRs, ATMs, and TORs are giant HEAT repeat proteins. Cell. 2003;112(2):151–155. doi: 10.1016/S0092-8674(03)00033-3. [DOI] [PubMed] [Google Scholar]
- 14.Pópulo H., Lopes J. M., Soares P. The mTOR signalling pathway in human cancer. International Journal of Molecular Sciences. 2012;13(2):1886–1918. doi: 10.3390/ijms13021886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou H., Huang S. MTOR signaling in cancer cell motility and tumor metastasis. Critical Reviews in Eukaryotic Gene Expression. 2010;20(1):1–16. doi: 10.1615/CritRevEukarGeneExpr.v20.i1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zoncu R., Efeyan A., Sabatini D. M. MTOR: from growth signal integration to cancer, diabetes and ageing. Nature Reviews Molecular Cell Biology. 2011;12(1):21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laplante M., Sabatini D. M. mTOR signaling at a glance. Journal of Cell Science. 2009;122(20):3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beauchamp E. M., Platanias L. C. The evolution of the TOR pathway and its role in cancer. Oncogene. 2013;32(34):3923–3932. doi: 10.1038/onc.2012.567. [DOI] [PubMed] [Google Scholar]
- 19.Fingar D. C., Salama S., Tsou C., Harlow E., Blenis J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes and Development. 2002;16(12):1472–1487. doi: 10.1101/gad.995802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim D.-H., Sarbassov D. D., Ali S. M., et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110(2):163–175. doi: 10.1016/S0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 21.Hara K., Maruki Y., Long X., Yoshino K.-I., Oshiro N., Hidayat S., Tokunaga C., Avruch J., Yonezawa K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110(2):177–189. doi: 10.1016/S0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- 22.Foster K. G., Acosta-Jaquez H. A., Romeo Y., Ekim B., Soliman G. A., Carriere A., Roux P. P., Ballif B. A., Fingar D. C. Regulation of mTOR complex 1 (mTORC1) by raptor Ser863 and multisite phosphorylation. The Journal of Biological Chemistry. 2010;285(1):80–94. doi: 10.1074/jbc.M109.029637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gwinn D. M., Shackelford D. B., Egan D. F., et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Molecular Cell. 2008;30(2):214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang L., Lawrence J. C., Jr., Sturgill T. W., Harris T. E. Mammalian target of rapamycin complex 1 (mTORC1) activity is associated with phosphorylation of raptorby mTOR. The Journal of Biological Chemistry. 2009;284(22):14693–14697. doi: 10.1074/jbc.C109.002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kovacina K. S., Park G. Y., Bae S. S., et al. Identification of a proline-rich Akt substrate as a 14-3-3 binding partner. The Journal of Biological Chemistry. 2003;278(12):10189–10194. doi: 10.1074/jbc.M210837200. [DOI] [PubMed] [Google Scholar]
- 26.Sancak Y., Thoreen C. C., Peterson T. R., Lindquist R. A., Kang S. A., Spooner E., Carr S. A., Sabatini D. M. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Molecular Cell. 2007;25(6):903–915. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 27.Oshiro N., Takahashi R., Yoshino K.-I., Tanimura K., Nakashima A., Eguchi S., Miyamoto T., Hara K., Takehana K., Avruch J., Kikkawa U., Yonezawa K. The proline-rich Akt substrate of 40 kDa (PRAS40) is a physiological substrate of mammalian target of rapamycin complex 1. The Journal of Biological Chemistry. 2007;282(28):20329–20339. doi: 10.1074/jbc.M702636200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guertin D. A., Stevens D. M., Thoreen C. C., Burds A. A., Kalaany N. Y., Moffat J., Brown M., Fitzgerald K. J., Sabatini D. M. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKC, but not S6K1. Developmental Cell. 2006;11(6):859–871. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 29.Vanhaesebroeck B., Leevers S. J., Panayotou G., Waterfield M. D. Phosphoinositide 3-kinases: a conserved family of signal transducers. Trends in Biochemical Sciences. 1997;22(7):267–272. doi: 10.1016/S0968-0004(97)01061-X. [DOI] [PubMed] [Google Scholar]
- 30.Franke T. F., Kaplan D. R., Cantley L. C. PI3K: downstream AKTion blocks apoptosis. Cell. 1997;88(4):435–437. doi: 10.1016/S0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- 31.Hemmings B. A. Akt signaling: linking membrane events to life and death decisions. Science. 1997;275(5300):628–630. doi: 10.1126/science.275.5300.628. [DOI] [PubMed] [Google Scholar]
- 32.Alessi D. R., James S. R., Downes C. P., Holmes A. B., Gaffney P. R. J., Reese C. B., Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα . Current Biology. 1997;7(4):261–269. doi: 10.1016/S0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 33.Sarbassov D. D., Guertin D. A., Ali S. M., Sabatini D. M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 34.Stambolic V., Suzuki A., de la Pompa J. L., et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95(1):29–39. doi: 10.1016/S0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 35.Inoki K., Li Y., Zhu T., Wu J., Guan K.-L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nature Cell Biology. 2002;4(9):648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 36.Garami A., Zwartkruis F. J. T., Nobukuni T., Joaquin M., Roccio M., Stocker H., Kozma S. C., Hafen E., Bos J. L., Thomas G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Molecular Cell. 2003;11(6):1457–1466. doi: 10.1016/S1097-2765(03)00220-X. [DOI] [PubMed] [Google Scholar]
