

Abstract
Patients with metastatic triple negative breast cancer (TNBC) have poor treatment outcomes. We reviewed the electronic records of consecutive patients with metastatic TNBC treated in phase I clinic at MD Anderson between August 2005 and May 2012. One hundred and six patients received at least 1 phase I trial. Twelve of 98 evaluable patients (12%) had either complete response (n=1); partial response (n=7); or, stable disease ≥6 months (n=4). Patients treated on matched therapy (n=16) compared to those on non-matched therapy (n=90) had improved SD≥6 months/PR/CR (33% vs 8%; p=0.018) and longer PFS (median, 6.4 vs 1.9 months; p=0.001). Eleven of 57 evaluable patients (19%) treated with combination chemotherapy and targeted therapy had SD≥ 6 months/PR/CR versus 1 of 41 evaluable patients (2%) treated on other phase I trials (p=0.013); and longer PFS (3.0 vs 1.6 months; p<0.0001). Patients with molecular alterations in the PI3K/AKT/mTOR pathway treated on matched therapy (n=16) had improved PFS compared to those with and without molecular alterations treated on non-matched therapy (n=27) (6.4 vs 3.2 months; p= 0.036). On multivariate analysis, improved PFS was associated with treatment with combined chemotherapy and targeted agents (p=0.0002); ≤2 metastatic sites (p=0.003); therapy with PI3K/AKT/mTOR inhibitors for those with cognate pathway abnormalities (p=0.018); and, treatment with anti-angiogenic agents (p=0.023). In summary, combinations of chemotherapy and angiogenesis and/or PI3K/AKT/mTOR inhibitors demonstrated improved outcomes in metastatic TNBC patients.
Keywords: next-generation sequencing, matched therapy, phase I trials, PI3K/AKT/mTOR pathway, triple negative breast cancer
Introduction
Patients with metastatic triple negative breast cancer (TNBC), comprising 12-17% of breast cancers, have poor outcomes due to the aggressive nature of the disease associated with a high proliferation index (1). The majority of these patients do not respond well to conventional systemic therapy (2) and to date, there have been no clear targets identified for effective treatment. Patients with TNBC have a median overall survival (OS) of 6 months from the time of initial diagnosis with metastatic disease versus 20 months for those patients with hormone receptor-positive and/or Her2-positive metastatic breast cancer (2). While impressive gains have been realized in the outcomes of metastatic Her2-positive and hormone receptor-positive breast cancer, TNBC remains an unmet need.
Recent studies have identified molecular subtypes within TNBC that have increased our understanding of the biologic heterogeneity of the disease and have suggested further therapeutic potential targets for evaluation (3). However, in this era of personalized medicine (4), there have been no trials of targeted agents demonstrating significant benefit for patients with any subtype of TNBC (5). A phase III trial of iniparib, failed to show benefit in unselected patients with TNBC (6). A subset analysis, as well as early stage poly (ADP-ribose) polymerase inhibitor (PARP) inhibitor trials suggest that BRCA1 or 2–mutant TNBC patients may benefit, contrary to those with BRCA 1 or 2 normal metastatic TNBC (7). Neo-adjuvant studies with mTOR inhibitors in TNBC have also failed to show benefit (8).
We analyzed the clinical, pathologic and molecular characteristics and treatment outcomes of patients with metastatic TNBC treated in the Clinical Center for Targeted Therapy (phase I clinic) at The University of Texas MD Anderson Cancer Center (MDACC). Our objectives were to analyze associations between treatments and outcomes including SD≥6 months/PR/CR in patients with metastatic TNBC and to identify potential biomarkers of clinical benefit. Herein, we report our experience with these patients.
Patients and Methods
Patients
We retrospectively reviewed the medical records of consecutive patients with advanced or metastatic TNBC, who were treated on at least one phase one clinical trial at the Clinical Center for Targeted Therapy at MDACC between August 2005 and May 2012. All patients provided written informed consent before enrollment on a clinical trial, and all clinical trials were approved by the MDACC Institutional Review Board.
Treatment
Patients were enrolled in a clinical trial after their clinical, laboratory and pathologic data were reviewed. The assignment of a patient to a clinical trial varied over time based on the availability of the protocol, eligibility criteria, molecular profile of tumor tissue, insurance coverage, patient's preference or physician's choice.
Definition of “Matched” Therapy
A phase I clinical trial was considered to be ‘matched’ to a patient if at least one drug in the regimen was known to inhibit the functional activity of one of the molecular alterations in the patient's tumor tissue at low nmol/L concentrations. Patients with actionable molecular alterations were preferably treated on ‘matched’ therapy when available, if they met the eligibility criteria and were willing to comply with study requirements. If patients did not have molecular alterations or were not tested, they were considered to be treated on ‘non-matched’ therapies.
Evaluation
Assessments, including history, physical examination, and laboratory evaluations, were performed as specified in each protocol, typically before the initiation of therapy and, at a minimum, at the beginning of each new treatment cycle. Response was assessed using computed tomography scans and/or magnetic resonance imaging at baseline before treatment initiation and then every two cycles or as specified by protocol. All radiographs were read in the Department of Radiology at MDACC and reviewed in the Department of Investigational Cancer Therapeutics tumor measurement clinic. Responses were categorized per Response Evaluation Criteria in Solid Tumors (RECIST) version 1.0 (9) and were reported as best response during the whole evaluation period. Patients enrolled in phase I trials continued treatment until disease progression, unacceptable toxicity, withdrawal of consent or, at the discretion of the treating physician.
For the purpose of this analysis, electronic patient records were reviewed for medical history, laboratory results, molecular profiling data and treatment outcome. Baseline characteristics collected were: age, gender, tumor histology, ECOG performance status, number of metastatic sites, serum albumin and LDH levels, prior systemic therapies for metastatic disease, best response to investigational phase I therapy based on RECIST, and date of death or date of last follow-up. The first phase I therapy received by the patient in our clinic was considered for this analysis.
The Royal Marsden Hospital score (RMH score) (10, 11) and the MD Anderson Cancer Center score (MDACC score) (12) were used to evaluate the prognostic status of the patients. The RMH score classified patients according to three variables: lactate dehydrogenase (LDH) normal (0) versus LDH >upper limit of normal (ULN) (+1); albumin >3.5 g/dL (0) versus albumin <3.5 g/dL (+1) and number of metastatic sites of disease ≤2 (0) versus metastatic sites of disease ≥3 (+1). The MDACC score included two additional variables namely, ECOG performance status <1 (0) versus ECOG performance status ≥1 (+1), and, non-gastrointestinal tumor type (0) versus gastrointestinal tumor type (1).