- 37.Inoki K., Li Y., Xu T., Guan K.-L. Rheb GTpase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes & Development. 2003;17(15):1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Um S. H., D'Alessio D., Thomas G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metabolism. 2006;3(6):393–402. doi: 10.1016/j.cmet.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 39.Inoki K., Zhu T., Guan K.-L. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–590. doi: 10.1016/S0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 40.Long X., Lin Y., Ortiz-Vega S., Yonezawa K., Avruch J. Rheb binds and regulates the mTOR kinase. Current Biology. 2005;15(8):702–713. doi: 10.1016/j.cub.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 41.Jacinto E., Loewith R., Schmidt A., Lin S., Rüegg M. A., Hall A., Hall M. N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nature Cell Biology. 2004;6(11):1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 42.Sarbassov D. D., Ali S. M., Sengupta S., Sheen J.-H., Hsu P. P., Bagley A. F., Markhard A. L., Sabatini D. M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Molecular Cell. 2006;22(2):159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 43.Peterson T. R., Laplante M., Thoreen C. C., et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137(5):873–886. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frias M. A., Thoreen C. C., Jaffe J. D., Schroder W., Sculley T., Carr S. A., Sabatini D. M. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Current Biology. 2006;16(18):1865–1870. doi: 10.1016/j.cub.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 45.Jacinto E., Facchinetti V., Liu D., Soto N., Wei S., Jung S. Y., Huang Q., Qin J., Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127(1):125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 46.Pearce L. R., Huang X., Boudeau J., et al. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochemical Journal. 2007;405(3):513–522. doi: 10.1042/BJ20070540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ikenoue T., Inoki K., Yang Q., Zhou X., Guan K.-L. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. The EMBO Journal. 2008;27(14):1919–1931. doi: 10.1038/emboj.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.García-Martínez J. M., Alessi D. R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1) Biochemical Journal. 2008;416(3):375–385. doi: 10.1042/BJ20081668. [DOI] [PubMed] [Google Scholar]
- 49.Yu Y., Yoon S.-O., Poulogiannis G., et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science. 2011;332(6035):1322–1326. doi: 10.1126/science.1199484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang Q., Inoki K., Kim E., Guan K.-L. TSC1/TSC2 and Rheb have different effects on TORC1 and TORC2 activity. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(18):6811–6816. doi: 10.1073/pnas.0602282103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dibble C. C., Asara J. M., Manning B. D. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Molecular and Cellular Biology. 2009;29(21):5657–5670. doi: 10.1128/MCB.00735-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Buck E., Eyzaguirre A., Rosenfeld-Franklin M., Thomson S., Mulvihill M., Barr S., Brown E., O'Connor M., Yao Y., Pachter J., Miglarese M., Epstein D., Iwata K. K., Haley J. D., Gibson N. W., Ji Q.-S. Feedback mechanisms promote cooperativity for small molecule inhibitors of epidermal and insulin-like growth factor receptors. Cancer Research. 2008;68(20):8322–8332. doi: 10.1158/0008-5472.CAN-07-6720. [DOI] [PubMed] [Google Scholar]
- 53.Carracedo A., Ma L., Teruya-Feldstein J., et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. The Journal of Clinical Investigation. 2008;118(9):3065–3074. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fenton T. R., Gout I. T. Functions and regulation of the 70 kDa ribosomal S6 kinases. International Journal of Biochemistry and Cell Biology. 2011;43(1):47–59. doi: 10.1016/j.biocel.2010.09.018. [DOI] [PubMed] [Google Scholar]
- 55.Dufner A., Thomas G. Ribosomal S6 kinase signaling and the control of translation. Experimental Cell Research. 1999;253(1):100–109. doi: 10.1006/excr.1999.4683. [DOI] [PubMed] [Google Scholar]
- 56.Alessi D., Kozlowski M. T., Weng Q.-P., Morrice N., Avruch J. 3-phosphoinositide-dependent protein kinase 1 (PDK1) phosphorylates and activates the p70 S6 kinase in vivo and in vitro. Current Biology. 1998;8(2):69–81. doi: 10.1016/S0960-9822(98)70037-5. [DOI] [PubMed] [Google Scholar]
- 57.Pullen N., Dennis P. B., Andjelkovic M., Dufner A., Kozma S. C., Hemmings B. A., Thomas G. Phosphorylation and activation of p70(s6k) by PDK1. Science. 1998;279(5351):707–710. doi: 10.1126/science.279.5351.707. [DOI] [PubMed] [Google Scholar]
- 58.Avruch J., Belham C., Weng Q., Hara K., Yonezawa K. The p70 S6 kinase integrates nutrient and growth signals to control translational capacity. Progress in Molecular and Subcellular Biology. 2001;26:115–154. doi: 10.1007/978-3-642-56688-2_5. [DOI] [PubMed] [Google Scholar]
- 59.Schalm S. S., Blenis J. Identification of a conserved motif required for mTOR signaling. Current Biology. 2002;12(8):632–639. doi: 10.1016/S0960-9822(02)00762-5. [DOI] [PubMed] [Google Scholar]
- 60.Schalm S. S., Tee A. R., Blenis J. Characterization of a conserved C-terminal motif (RSPRR) in ribosomal protein S6 kinase 1 required for its mammalian target of rapamycin-dependent regulation. The Journal of Biological Chemistry. 2005;280(12):11101–11106. doi: 10.1074/jbc.M413995200. [DOI] [PubMed] [Google Scholar]
- 61.Ali S. M., Sabatini D. M. Structure of S6 kinase 1 determines whether raptor-mTOR or rictor-mTOR phosphorylates its hydrophobic motif site. The Journal of Biological Chemistry. 2005;280(20):19445–19448. doi: 10.1074/jbc.C500125200. [DOI] [PubMed] [Google Scholar]
- 62.Keshwani M. M., von Daake S., Newton A. C., Harris T. K., Taylor S. S. Hydrophobic motif phosphorylation is not required for activation loop phosphorylation of p70 ribosomal protein S6 kinase 1 (S6K1) The Journal of Biological Chemistry. 2011;286(26):23552–23558. doi: 10.1074/jbc.M111.258004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Beigh M. A., Showkat M., Hussain M. U., Andrabi K. I. Loss of hydrophobic motif and activation loop phosphorylation is a consequence and not the mechanism of S6 kinase 1 inhibition by rapamycin. Journal of Biological Regulators and Homeostatic Agents. 2013;27(2):399–408. [PubMed] [Google Scholar]
- 64.Beigh M. A., Showkat M., Ul Hussain M., Latoo S. A., Majeed S. T., Andrabi K. I. Rapamycin inhibition of baculovirus recombinant (BVr) ribosomal protein S6 kinase (S6K1) is mediated by an event other than phosphorylation. Cell Communication and Signaling. 2012;10, article 4 doi: 10.1186/1478-811X-10-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ferrari S., Bandi H. R., Hofsteenge J., Russian B. M., Thomas G. Mitogen-activated 70K S6 kinase: identification of in vitro 40 S ribosomal S6 phosphorylation sites. The Journal of Biological Chemistry. 1991;266(33):22770–22775. [PubMed] [Google Scholar]
- 66.Shima H., Pende M., Chen Y., Fumagalli S., Thomas G., Kozma S. C. Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. The EMBO Journal. 1998;17:6649–6659. doi: 10.1093/emboj/17.22.6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pende M., Kozma S. C., Jaquet M., Oorschot V., Burcelin R., Le Marchand-Brustel Y., Klumperman J., Thorens B., Thomas G. Hypoinsulinaemia, glucose intolerance and diminished -cell size in S6K1-deficient mice. Nature. 2000;408(6815):994–997. doi: 10.1038/35050135. [DOI] [PubMed] [Google Scholar]
- 68.Montagne J., Stewart M. J., Stocker H., Hafen E., Kozma S. C., Thomas G. Drosophila S6 kinase: a regulator of cell size. Science. 1999;285(5436):2126–2129. doi: 10.1126/science.285.5436.2126. [DOI] [PubMed] [Google Scholar]
- 69.Ruvinsky I., Sharon N., Lerer T., et al. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes & Development. 2005;19(18):2199–2211. doi: 10.1101/gad.351605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Raught B., Peiretti F., Gingras A.-C., et al. Phosphorylation of eucaryotic translation initiation factor 4B Ser422 is modulated by S6 kinases. The EMBO Journal. 2004;23(8):1761–1769. doi: 10.1038/sj.emboj.7600193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang H.-S., Jansen A. P., Komar A. A., Zheng X., Merrick W. C., Costes S., Lockett S. J., Sonenberg N., Colburn N. H. The transformation suppressor Pdcd4 is a novel eukaryotic translation initiation factor 4A binding protein that inhibits translation. Molecular and Cellular Biology. 2003;23(1):26–37. doi: 10.1128/MCB.23.1.26-37.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dorrello N. V., Peschiaroli A., Guardavaccaro D., Colburn N. H., Sherman N. E., Pagano M. S6k1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science. 2006;314(5798):467–471. doi: 10.1126/science.1130276. [DOI] [PubMed] [Google Scholar]
- 73.Wang X., Li W., Williams M., Terada N., Alessi D. R., Proud C. G. Regulation of elongation factor 2 kinase by p90RSK1 and p70 S6 kinase. The EMBO Journal. 2001;20(16):4370–4379. doi: 10.1093/emboj/20.16.4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Holz M. K., Ballif B. A., Gygi S. P., Blenis J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 2005;123(4):569–580. doi: 10.1016/j.cell.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 75.Hannan K. M., Brandenburger Y., Jenkins A., Sharkey K., Cavanaugh A., Rothblum L., Moss T., Poortinga G., McArthur G. A., Pearson R. B., Hannan R. D. mTOR-dependent regulation of ribosomal gene transcription requires S6K1 and is mediated by phosphorylation of the carboxy-terminal activation domain of the nucleolar transcription factor UBF. Molecular and Cellular Biology. 2003;23(23):8862–8877. doi: 10.1128/MCB.23.23.8862-8877.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.de Groot R. P., Ballou L. M., Sassone-Corsi P. Positive regulation of the cAMP-responsive activator CREM by the p70 S6 kinase: an alternative route to mitogen-induced gene expression. Cell. 1994;79(1):81–91. doi: 10.1016/0092-8674(94)90402-2. [DOI] [PubMed] [Google Scholar]
- 77.Yamnik R. L., Digilova A., Davis D. C., Brodt Z. N., Murphy C. J., Holz M. K. S6 kinase 1 regulates estrogen receptor in control of breast cancer cell proliferation. Journal of Biological Chemistry. 2009;284(10):6361–6369. doi: 10.1074/jbc.M807532200. [DOI] [PubMed] [Google Scholar]