Molecular profiling studies
“Hotspot” mutation analyses were carried out in select patients where tissue was available. These were done on archival formalin-fixed, paraffin-embedded tissue blocks or material from fine needle aspiration biopsy obtained from diagnostic and/or therapeutic procedures. Pathology was centrally reviewed at MDACC. All testing was carried out in a Clinical Laboratory Improvement Amendment (CLIA)–certified Molecular Diagnostic Laboratory within the Division of Pathology and Laboratory Medicine at MDACC. DNA was extracted from micro-dissected, paraffin-embedded tumor sections and further studied using a polymerase chain reaction (PCR)–based DNA sequencing method for PIK3CA mutations in codons (c) 532 to c554 of exon 9 (helical domain) and c1011-1062 of exon 20 (kinase domain), which included the mutation hotspot region of the PIK3CA proto-oncogene by Sanger sequencing after amplification of 276 and 198 base pair amplicons, respectively, using primers designed by the MD Anderson Molecular Diagnostic Laboratory. After January 2011, the assay used was mass spectrometric detection (Sequenom MassARRAY) to screen for the mutational hot spots in exon 1 (Q60K, R88Q, E110K and K111N), exon 4 (N345K), exon 6 (S405S), exon 7 (E418K, C420R, E453K), exon 9 (P539R, E542 [bases 1 and 2], E545 [all 3 bases] and Q546 [base 1 and 2]), exon 18 (F909L) and exon 20 (Y1021 [base 1 and 2], T1025 [base 1], M1043I, M1043V, A1046V, H1047Y, H1047R, H1047L, G1049R). The mutations identified during the initial screening were confirmed by Sanger sequencing assay. The lower limit of detection is approximately 10%. Archival samples were tested for PTEN expression by immunohistochemistry (IHC). PTEN immunostaining was performed in the MD Anderson IHC CLIA laboratory with the following antibody: PTEN (Dako #M3627, 1:100, 15 min). Samples with complete loss of PTEN staining were considered as PTEN loss. Additionally, whenever possible, mutation analyses for BRAF (exon 15: codons 595–600); KRAS and NRAS (exon 2: codons 12, 13 and 61); KIT (exons 9, 11, 13 and 17); and GNAQ (exon 5); and TP53 (exons 4–9) were carried out using PCR-based DNA sequencing mutation, as previously described (13).
In addition, tissue from 9 patients was submitted to CLIA-certified laboratory where next-generation sequencing of 3,320 exons of 182 cancer-related genes and the introns of 14 genes frequently rearranged in cancer was performed (Foundation Medicine, Cambridge, MA).
Statistical Analysis
Statistical analysis was verified by our statistician (JJL). Patient characteristics were summarized using descriptive statistics. The Fisher's exact test was used to determine associations between categorical variables and responses (SD ≥6 months/PR/CR). Multivariable logistic regression was used to identify predictors of response. Progression-free survival (PFS) was defined as the time interval from the start of therapy to the first observation of disease progression or death, whichever occurred first. For PFS, patients were censored at the time of their last follow-up date if they were progression-free. Overall survival (OS) was measured from the date of starting treatment on the first phase I therapy until death from any cause or last follow-up. Patients were censored at the time of their last follow-up if they were alive. PFS and OS were estimated using the Kaplan–Meier method (14), and the survival function between groups was compared using a two-sided log-rank test. The multivariable Cox proportional hazards regression model was used to examine risk factors related to PFS and OS, after adjusting for other factors (15).
The following covariates were included in the analyses: Age (≤60 vs >60); histology (invasive ductal carcinoma vs non-invasive ductal carcinoma); number of prior therapies in metastatic setting (<3 vs ≥3); history of thromboembolism (yes vs no); metastatic sites (≤2 vs >2); ECOG performance status (0 vs ≥1); LDH levels (≤618 vs >618 IU/L); albumin levels (<3.5 vs ≥3.5 g/dL); RMH score (≤1 vs >1); MDACC score (≤2 vs >2); phase I therapy (combination that included chemotherapeutic and targeted agent vs chemotherapeutic or targeted agent only); use of PI3K pathway inhibitors (yes vs. no); use of anti-angiogenic agents (yes vs no); and, type of phase I therapy (matched vs non-matched therapy). All statistical tests were 2-sided, and p< 0.05 was considered statistically significant. Waterfall plot analysis was used to graph individual patients' best response on protocol treatments. Statistical analyses were carried out using SPSS (version 19.0; SPSS, Chicago, IL, USA).
Results
Baseline patient characteristics
A total of 106 consecutive patients (105 female and 1 male) with advanced or metastatic TNBC, who were treated on at least one phase I clinical trial were included in the analysis. Baseline characteristics are presented in Table 1. The median age was 51 years (range, 27-81 years). The median number of previous systemic anticancer treatments in the metastatic setting was 2 (range, 0-10) and the median number of metastatic sites present at the time of phase I trial initiation was 3 (range, 1-8).
Table 1.
Baseline patient characteristics of 106 patients with metastatic TNBC.
| Variable | Group | Chemo+ targeted agents (n=63) | % | Othersd (n=43) | % | Total (n=106) | % |
|---|---|---|---|---|---|---|---|
| Age | ≤60 years | 49 | 78 | 31 | 72 | 80 | 75 |
| >60 years | 14 | 22 | 12 | 28 | 26 | 25 | |
|
| |||||||
| Sex | Female | 63 | 100 | 42 | 98 | 105 | 99 |
| Male | 0 | 0 | 1 | 2 | 1 | 1 | |
|
| |||||||
| Ethnicity | White | 50 | 79 | 35 | 81 | 85 | 80 |
| African-American | 9 | 14 | 2 | 5 | 11 | 10 | |
| Hispanic | 3 | 5 | 6 | 14 | 9 | 8 | |
| Asian | 1 | 2 | 0 | 0 | 1 | 1 | |
|
| |||||||
| Histology | Invasive ductal carcinoma | 44 | 70 | 39 | 91 | 83 | 78 |
| Metaplastic | 15 | 24 | 2 | 5 | 17 | 16 | |
| Inflammatory | 3 | 5 | 1 | 2 | 4 | 4 | |
| Otherc | 1 | 2 | 1 | 2 | 2 | 2 | |
|
| |||||||
| Metastatic sites | ≤2 | 27 | 43 | 19 | 44 | 46 | 43 |
| >2 | 36 | 57 | 24 | 56 | 60 | 57 | |
|
| |||||||
| ECOG PS | 0 | 4 | 6 | 9 | 21 | 13 | 12 |
| 1 | 58 | 92 | 33 | 77 | 91 | 86 | |
| 2 | 1 | 2 | 1 | 2 | 2 | 2 | |
|
| |||||||
| Serum LDH | ≤ 618U/L | 33 | 52 | 21 | 49 | 54 | 51 |
| >618U/L | 30 | 48 | 22 | 51 | 52 | 49 | |
|
| |||||||
| Serum albumin | <3.5 g/dL | 10 | 16 | 2 | 5 | 12 | 11 |
| ≥3.5 g/dL | 53 | 84 | 41 | 95 | 94 | 89 | |
|
| |||||||
| RMH scorea | ≤1 (low risk) | 42 | 67 | 29 | 67 | 71 | 67 |
| >1 (high risk) | 21 | 33 | 14 | 33 | 35 | 33 | |
|
| |||||||
| MDACC scoreb | ≤2 (low risk) | 42 | 67 | 30 | 70 | 72 | 68 |
| >2 (high risk) | 21 | 33 | 13 | 30 | 34 | 32 | |
|
| |||||||
| History of thromboembolism | No | 59 | 94 | 38 | 88 | 97 | 92 |
| Yes | 4 | 6 | 5 | 12 | 9 | 8 | |
|
| |||||||
| Prior therapies in metastatic setting | <3 | 29 | 46 | 16 | 37 | 45 | 42 |
| ≥ 3 | 34 | 54 | 27 | 63 | 61 | 58 | |
RMH score classified patients according to three variables: lactate dehydrogenase (LDH) normal (0) versus LDH >upper limit of normal (ULN) (+1); albumin >3.5 g/dL (0) versus albumin <3.5 g/dL (+1) and number of metastatic sites of disease ≤2 (0) versus metastatic sites of disease ≥3 (+1).
MDACC score included two additional variables to that of RMH score namely, ECOG performance status <1 (0) versus ECOG performance status ≥1 (+1), and, non-gastrointestinal tumor type (0) versus gastrointestinal tumor type (1).
one patient with adenoid cystic breast carcinoma and another with lobular carcinoma
includes patients treated with either chemotherapeutic agents or targeted agents only
Abbreviations: ECOG, Eastern Cooperative Oncology Group; LDH, lactate dehydrogenase; MDACC, MD Anderson Cancer Center; PS, performance status; RMH, Royal Marsden Hospital.
Molecular testing and next-generation sequencing
Molecular testing was not done in all patients due to limited tissue availability. Molecular testing data was available in 47 patients including 9 with next generation sequencing (NGS) analysis. Molecular alterations in the PI3K/AKT/mTOR pathway were noted in 21 out of 43 patients (49%) tested including: PTEN loss by IHC (n=8 out of 30 tested); PIK3CA mutation (n=7 out of 40 tested); PTEN mutation (n=3 out of 12 tested); PTEN deletion (n=2 out of 12 tested); PIK3R1 mutation (n=2 out of 9 tested); and, NF2 mutation (n=1 out of 9 tested). These patients had at least one gene in this pathway evaluated.
Molecular evaluation of the 9 patients with NGS profiling of their tumors demonstrated a median of 3 (range, 0-6) alterations per patient with 4 of 9 having ≥5 molecular alterations including: TP53 mutation (n=8); MYC amplification (n=4); PIK3R1 mutation (n=2); FGFR2, MCL1, and CCND1 amplification (n=2 each); mutations in NF2, PTEN, KDM6A, and RB1 (n=1 each); amplifications in FGFR1, PIK3CA, CDK8, MAP2K2, and KRAS (n=1 each); and, PTEN deletion (n=1).
Other molecular alterations seen in these patients were: NRAS mutation in 1 out of 24, and, TP53 mutation in 10 out of 13. Thirty-five patients assessed for KRAS mutation, 30 for BRAF mutation, 21 for c-KIT mutation, 12 for GNAQ mutation, and, 32 for EGFR mutation were all negative for alterations.
Treatment
All 106 patients were treated on at least 1 phase I clinical trial including: chemotherapy only (n=8); combination chemotherapy and targeted therapy (n=62); single-agent targeted therapy (n=16); and, targeted therapy with 2 or more agents (n=20) (Table 2 and Supplementary Table S1). Nineteen patients (18%) patients received treatment on more than 1 phase trial. Fourteen patients (13%) received 2 trials; 4 (4%) received 3 trials; and, 1 received 5 trials. Sixty-three of 106 patients (59%) received a phase I trial with combination chemotherapy and targeted therapy. Of the 106 patients, 16 (15%) were treated on matched therapies. Thirty-nine of 106 patients (37%) received protocols containing drugs targeting the PI3K/AKT/mTOR pathway [37/39 (95%) included mTOR inhibitors, 1 (3%) a PI3K inhibitor, and 1 (3%) a combination of FGFR and AKT inhibitors]. Thirty-eight of 106 patients (36%) received protocols containing an anti-angiogenic agent [34/38 (89%) bevacizumab, 2/38 (5%) pazopanib-based therapy, 1/38(3%) sorafenib-based therapy and 1/38 (3%) anti-HIF-1 therapy].
Table 2.
Therapies received in the phase I clinical trials program.