- 78.Fang J., Meng Q., Vogt P. K., Zhang R., Jiang B.-H. A downstream kinase of the mammalian target of rapamycin, p70s6k1, regulates human double minute 2 protein phosphorylation and stability. Journal of Cellular Physiology. 2006;209(2):261–265. doi: 10.1002/jcp.20749. [DOI] [PubMed] [Google Scholar]
- 79.Harada H., Andersen J. S., Mann M., Terada N., Korsmeyer S. J. p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(17):9666–9670. doi: 10.1073/pnas.171301998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Richardson C. J., Bröenstrup M., Fingar D. C., Jülich K., Ballif B. A., Gygi S., Blenis J. SKAR is a specific target of S6 kinase 1 in cell growth control. Current Biology. 2004;14(17):1540–1549. doi: 10.1016/j.cub.2004.08.061. [DOI] [PubMed] [Google Scholar]
- 81.Sonenberg N., Hinnebusch A. G. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136(4):731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mader S., Lee H., Pause A., Sonenberg N. The translation initiation factor eIF-4E binds to a common motif shared by the translation factor eIF-4γ and the translational reprossers 4E-binding proteins. Molecular and Cellular Biology. 1995;15(9):4990–4997. doi: 10.1128/mcb.15.9.4990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Richter J. D., Sonenberg N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature. 2005;433(7025):477–480. doi: 10.1038/nature03205. [DOI] [PubMed] [Google Scholar]
- 84.Pyronnet S. Phosphorylation of the cap-binding protein eIF4E by the MAPK-activated protein kinase Mnk1. Biochemical Pharmacology. 2000;60(8):1237–1243. doi: 10.1016/S0006-2952(00)00429-9. [DOI] [PubMed] [Google Scholar]
- 85.Heesom K. J., Denton R. M. Dissociation of the eukaryotic initiation factor-4E/4E-BP1 complex involves phosphorylation of 4E-BP1 by an mTOR-associated kinase. FEBS Letters. 1999;457(3):489–493. doi: 10.1016/S0014-5793(99)01094-7. [DOI] [PubMed] [Google Scholar]
- 86.Fadden P., Haystead T. A. J., Lawrence J. C., Jr. Identification of phosphorylation sites in the translational regulator, PHAS-I, that are controlled by insulin and rapamycin in rat adipocytes. Journal of Biological Chemistry. 1997;272(15):10240–10247. doi: 10.1074/jbc.272.15.10240. [DOI] [PubMed] [Google Scholar]
- 87.Gingras A.-C., Gygi S. P., Raught B., Polakiewicz R. D., Abraham R. T., Hoekstra M. F., Aebersold R., Sonenberg N. Regulation of 4E-BP1 phosphorylation: a novel two step mechanism. Genes and Development. 1999;13(11):1422–1437. doi: 10.1101/gad.13.11.1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ayuso M. I., Hernández-Jiménez M., Martín M. E., Salinas M., Alcázar A. New hierarchical phosphorylation pathway of the translational repressor eIF4E-binding protein 1 (4E-BP1) in ischemia-reperfusion stress. The Journal of Biological Chemistry. 2010;285(45):34355–34363. doi: 10.1074/jbc.M110.135103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Martineau Y., Azar R., Bousquet C., Pyronnet S. Anti-oncogenic potential of the eIF4E-binding proteins. Oncogene. 2013;32(6):671–677. doi: 10.1038/onc.2012.116. [DOI] [PubMed] [Google Scholar]
- 90.Tee A. R., Proud C. G. Caspase cleavage of initiation factor 4E-binding protein 1 yields a dominant inhibitor of cap-dependent translation and reveals a novel regulatory motif. Molecular and Cellular Biology. 2002;22(6):1674–1683. doi: 10.1128/MCB.22.6.1674-1683.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Beugnet A., Wang X., Proud C. G. Target of rapamycin (TOR)-signaling and RAIP motifs play distinct roles in the mammalian TOR-dependent phosphorylation of initiation factor 4e-binding protein. Journal of Biological Chemistry. 2003;278(42):40717–40722. doi: 10.1074/jbc.M308573200. [DOI] [PubMed] [Google Scholar]
- 92.Schalm S. S., Fingar D. C., Sabatini D. M., Blenis J. TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Current Biology. 2003;13(10):797–806. doi: 10.1016/S0960-9822(03)00329-4. [DOI] [PubMed] [Google Scholar]
- 93.Choi K. M., McMahon L. P., Lawrence J. C., Jr. Two motifs in the translational repressor PHAS-I required for efficient phosphorylation by mammalian target of rapamycin and for recognition by raptor. The Journal of Biological Chemistry. 2003;278(22):19667–19673. doi: 10.1074/jbc.M301142200. [DOI] [PubMed] [Google Scholar]
- 94.Wang X., Beugnet A., Murakami M., Yamanaka S., Proud C. G. Distinct signaling events downstream of mTOR cooperate to mediate the effects of amino acids and insulin on initiation factor 4E-binding proteins. Molecular & Cellular Biology. 2005;25(7):2558–2572. doi: 10.1128/MCB.25.7.2558-2572.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gosselin P., Oulhen N., Jam M., Ronzca J., Cormier P., Czjzek M., Cosson B. The translational repressor 4E-BP called to order by eIF4E: new structural insights by SAXS. Nucleic Acids Research. 2011;39(8):3496–3503. doi: 10.1093/nar/gkq1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Paku K. S., Umenaga Y., Usui T., Fukuyo A., Mizuno A., In Y., Ishida T., Tommo K. A conserved motif within the flexible C-terminus of the translational regulator 4E-BP is required for tight binding to the mRNA cap-binding protein eIF4E. Biochemical Journal. 2012;441(1):237–245. doi: 10.1042/BJ20101481. [DOI] [PubMed] [Google Scholar]