| Phase I therapy | No. of patients | Evaluable (n) | CR (n) | PR (n) | SD≥6mo (n) | SD≥6mo/PR/CR n (%) | Median PFS mo (95%CI) |
|---|---|---|---|---|---|---|---|
| Chemotherapy alonea | 8(8) | 7 | 0 | 0 | 0 | 0/7 (0) | 2.1 (0.9-3.3)c |
| Chemotherapy and targeted agentb | 63 (59) | 57 | 1 | 6 | 4 | 11/57 (19) | 3.0 (1.9-4.1)c |
| Single agent targeted drug | 15(14) | 15 | 0 | 0 | 0 | 0/15 (0) | 1.1 (0.7-1.4)c |
| ≥2 targeted agents | 20 (19) | 19 | 0 | 1 | 0 | 1/19 (5) | 1.9 (1.4-2.4)c |
| Total | 106 (100) | 98 | 1 | 7 | 4 | 12/98 (12) | 2.1 (1.5-2.6) |
one or more agents
one or more of each
p value <0.0001for comparison of PFS by log rank test across all 4 groups
Abbreviations: CR, complete response; CI, confidence interval; mo, months; PR, partial response; PFS, progression-free survival; SD, stable disease
Response to phase I clinical trial
Out of 106 patients who initiated phase I therapy, 98 were evaluable for response. Response assessments were not carried out in 8 patients (5 withdrew consent and 3 stopped therapy in less than a month because of toxicity]. Twelve of 98 patients (12%) had SD ≥6 months/PR/CR including: 1 CR, 7 PRs and 4 SD ≥ 6 months (Figure 1). Eleven of 57 evaluable patients (19%) who received a combination of chemotherapy and targeted agent(s) had SD≥6 months/PR/CR versus 1 of 41 evaluable patients (2%) who received either targeted agents alone or chemotherapy alone (p value = 0.013). Five of 15 evaluable patients (33%) treated on matched therapies had SD≥6 months/PR/CR compared to 7 of 83 (8%) patients who were treated on non-matched therapy (p=0.018). Among the 12 patients who had SD≥6 months/PR/CR (Table 3), 5 received the same combination of chemotherapy (liposomal doxorubicin), anti-angiogenic agent (bevacizumab) and mTOR inhibitor (temsirolimus). Three of these 5 patients had metaplastic histology (16-18). Eleven of 12 patients with SD≥6 months/PR/CR received a combination of chemotherapy and targeted agent(s) while one received a combination of two targeted agents.
Figure 1.
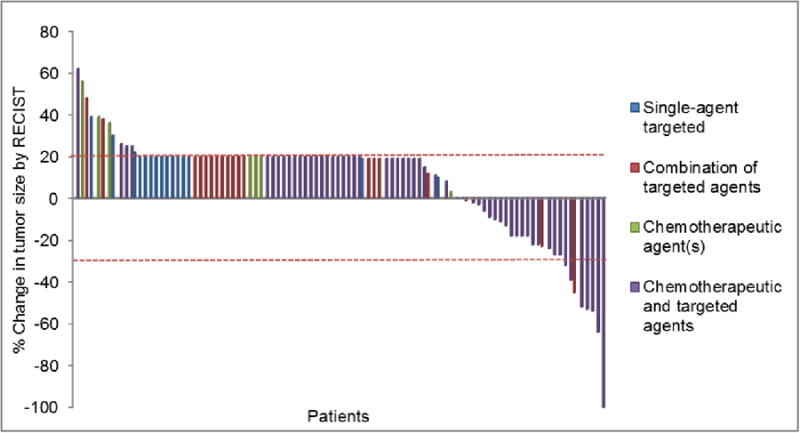
Waterfall plot. Best response by RECIST of 98 evaluable patients with triple negative breast cancer by treatment received in the phase I clinical trials program. Patients with clinical progression or with new metastasis were graphed as 20% progression. Dotted horizontal line at -30% and 20% indicates border for partial response and progression respectively.
Table 3. Profile of patients with SD≥6months/PR/CR (n=12).
| Case No. |
Histopathology | Age (years) |
No. of metastatic sites |
Molecular alterations |
ECOG PS |
RMH score |
MDACC score |
Prior therapies in metastatic setting |
Phase I therapy | PFS on phase I therapy (mo) |
Best response (RECIST %) |
OS from phase I therapy |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Invasive ductal carcinoma | 46 | 1 | TP53a (splice site 559 + 1 G>A) | 1 | 0 | 1 | 1 | PI3K and AKT inhibitor | 2.5 | PR (-45) | 4.9 |
| 2 | Metaplastic | 60 | 2 | PIK3CA (H1047R) | 1 | 0 | 1 | 1 | DAT | 11.7 | PR (-64) | 24.3 |
| 3 | Metaplastic | 52 | 4 | NF2a (p.K159fs*16) TP53 a (p.F109fs*39) | 0 | 1 | 1 | 0 | DAT | 19.1 | CR (-100) | 48.3 |
| 4 | Invasive ductal carcinoma | 51 | 2 | NA | 0 | 0 | 0 | 3 | Dasatinib, gemcitabine | 4.2 | PR (-54) | 5.3 |
| 5 | Invasive ductal carcinoma | 65 | 1 | TP53a (p.R273H) CDK8 ampa MYC amp a | 1 | 0 | 1 | 1 | Aurorakinase inhibitor, taxol | 8.0 | PR (-52) | 16.2 |
| 6 | Invasive ductal carcinoma | 56 | 1 | NA | 1 | 1 | 2 | 0 | DAT | 9.3 | PR (-39) | 10.7 |
| 7 | Metaplastic | 70 | 1 | PTEN loss (IHC) | 1 | 1 | 2 | 0 | DAT | 11.0 | PR (-32) | 11.4 |
| 8 | Invasive ductal carcinoma | 37 | 1 | NA | 1 | 1 | 2 | 4 | Carboplatin, etoposide, IGFR inhibitor | 6.1 | SD (-24) | 13.6 |
| 9 | Invasive ductal carcinoma | 72 | 3 | PIK3CA (H1047R) | 1 | 2 | 3 | 0 | DAT | 6.4 | SD (+8) | 6.4 |
| 10 | Adenoid Cystic | 47 | 1 | NA | 1 | 0 | 1 | 1 | HAI abraxane, IV gemcitabine, IV avastin | 7.5 | SD (-18) | 11.8 |
| 11 | Invasive ductal carcinoma | 57 | 1 | PTEN mutation (D92G) | 1 | 0 | 1 | 3 | DAT | 7.8 | SD (-18) | 9.6 |
| 12 | Invasive ductal carcinoma | 48 | 2 | PTEN loss (IHC) | 1 | 1 | 2 | 0 | mTOR inhibitor, taxol | 3.0 | PR (-53) | 10.7 |
next generation sequencing data
Abbreviations: CR, complete response; DAT, liposomal doxorubicin, bevacizumab and temsirolimus; ECOG, Eastern Cooperative Oncology Group; HAI, hepatic arterial infusion; IHC, immunohistochemistry; IV, intravenous; MDACC, MD Anderson Cancer Center; mo, months; NA, not available; OS, overall survival; PR, partial response; PS, performance status; PFS, progression-free survival; RECIST, response evaluation criteria in solid tumors; RMH, Royal Marsden Hospital; SD, stable disease.