- 97.Tamburini J., Green A. S., Bardet V., Chapuis N., Park S., Willems L., Uzunov M., Ifrah N., Dreyfus F., Lacombe C., Mayeux P., Bouscary D. Protein synthesis is resistant to rapamycin and constitutes a promising therapeutic target in acute myeloid leukemia. Blood. 2009;114(8):1618–1627. doi: 10.1182/blood-2008-10-184515. [DOI] [PubMed] [Google Scholar]
- 98.Nawijn M. C., Alendar A., Berns A. For better or for worse: the role of Pim oncogenes in tumorigenesis. Nature Reviews Cancer. 2011;11(1):23–34. doi: 10.1038/nrc2986. [DOI] [PubMed] [Google Scholar]
- 99.Burnett P. E., Barrow R. K., Cohen N. A., Snyder S. H., Sabatini D. M. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(4):1432–1437. doi: 10.1073/pnas.95.4.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lin T.-A., Kong X., Haystead T. A. J., Pause A., Belsham G., Sonenberg N., Lawrence J. C., Jr. PHAS-I as a link between mitogen-activated protein kinase and translation initiation. Science. 1994;266(5185):653–656. doi: 10.1126/science.7939721. [DOI] [PubMed] [Google Scholar]
- 101.Oulhen N., Boulben S., Bidinosti M., Morales J., Cormier P., Cosson B. A variant mimicking hyperphosphorylated 4E-BP inhibits protein synthesis in a sea urchin cell-free, cap-dependent translation system. PLoS ONE. 2009;4(3) doi: 10.1371/journal.pone.0005070.e5070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jiang Y.-P., Ballou L. M., Lin R. Z. Rapamycin-insensitive regulation of 4E-BP1 in regenerating rat liver. The Journal of Biological Chemistry. 2001;276(14):10943–10951. doi: 10.1074/jbc.M007758200. [DOI] [PubMed] [Google Scholar]
- 103.Choo A. Y., Yoon S.-O., Kim S. G., Roux P. P., Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(45):17414–17419. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Choo A. Y., Blenis J. Not all substrates are treated equally: implications for mTOR, rapamycin-resistance and cancer therapy. Cell Cycle. 2009;8(4):567–572. doi: 10.4161/cc.8.4.7659. [DOI] [PubMed] [Google Scholar]
- 105.Thoreen C. C., Kang S. A., Chang J. W., et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. The Journal of Biological Chemistry. 2009;284(12):8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Constantinou C., Elia A., Clemens M. J. Activation of p53 stimulates proteasome-dependent truncation of elF4E-binding protein 1 (4E-BP1) Biology of the Cell. 2008;100(5):279–289. doi: 10.1042/BC20070121. [DOI] [PubMed] [Google Scholar]
- 107.Constantinou C., Clemens M. J. Regulation of the phosphorylation and integrity of protein synthesis initiation factor eIF4GI and the translational repressor 4E-BP1 by p53. Oncogene. 2005;24(30):4839–4850. doi: 10.1038/sj.onc.1208648. [DOI] [PubMed] [Google Scholar]
- 108.Tee A. R., Proud C. G. DNA-damaging agents cause inactivation of translational regulators linked to mTOR signalling. Oncogene. 2000;19(26):3021–3031. doi: 10.1038/sj.onc.1203622. [DOI] [PubMed] [Google Scholar]
- 109.Horton L. E., Bushell M., Barth-Baus D., Tilleray V. J., Clemens M. J., Hensold J. O. p53 activation results in rapid dephosphorylation of the eIF4E-binding protein 4E-BP1, inhibition of ribosomal protein S6 kinase and inhibition of translation initiation. Oncogene. 2002;21(34):5325–5334. doi: 10.1038/sj.onc.1205662. [DOI] [PubMed] [Google Scholar]
- 110.Kumar V., Pandey P., Sabatini D., Kumar M., Majumder P. K., Bharti A., Carmichael G., Kufe D., Kharbanda S. Functional interaction between RAFT1/FRAP/mTOR and protein kinase C in the regulation of cap-dependent initiation of translation. The EMBO Journal. 2000;19(5):1087–1097. doi: 10.1093/emboj/19.5.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mamane Y., Petroulakis E., LeBacquer O., Sonenberg N. mTOR, translation initiation and cancer. Oncogene. 2006;25(48):6416–6422. doi: 10.1038/sj.onc.1209888. [DOI] [PubMed] [Google Scholar]
- 112.Kantidakis T., Ramsbottom B. A., Birch J. L., Dowding S. N., White R. J. mTOR associates with TFIIIC, is found at tRNA and 5S rRNA genes, and targets their repressor Maf1. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:11823–11828. doi: 10.1073/pnas.1005188107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shor B., Wu J., Shakey Q., Toral-Barza L., Shi C., Follettie M., Yu K. Requirement of the mTOR kinase for the regulation of Maf1 phosphorylation and control of RNA polymerase III-dependent transcription in cancer cells. The Journal of Biological Chemistry. 2010;285(20):15380–15392. doi: 10.1074/jbc.M109.071639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mayer C., Zhao J., Yuan X., Grummt I. mTOR-dependent activation of the transcription factor TIF-IA links rRNA synthesis to nutrient availability. Genes & Development. 2004;18(4):423–434. doi: 10.1101/gad.285504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Laplante M., Sabatini D. M. An emerging role of mTOR in lipid biosynthesis. Current Biology. 2009;19(22):R1046–R1052. doi: 10.1016/j.cub.2009.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Düvel K., Yecies J. L., Menon S., Raman P., Lipovsky A. I., Souza A. L., Triantafellow E., Ma Q., Gorski R., Cleaver S., Vander Heiden M. G., MacKeigan J. P., Finan P. M., Clish C. B., Murphy L. O., Manning B. D. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Molecular Cell. 2010;39(2):171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Porstmann T., Santos C. R., Griffiths B., Cully M., Wu M., Leevers S., Griffiths J. R., Chung Y.