Response assessment in patients with evidence of PI3K/AKT/mTOR pathway activation
Of 43 patients evaluated for alterations in the PI3K/AKT/mTOR pathway, 21 (49%) demonstrated at least one alteration (including mutations in PIK3CA, PIK3R1, PTEN, NF2, deletion in PTEN, PIK3CA amplification and PTEN loss on IHC). Sixteen of these 21 patients received protocols with at least 1 drug targeting the PI3K/AKT/mTOR pathway, and 15 were evaluable for response. Five of 15 evaluable patients (33%) treated with matched therapy had SD≥6 months/PR/CR, versus 6 of the 25 evaluable patients (20%) treated on non-matched therapy (p=0.716).
Factors predicting response to treatment
Factors that were associated with improved response (SD≥6 months/PR/CR) on univariate analysis included: ≤2 metastatic sites (p=0.004), treatment that included both chemotherapeutic and targeted agent(s) (p=0.013), treatment with a matched therapy (0.018), and, <3 prior therapies in metastatic setting (p=0.032) (Supplementary Table S2). On multivariate analysis, independent factors that predicted greater response (SD≥6 months/PR/CR) were ≤2 metastatic sites (p=0.017) and combination therapies that included chemotherapeutic and targeted agents (p=0.028) (Table 4).
Table 4. Summary of multivariate analysis for response, progression-free survival, and, overall survival.
| Variable | Estimated Effect | 95% CI | p-value |
|---|---|---|---|
| Response (SD ≥6 months/PR/CR) | ORa | ||
| Metastatic sites ≤2 (vs >2) | 10.62 | 1.52-74.09 | 0.017 |
| Chemotherapeutic and targeted agents (vs chemotherapeutic or targeted agent only) | 27.02 | 1.43-511.4 | 0.028 |
| Progression-free survival | HRb | ||
| Metastatic sites ≤2 (vs >2) | 0.44 | 0.26-0.75 | 0.003 |
| Chemotherapeutic and targeted agents (vs chemotherapeutic or targeted agent only) | 0.38 | 0.22-0.633 | 0.0002 |
| PI3K pathway inhibitors, yes (vs no) | 0.49 | 0.27-0.88 | 0.018 |
| Anti-angiogenic agents, yes (vs no) | 0.52 | 0.29-0.91 | 0.023 |
| Overall survival | HRb | ||
| MDACC score ≤2 (vs >2) | 0.25 | 0.15-0.41 | <0.0001 |
OR>1 is associated with higher response.
HR<1 is associated with longer progression-free survival or overall survival.
Abbreviations: CR, complete response; CI, confidence interval; HR, hazards ratio; OR, odds ratio; PR, partial response; SD, stable disease; MDACC, MD Anderson Cancer Center Figure Legend
PFS on the first phase I trial and prognostic factors
The median PFS of 106 patients treated on a first phase I trial was 2.1 months (95% CI: 1.6-2.6 months). The median PFS was significantly longer for 16 patients treated on matched therapy (6.4 months) versus 1.9 months for the 90 patients treated on non-matched therapy (p=0.001). Of 43 patients tested for molecular alterations in the PI3K/AKT/mTOR signaling pathway, the median PFS was significantly longer for 16 patients with molecular alterations in PI3K pathway treated on matched therapy (6.4 months) versus 3.2 months for 27 patients with or without molecular alterations treated on non-matched therapy (p=0.036).
Baseline characteristics associated with longer PFS on univariate analysis (Supplementary Table S2) included: RMH score ≤1 (p=0.009); non-invasive ductal carcinoma histology (p=0.010); ≤2 metastatic sites (p=0.014); and, MDACC score ≤2 (p=0.017). Improved PFS was associated with combinations treatment with chemotherapy and targeted agents (p <0.0001), PI3K/AKT/mTOR pathway inhibitors (p <0.0001) (regardless of molecular alterations), therapies that included anti-angiogenic agents (p <0.0001), and, matched therapies (p=0.002). Combination treatment with chemotherapy and targeted agents demonstrated improved PFS (3.0 months; 95% CI: 1.5-4.4 months) when compared to other therapies (either chemotherapy or targeted therapies alone) (1.6, 95% CI: 0.9-2.3 months; p value <0.0001) (Supplementary Table S2, Figure 2). On multivariate analysis (Table 4), factors which were predictive of improved PFS were therapies containing chemotherapeutic and targeted agents (p=0.0002), ≤2 metastatic sites (p=0.003), use of PI3K/AKT/mTOR pathway inhibitors (p=0.018), and, combinations that included an anti-angiogenic agent (p=0.023).
Figure 2.
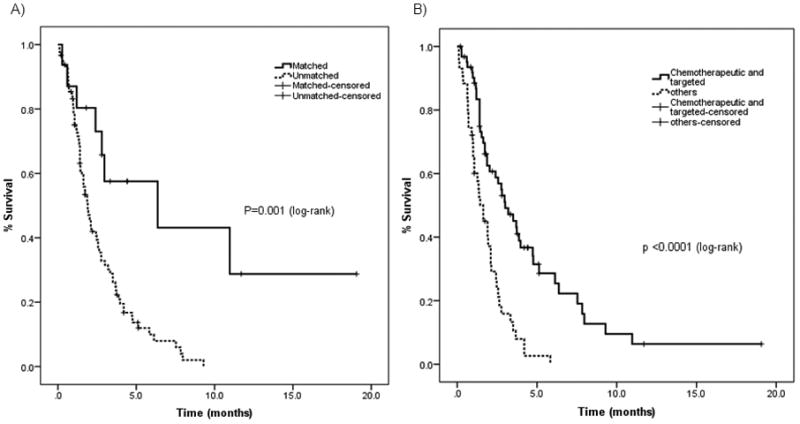
Kaplan-Meier estimates of PFS in 106 triple negative breast cancer patients A) PFS of 16 patients treated on matched therapy (6.4 months) versus those treated on other therapies (1.9 months), p value <0.001; log-rank test. B) PFS of 63 patients treated on combination therapies that included chemotherapy and targeted agents (3.0 months) versus 43 treated on other therapies (1.6 months), p value <0.0001; log-rank test.