-L., Schulze A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metabolism. 2008;8(3):224–236. doi: 10.1016/j.cmet.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang B. T., Ducker G. S., Barczak A. J., Barbeau R., Erle D. J., Shokat K. M. The mammalian target of rapamycin regulates cholesterol biosynthetic gene expression and exhibits a rapamycin-resistant transcriptional profile. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(37):15201–15206. doi: 10.1073/pnas.1103746108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Peterson T. R., Sengupta S. S., Harris T. E., et al. MTOR complex 1 regulates lipin 1 localization to control the srebp pathway. Cell. 2011;146(3):408–420. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kim J. E., Chen J. Regulation of peroxisome proliferator-activated receptor-γ activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes. 2004;53(11):2748–2756. doi: 10.2337/diabetes.53.11.2748. [DOI] [PubMed] [Google Scholar]
- 121.Zhang H. H., Huang J., Düvel K., Boback B., Wu S., Squillance R. M., Wu C.-L., Manning B. D. Insulin stimulates adipogenesis through the Akt-TSC2-mTORC1 pathway. PLoS ONE. 2009;4(7) doi: 10.1371/journal.pone.0006189.e6189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hudson C. C., Liu M., Chiang G. G., Otterness D. M., Loomis D. C., Kaper F., Giaccia A. J., Abraham R. T. Regulation of hypoxia-inducible factor 1α expression and function by the mammalian target of rapamycin. Molecular and Cellular Biology. 2002;22(20):7004–7014. doi: 10.1128/MCB.22.20.7004-7014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Levine B., Abrams J. p53: the Janus of autophagy? Nature Cell Biology. 2008;10(6):637–639. doi: 10.1038/ncb0608-637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jung C. H., Ro S. H., Cao J., Otto N. M., Kim D. H. mTOR regulation of autophagy. FEBS Letters. 2010;584(7):1287–1295. doi: 10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tooze S. A., Schiavo G. Liaisons dangereuses: autophagy, neuronal survival and neurodegeneration. Current Opinion in Neurobiology. 2008;18(5):504–515. doi: 10.1016/j.conb.2008.09.015. [DOI] [PubMed] [Google Scholar]
- 126.Hosokawa N., Hara T., Kaizuka T., Kishi C., Takamura A., Miura Y., Iemura S.-I., Natsume T., Takehana K., Yamada N., Guan J.-L., Oshiro N., Mizushima N. Nutrient-dependent mTORCl association with the ULK1-Atg13-FIP200 complex required for autophagy. Molecular Biology of the Cell. 2009;20(7):1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jung C. H., Jun C. B., Ro S.-H., Kim Y.-M., Otto N. M., Cao J., Kundu M., Kim D.-H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Molecular Biology of the Cell. 2009;20(7):1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Polson H. E. J., De Lartigue J., Rigden D. J., Reedijk M., Urbé S., Clague M. J., Tooze S. A. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy. 2010;6(4):506–522. doi: 10.4161/auto.6.4.11863. [DOI] [PubMed] [Google Scholar]
- 129.Koren I., Reem E., Kimchi A. DAP1, a novel substrate of mTOR, negatively regulates autophagy. Current Biology. 2010;20(12):1093–1098. doi: 10.1016/j.cub.2010.04.041. [DOI] [PubMed] [Google Scholar]
- 130.Mizushima N., Levine B. Autophagy in mammalian development and differentiation. Nature Cell Biology. 2010;12(9):823–830. doi: 10.1038/ncb0910-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Vivanco I., Sawyers C. L. The phosphatidylinositol 3-kinase-AKT pathway in human cancer. Nature Reviews Cancer. 2002;2(7):489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 132.Laplante M., Sabatini D. M. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ruggero D., Montanaro L., Ma L., Xu W., Londei P., Cordon-Cardo C., Pandolfi P. P. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nature Medicine. 2004;10(5):484–486. doi: 10.1038/nm1042. [DOI] [PubMed] [Google Scholar]
- 134.Wendel H.-G., de Stanchina E., Fridman J. S., Malina A., Ray S., Kogan S., Cordon-Cardo C., Pelletier J., Lowe S. W. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature. 2004;428(6980):332–337. doi: 10.1038/nature02369. [DOI] [PubMed] [Google Scholar]
- 135.Mamane Y., Petroulakis E., Martineau Y., et al. Epigenetic activation of a subset of mRNAs by elF4E explains its effects on cell proliferation. PLoS ONE. 2007;2(2, article e242) doi: 10.1371/journal.pone.0000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rojo F., Najera L., Lirola J., Jiménez J., Guzmán M., Dolors Sabadell M., Baselga J., Ramon Y Cajal S. 4E-binding protein 1, a cell signaling hallmark in breast cancer that correlates with pathologic grade and prognosis. Clinical Cancer Research. 2007;13(1):81–89. doi: 10.1158/1078-0432.CCR-06-1560. [DOI] [PubMed] [Google Scholar]
- 137.Dowling R. J. O., Topisirovic I., Alain T., Bidinosti M., Fonseca B. D., Petroulakis E., Wang X., Larsson O., Selvaraj A., Liu Y., Kozma S. C., Thomas G., Sonenberg N. mTORCI-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science. 2010;328(5982):1172–1176. doi: 10.1126/science.1187532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lynch M., Fitzgerald C., Johnston K. A., Wang S., Schmidt E. V. Activated eIF4E-binding protein slows G1 progression and blocks transformation by c-myc without inhibiting cell growth. The Journal of Biological Chemistry. 2004;279(5):3327–3339. doi: 10.1074/jbc.M310872200. [DOI] [PubMed] [Google Scholar]