Overall survival on the first phase I trial and prognostic factors
The median OS of the 106 TNBC patients starting from the beginning of a phase I trial was 7.7 months (95% CI: 6.3-9.0 months).Factors that were associated with improved OS on univariate analysis (Supplementary Table S1) were MDACC score ≤2 (p<0.0001); RMH score ≤1 (p<0.0001); serum albumin ≥3.5 g/dL (p=0.001); ≤2 metastatic sites (p=0.007); serum LDH ≤ 618 IU/L (p=0.026); less than 3 prior therapies in metastatic setting (p=0.026); non-invasive ductal carcinoma histology (p=0.033); and, ECOG performance status of 0 (p=0.040). On multivariate analysis, MDACC score ≤2 was predictive for overall survival (p <0.0001; Table 4).
Discussion
Historically, patients treated on Phase I studies have response rates of 4-10.6% (19, 20). With the evolving paradigm shift favoring personalized medicine that ‘matches’ molecular profiles of patients' tumors with targeted therapies, response rates on phase I trials are higher. A recent study demonstrated that patients treated with molecularly matched-targeted therapy compared to non-matched therapy had improved clinical benefit (4). In general, patients with metastatic TNBC have very poor outcomes and limited treatment options. Metastatic TNBC, therefore, remains an urgent, unmet need. (21). Nevertheless, certain patients with TNBC characterized by low proliferation index, tumor with lymphocytic infiltration and absence of central fibrosis, or, a rare type of TNBC such as adenoid cystic and secretory carcinoma with ETV6-NTRK3 and MYB-NFIB fusion genes and KIT positivity, may demonstrate indolent disease, excellent response to therapy and good prognosis (22-24). The purpose of this study was to systematically analyze the clinical outcomes of 106 patients with metastatic TNBC, who were mostly referred to the phase I clinic to receive second, third or salvage line treatments.
We demonstrated that patients with TNBC treated on phase I trials with combinations of chemotherapy and targeted agents had a significant improvement in PFS versus those patients treated on either chemotherapeutic or targeted agents only (3.0 vs 1.6 months; p<0.0001). In particular, the median PFS was significantly longer in patients who were treated on a combination that included an anti-angiogenic agent (3.7 vs 1.7 months; p<0.0001) and/or a PI3K/AKT/mTOR inhibitor (3.5 vs 1.6 months; p<0.0001) than patients who did not receive these agents. On multivariate analysis (Table 4) the most significant independent predictor of longer PFS was treatment with both chemotherapy and targeted agent(s) (p=0.0002). Treatment with a PI3K pathway inhibitor (p=0.018); or an ant-angiogenic agent (p=0.023) were also significant predictors of increased PFS.
Our results are consistent with previously published studies of patients with TNBC treated with a combination including an anti-angiogenic agent that reported improved response (SD≥6 months/PR/CR) in the neoadjuvant setting and prolonged PFS in patients with metastatic disease (25-28). Several studies have demonstrated that the addition of bevacizumab to neoadjuvant chemotherapy improved pathological CR rate in early and locally resectable TNBC (25, 26). A meta-analysis of three phase III trials with first-line bevacizumab-containing combinations in patients with metastatic TNBC showed a 35% reduction in risk of disease progression and longer PFS than those treated with regimens that included a chemotherapeutic agent without an ant-angiogenic agent (8.1 vs 5.4 months; HR=0.680; p=0.0002) (27). Similarly, RIBBON-2 trial demonstrated 51% reduction in risk of progression with second-line bevacizumab-containing combinations in patients with metastatic TNBC, compared to chemotherapy alone (6.0 vs 2.7 months; HR=0.494; p=0.0006) (28). The effectiveness of therapies that target VEGF may be explained by the higher expression of intra-tumoral vascular endothelial growth factor (VEGF) by 1.5-3 times in TNBC compared to non-TNBC and dysregulation of VEGF-related genes (29, 30). There are also studies, however, that failed to demonstrate benefit with adjuvant anti-angiogenic therapy (31). Unfortunately, biomarkers to identify angiogenesis inhibitor responders remain speculative (32, 33) and there are no validated biomarkers to identify potential responders to anti-angiogenic therapy and improvement in overall survival has not yet been demonstrated (34).
Treatment of advanced cancer with inhibitors of the PI3K/AKT/mTOR axis is supported by both preclinical studies (35) and early clinical data (17, 18, 36). Pre-clinical models have shown that the PI3K/AKT/mTOR is activated in TNBC and that blocking this pathway can induce tumor regression (1, 37). While clinical data is still limited (38) and early results are mixed, there is some indication that treatment with a PI3K/AKT/mTOR inhibitor in combination may be effective in TNBC (39).
Though limited in sample size, our molecular data was revealing of several trends. We report that 21 of 43 patients (49%) demonstrated at least one direct PI3K/AKT/mTOR pathway alteration (including mutations in PIK3CA, PIK3R1, PTEN, NF2, deletion in PTEN, PIK3CA amplification and PTEN loss on IHC). Seven of 43 patients (16%) tested for the PIK3CA mutation was positive. Among 37 patients tested for either a genomic or proteomic alteration in PTEN, 8 had PTEN loss by IHC, 5 had either PTEN mutation or deletion, and, 1 had both PTEN loss by IHC and PTEN mutation. Previous studies have reported on the prevalence of PI3K molecular alterations in breast cancer including different subtypes (36, 38). Prior data suggests that 8% of TNBC patients harbor a PIK3CA mutation (40). Higher rates of these alterations reported in our analysis may be explained by a higher representation of patients with metaplastic breast cancer, 17 of 106 (16%) patients in our study, a rare subtype known to harbor PI3K alterations at a higher rate than other TNBC (16, 41). In addition, the higher overall percentage of PI3K/AKT/mTOR pathway alterations in our study versus other analyses may be due to inclusion of patients with PIK3R1 and PTEN mutations and PTEN loss on IHC.
NGS profiling demonstrated a high number of alterations. Four of 9 patients who underwent NGS profiling had at least 5 alterations. Previous studies have demonstrated that an increased number of molecular alterations may be associated with more aggressive disease and worse outcomes (4). Consistent with these reports, we noted that the 3 of 9 patients in our study with NGS testing who demonstrated response (including 2 patients with PR, cases #1 and #5; and one with a CR, case #3, Table 3) had ≤3 molecular alterations. Of the two patients with PR, case #1 demonstrated only a TP53 mutation, case #5 had two amplifications in addition to the TP53 mutation, and the patient with a CR (case #3) demonstrated TP53 and NF2 mutations.
There are several limitations to this retrospective study. Of 106 patients, only 47 (44%) had molecular profiling including 9 with NGS. The tissue used for analysis was also not consistent among patients. That is, some of the tissue was from the primary tumor while others were from a metastatic site. Unfortunately, none included pre- and post-treatment biopsies, ideal for molecular analysis. In terms of the patient population in this study, patients with metaplastic TNBC were overrepresented; thus our results may be less generalizable to other metastatic TNBC subtypes. Further, different imaging studies such as computed tomography scans and/or magnetic resonance imaging were used to evaluate therapeutic response in these patients with TNBC. Despite these shortcomings, this is one of the largest reports to date describing outcomes in patients with metastatic TNBC treated on phase I studies including targeted therapy.
In conclusion, our study suggests that identification of molecular alterations and optimizing treatment with agents that targeted these molecular alterations (matched therapy) enhanced response (SD≥6 months/PR/CR) and PFS in this heavily pretreated group of patients with metastatic TNBC. Furthermore, response (SD≥6 months/PR/CR) and PFS was significantly longer in patients on a treatment that included either an anti-angiogenic and/or a PI3K/AKT/mTOR inhibitor in addition to a chemotherapeutic agent. Our data showed higher frequency of molecular alterations in patients with metastatic TNBC than has previously been reported. We currently are exploring NGS in a large prospective study. The study also demonstrated that patients with metastatic TNBC treated on phase I trials have comparable overall outcomes to those patients with TNBC treated with chemotherapy. Therefore, our data supports participation in phase I clinical trials with novel agents to develop new options for these patients with limited therapeutic options and poor prognosis.
Supplementary Material
Acknowledgments
Financial Support: This work was supported in part by the National Center for Research Resources Grants 3UL1RR024148 (D.D. McPherson, F. Meric-Bernstam), the National Cancer Institute through UL1TR000371(D.D. Karp, F. Meric-Bernstam), U01CA062461 (J.C. Yao, F. Meric-Bernstam), 1R21CA159270 (F. Meric-Bernstam), The MD Anderson Cancer Center Sheikh Khalifa Bin Zayed Al Nahyan Institute for Personalized Cancer Therapy (F. Meric-Bernstam), and The University of Texas MD Anderson Cancer Center Support Grant P30 CA016672 (F. Meric-Bernstam, J. Wheler, F. Janku).
Abbreviations
- CLIA
Clinical Laboratory Improvement Amendment
- CR
complete response
- CI
confidence interval
- DAT
liposomal doxorubicin, bevacizumab and temsirolimus
- ECOG
Eastern Cooperative Oncology Group
- HR
hazards ratio
- HAI
hepatic arterial infusion
- IHC
immunohistochemistry
- IV
intravenous
- LDH
lactate dehydrogenase
- MDACC
MD Anderson Cancer Center
- mo
months
- NGS
next generation sequencing
- NA
not available
- OR
odds ratio
- OS
overall survival
- PR
partial response
- PS
performance status
- PARP
poly (ADP-ribose) polymerase inhibitor
- PCR
polymerase chain reaction
- PFS
progression-free survival
- RECIST
response evaluation criteria in solid tumors
- RMH
Royal Marsden Hospital
- SD
stable disease
- TNBC
triple negative breast cancer
- VEGF
vascular endothelial growth factor
Footnotes
Previous presentation: Poster presentation at 2013 San Antonio Breast Cancer Symposium, San Antonio, Texas
Conflict of Interest: Roman Yelensky and Philip Stephens are employees and stock holders in Foundation Medicine Inc. Filip Janku received research funding from Novartis. Funda Meric-Bernstam was a consultant for Novartis and Roche and was on the advisory board of Genentech. All remaining authors declare no conflicts of interest.
References
- 1.Sohn J, Do KA, Liu S, Chen H, Mills GB, Hortobagyi GN, et al. Functional proteomics characterization of residual triple-negative breast cancer after standard neoadjuvant chemotherapy. Ann Oncol. 2013;24:2522–6. doi: 10.1093/annonc/mdt248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O'Toole SA, Beith JM, Millar EK, West R, McLean A, Cazet A, et al. Therapeutic targets in triple negative breast cancer. J Clin Pathol. 2013;66:530–42. doi: 10.1136/jclinpath-2012-201361. [DOI] [PubMed] [Google Scholar]
- 3.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750–67. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsimberidou AM, Iskander NG, Hong DS, Wheler JJ, Falchook GS, Fu S, et al. Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center initiative. Clin Cancer Res. 2012;18:6373–83. doi: 10.1158/1078-0432.CCR-12-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gelmon K, Dent R, Mackey JR, Laing K, McLeod D, Verma S. Targeting triple-negative breast cancer: optimising therapeutic outcomes. Ann Oncol. 2012;23:2223–34. doi: 10.1093/annonc/mds067. [DOI] [PubMed] [Google Scholar]
- 6.O'Shaughnessy J, Schwartzberg LS, Danso MA, Rugo HS, Miller K, Yardley DA, et al. A randomized phase III study of iniparib (BSI-201) in combination with gemcitabine/carboplatin (G/C) in metastatic triple-negative breast cancer (TNBC) J Clin Oncol. 2011;29 doi: 10.1200/JCO.2014.55.2984. suppl; abstr 1007. [DOI] [PubMed] [Google Scholar]
- 7.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–34. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 8.Gonzalez-Angulo AM, Green MC, Murray JL, Palla SL, Koenig KH, Brewster AM, et al. Open label, randomized clinical trial of standard neoadjuvant chemotherapy with paclitaxel followed by FEC (T-FEC) versus the combination of paclitaxel and RAD001 followed by FEC (TR-FEC) in women with triple receptor-negative breast cancer (TNBC) J Clin Oncol. 2011;29 doi: 10.1093/annonc/mdu124. suppl; abstr 1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 10.Arkenau HT, Olmos D, Ang JE, de Bono J, Judson I, Kaye S. Clinical outcome and prognostic factors for patients treated within the context of a phase I study: the Royal Marsden Hospital experience. Br J Cancer. 2008;98:1029–33. doi: 10.1038/sj.bjc.6604218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garrido-Laguna I, Janku F, Vaklavas C, Falchook GS, Fu SQ, Hong DS, et al. Validation of the royal marsden hospital prognostic score in patients treated in the phase I clinical trials program at the MD Anderson Cancer Center. Cancer. 2012;118:1422–8. doi: 10.1002/cncr.26413. [DOI] [PubMed] [Google Scholar]