- 139.She Q.-B., Halilovic E., Ye Q., Zhen W., Shirasawa S., Sasazuki T., Solit D. B., Rosen N. 4E-BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors. Cancer Cell. 2010;18(1):39–51. doi: 10.1016/j.ccr.2010.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hsieh A. C., Liu Y., Edlind M. P., Ingolia N. T., Janes M. R., Sher A., Shi E. Y., Stumpf C. R., Christensen C., Bonham M. J., Wang S., Ren P., Martin M., Jessen K., Feldman M. E., Weissman J. S., Shokat K. M., Rommel C., Ruggero D. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485(7396):55–61. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Barlund M., Monni O., Kononen J., Cornelison R., Torhorst J., Sauter G., Kallioniemi O.-P., Kallioniemi A. Multiple genes at 17q23 undergo amplification and overexpression in breast cancer. Cancer Research. 2000;60(19):5340–5344. [PubMed] [Google Scholar]
- 142.Ismail H. M. Overexpression of S6 kinase 1 in brain tumours is associated with induction of hypoxia-responsive genes and predicts patients' survival. Journal of Oncology. 2012;2012 doi: 10.1155/2012/416927.416927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Masri J., Bernath A., Martin J., et al. mTORC2 activity is elevated in gliomas and promotes growth and cell motility via overexpression of rictor. Cancer Research. 2007;67(24):11712–11720. doi: 10.1158/0008-5472.CAN-07-2223. [DOI] [PubMed] [Google Scholar]
- 144.Hietakangas V., Cohen S. M. TOR complex 2 is needed for cell cycle progression and anchorage-independent growth of MCF7 and PC3 tumor cells. BMC Cancer. 2008;8, article 282 doi: 10.1186/1471-2407-8-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sansal I., Sellers W. R. The biology and clinical relevance of the PTEN tumor suppressor pathway. Journal of Clinical Oncology. 2004;22(14):2954–2963. doi: 10.1200/JCO.2004.02.141. [DOI] [PubMed] [Google Scholar]
- 146.Guertin D. A., Stevens D. M., Saitoh M., Kinkel S., Crosby K., Sheen J.-H., Mullholland D. J., Magnuson M. A., Wu H., Sabatini D. M. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell. 2009;15(2):148–159. doi: 10.1016/j.ccr.2008.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Johnstone C. N., Castellví-Bel S., Chang L. M., Sung R. K., Bowser M. J., Piqué J. M., Castells A., Rustgi A. K. PRR5 encodes a conserved proline-rich protein predominant in kidney: analysis of genomic organization, expression, and mutation status in breast and colorectal carcinomas. Genomics. 2005;85(3):338–351. doi: 10.1016/j.ygeno.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 148.Yen C.-H., Lu Y.-C., Li C.-H., Lee C.-M., Chen C.-Y., Cheng M.-Y., Huang S.-F., Chen K.-F., Cheng A.-L., Liao L.-Y., Lee Y.-H. W., Chen Y.-M. A. Functional characterization of glycine N-methyltransferase and its interactive protein DEPDC6/DEPTOR in hepatocellular carcinoma. Molecular Medicine. 2012;18(2):286–296. doi: 10.2119/molmed.2011.00331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Mavrakis K. J., Zhu H., Silva R. L. A., Mills J. R., Teruya-Feldstein J., Lowe S. W., Tam W., Pelletier J., Wendel H.-G. Tumorigenic activity and therapeutic inhibition of Rheb GTPase. Genes and Development. 2008;22(16):2178–2188. doi: 10.1101/gad.1690808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lu Z. H., Shvartsman M. B., Lee A. Y., et al. Mammalian target of rapamycin activator RHEB is frequently overexpressed in human carcinomas and is critical and sufficient for skin epithelial carcinogenesis. Cancer Research. 2010;70:3287–3298. doi: 10.1158/0008-5472.CAN-09-3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Menendez J. A., Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nature Reviews Cancer. 2007;7(10):763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- 152.Woods A., Johnstone S. R., Dickerson K., Leiper F. C., Fryer L. G. D., Neumann D., Schlattner U., Wallimann T., Carlson M., Carling D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Current Biology. 2003;13(22):2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 153.Hemminki A., Markie D., Tomlinson I., Avizienyte E., Roth S., Loukola A., Bignell G., Warren W., Aminoff M., Höglund P., Järvinen H., Kristo P., Pelin K., Ridanpää M., Salovaara R., Toro T., Bodmer W., Olschwang S., Olsen A. S., Stratton M. R., De La Chapelle A., Aaltonen L. A. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391(6663):184–187. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- 154.Inoki K., Corradetti M. N., Guan K.-L. Dysregulation of the TSC-mTOR pathway in human disease. Nature Genetics. 2005;37(1):19–24. doi: 10.1038/ng1494. [DOI] [PubMed] [Google Scholar]
- 155.Liaw D., Marsh D. J., Li J., et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nature Genetics. 1997;16(1):64–67. doi: 10.1038/ng0597-64. [DOI] [PubMed] [Google Scholar]
- 156.Khokhar N. Z., Altman J. K., Platanias L. C. Emerging roles for mammalian target of rapamycin inhibitors in the treatment of solid tumors and hematological malignancies. Current Opinion in Oncology. 2011;23(6):578–586. doi: 10.1097/CCO.0b013e32834b892d. [DOI] [PubMed] [Google Scholar]
- 157.Mendoza M. C., Er E. E., Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends in Biochemical Sciences. 2011;36(6):320–328. doi: 10.1016/j.tibs.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chresta C. M., Davies B. R., Hickson I., Harding T., Cosulich S., Critchlow S. E., Vincent J. P., Ellston R., Jones D., Sini P., James D., Howard Z., Dudley P., Hughes G., Smith L., Maguire S., Hummersone M., Malagu K., Menear K., Jenkins R., Jacobsen M., Smith G. C. M., Guichard S., Pass M. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Research. 2010;70(1):288–298. doi: 10.1158/0008-5472.CAN-09-1751. [DOI] [PubMed] [Google Scholar]
- 159.Feldman M. E., Apsel B., Uotila A., et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biology. 2009;7, article e38 doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.García-Martínez J. M., Moran J., Clarke R. G., Gray A., Cosulich S. C., Chresta C. M., Alessi D. R. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR) Biochemical Journal. 2009;421(1):29–42. doi: 10.1042/BJ20090489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Gentzler R. D., Altman J. K., Platanias L. C. An overview of the mTOR pathway as a target in cancer therapy. Expert Opinion on Therapeutic Targets. 2012;16(5):481–489. doi: 10.1517/14728222.2012.677439. [DOI] [PubMed] [Google Scholar]
- 162.Benjamin D., Colombi M., Moroni C., Hall M. N. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nature Reviews Drug Discovery. 2011;10(11):868–880. doi: 10.1038/nrd3531. [DOI] [PubMed] [Google Scholar]
- 163.Carayol N., Vakana E., Sassano A., Kaur S., Goussetis D. J., Glaser H., Druker B. J., Donato N. J., Altman J. K., Barr S., Platanias L. C. Critical roles for mTORC2- and rapamycin-insensitive mTORC1-complexes in growth and survival of BCR-ABL-expressing leukemic cells. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(28):12469–12474. doi: 10.1073/pnas.1005114107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Altman J. K., Sassano A., Kaur S., Glaser H., Kroczynska B., Redig A. J., Russo S., Barr S., Platanias L. C. Dual mTORC2/mTORC1 targeting results in potent suppressive effects on Acute Myeloid Leukemia (AML) progenitors. Clinical Cancer Research. 2011;17(13):4378–4388. doi: 10.1158/1078-0432.CCR-10-2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Holt S. V., Logié A., Odedra R., Heier A., Heaton S. P., Alferez D., Davies B. R., Wilkinson R. W., Smith P. D. The MEK1/2 inhibitor, selumetinib (AZD6244; ARRY-142886), enhances anti-tumour efficacy when combined with conventional chemotherapeutic agents in human tumour xenograft models. British Journal of Cancer. 2012;106(5):858–866. doi: 10.1038/bjc.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.O'Reilly K. E., Rojo F., She Q.-B., Solit D., Mills G. B., Smith D., Lane H., Hofmann F., Hicklin D. J., Ludwig D. L., Baselga J., Rosen N. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Research. 2006;66(3):1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nature Reviews Cancer. 2012;12(6):401–410. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Bray K., Mathew R., Lau A., Kamphorst J. J., Fan J., Chen J., Chen H.-Y., Ghavami A., Stein M., DiPaola R. S., Zhang D., Rabinowitz J. D., White E. Autophagy suppresses RIP kinase-dependent necrosis enabling survival to mTOR inhibition. PLoS ONE. 2012;7(7) doi: 10.1371/journal.pone.0041831.e41831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Alain T., Morita M., Fonseca B. D., Yanagiya A., Siddiqui N., Bhat M., Zammit D., Marcus V., Metrakos P., Voyer L.-A., Gandin V., Liu Y., Topisirovic I., Sonenberg N. eIF4E/4E-BP ratio predicts the efficacy of mTOR targeted therapies. Cancer Research. 2012;72(24):6468–6476. doi: 10.1158/0008-5472.CAN-12-2395. [DOI] [PubMed] [Google Scholar]
- 170.Brachmann S. M., Hofmann I., Schnell C., Fritsch C., Wee S., Lane H., Wang S., Garcia-Echeverria C., Maira S.-M. Specific apoptosis induction by the dual PI3K/mTor inhibitor NVP-BEZ235 in HER2 amplified and PIK3CA mutant breast cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(52):22299–22304. doi: 10.1073/pnas.0905152106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Molckovsky A., Siu L. L. First-in-class, first-in-human phase I results of targeted agents: highlights of the 2008 American Society of Clinical Oncology meeting. Journal of Hematology and Oncology. 2008;1(1, article 20) doi: 10.1186/1756-8722-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Yap T. A., Garrett M. D., Walton M. I., Raynaud F., de Bono J. S., Workman P. Targeting the PI3K-AKT-mTOR pathway: progress, pitfalls, and promises. Current Opinion in Pharmacology. 2008;8(4):393–412. doi: 10.1016/j.coph.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 173.Vilar E., Perez-Garcia J., Tabernero J. Pushing the envelope in the mTOR pathway: the second generation of inhibitors. Molecular Cancer Therapeutics. 2011;10(3):395–403. doi: 10.1158/1535-7163.MCT-10-0905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Janes M. R., Limon J. J., So L., Chen J., Lim R. J., Chavez M. A., Vu C., Lilly M. B., Mallya S., Ong S. T., Konopleva M., Martin M. B., Ren P., Liu Y., Rommel C., Fruman D. A. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nature Medicine. 2010;16(2):205–213. doi: 10.1038/nm.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]