- 12.Wheler J, Tsimberidou AM, Hong D, Naing A, Falchook G, Piha-Paul S, et al. Survival of 1,181 patients in a phase I clinic: the MD Anderson Clinical Center for targeted therapy experience. Clin Cancer Res. 2012;18:2922–9. doi: 10.1158/1078-0432.CCR-11-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zuo Z, Chen SS, Chandra PK, Galbincea JM, Soape M, Doan S, et al. Application of COLD-PCR for improved detection of KRAS mutations in clinical samples. Mod Pathol. 2009;22:1023–31. doi: 10.1038/modpathol.2009.59. [DOI] [PubMed] [Google Scholar]
- 14.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. Journal of the American Statistical Association. 1958;53:457–81. [Google Scholar]
- 15.Cox DR. Regression Models and Life-Tables. Journal of the Royal Statistical Society Series B-Statistical Methodology. 1972;34:187–220. [Google Scholar]
- 16.Moulder S, Moroney J, Helgason T, Wheler J, Booser D, Albarracin C, et al. Responses to liposomal doxorubicin, bevacizumab, and temsirolimus in metaplastic carcinoma of the breast: biologic rationale and implications for stem-cell research in breast cancer. J Clin Oncol. 2011;29:e572–5. doi: 10.1200/JCO.2010.34.0604. [DOI] [PubMed] [Google Scholar]
- 17.Moroney JW, Schlumbrecht MP, Helgason T, Coleman RL, Moulder S, Naing A, et al. A phase I trial of liposomal doxorubicin, bevacizumab, and temsirolimus in patients with advanced gynecologic and breast malignancies. Clin Cancer Res. 2011;17:6840–6. doi: 10.1158/1078-0432.CCR-11-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moroney J, Fu S, Moulder S, Falchook G, Helgason T, Levenback C, et al. Phase I study of the antiangiogenic antibody bevacizumab and the mTOR/hypoxia-inducible factor inhibitor temsirolimus combined with liposomal doxorubicin: tolerance and biological activity. Clin Cancer Res. 2012;18:5796–805. doi: 10.1158/1078-0432.CCR-12-1158. [DOI] [PubMed] [Google Scholar]
- 19.Horstmann E, McCabe MS, Grochow L, Yamamoto S, Rubinstein L, Budd T, et al. Risks and benefits of phase 1 oncology trials, 1991 through 2002. N Engl J Med. 2005;352:895–904. doi: 10.1056/NEJMsa042220. [DOI] [PubMed] [Google Scholar]
- 20.Kurzrock R, Benjamin RS. Risks and benefits of phase 1 oncology trials, revisited. N Engl J Med. 2005;352:930–2. doi: 10.1056/NEJMe058007. [DOI] [PubMed] [Google Scholar]
- 21.Fornier M, Fumoleau P. The paradox of triple negative breast cancer: novel approaches to treatment. Breast J. 2012;18:41–51. doi: 10.1111/j.1524-4741.2011.01175.x. [DOI] [PubMed] [Google Scholar]
- 22.Kreike B, van Kouwenhove M, Horlings H, Weigelt B, Peterse H, Bartelink H, et al. Gene expression profiling and histopathological characterization of triple-negative/basal-like breast carcinomas. Breast Cancer Res. 2007;9:R65. doi: 10.1186/bcr1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Azoulay S, Lae M, Freneaux P, Merle S, Al Ghuzlan A, Chnecker C, et al. KIT is highly expressed in adenoid cystic carcinoma of the breast, a basal-like carcinoma associated with a favorable outcome. Mod Pathol. 2005;18:1623–31. doi: 10.1038/modpathol.3800483. [DOI] [PubMed] [Google Scholar]
- 24.Brouckaert O, Wildiers H, Floris G, Neven P. Update on triple-negative breast cancer: prognosis and management strategies. Int J Womens Health. 2012;4:511–20. doi: 10.2147/IJWH.S18541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerber B, Eidtmann H, Rezai M, Fasching PA, Tesch H, Eggemann H, et al. Neoadjuvant bevacizumab and anthracycline–taxane-based chemotherapy in 686 triple-negative primary breast cancers: seconday endpoint analysis of the GeparQuinto study (GBG 44) J Clin Oncol. 2011;29 doi: 10.1093/annonc/mdt361. suppl; abstr 1006. [DOI] [PubMed] [Google Scholar]
- 26.Amos KD, Adamo B, Anders CK. Triple-negative breast cancer: an update on neoadjuvant clinical trials. Int J Breast Cancer. 2012;2012:385978. doi: 10.1155/2012/385978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O'Shaughnessy J, Romieu G, Diéras V, Byrtek M, Duenne AA, Miles D. Meta-analysis of patients with triple-negative breast cancer (TNBC) from three randomized trials of first-line bevacizumab (BV) and chemotherapy treatment for metastatic breast cancer (MBC) Cancer Res. 2010;70 suppl; abstr P6-12-03. [Google Scholar]
- 28.Brufsky A, Valero V, Tiangco B, Dakhil S, Brize A, Rugo HS, et al. Second-line bevacizumab-containing therapy in patients with triple-negative breast cancer: subgroup analysis of the RIBBON-2 trial. Breast Cancer Res Treat. 2012;133:1067–75. doi: 10.1007/s10549-012-2008-6. [DOI] [PubMed] [Google Scholar]
- 29.Bender RJ, Mac Gabhann F. Expression of VEGF and semaphorin genes define subgroups of triple negative breast cancer. PLoS ONE. 2013;8:e61788. doi: 10.1371/journal.pone.0061788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Linderholm BK, Hellborg H, Johansson U, Elmberger G, Skoog L, Lehtio J, et al. Significantly higher levels of vascular endothelial growth factor (VEGF) and shorter survival times for patients with primary operable triple-negative breast cancer. Ann Oncol. 2009;20:1639–46. doi: 10.1093/annonc/mdp062. [DOI] [PubMed] [Google Scholar]
- 31.Cameron D, Brown J, Dent R, Jackisch C, Mackey J, Pivot X, et al. Adjuvant bevacizumab-containing therapy in triple-negative breast cancer (BEATRICE): primary results of a randomised, phase 3 trial. Lancet Oncol. 2013;14:933–42. doi: 10.1016/S1470-2045(13)70335-8. [DOI] [PubMed] [Google Scholar]
- 32.Said R, Hong D, Wheler J, Naing A, Falchook G, Fu S, et al. p53 mutations in advanced cancers: clinical characteristics and outcomes in a phase I setting. J Clin Oncol. 2012;30 suppl; abstr 10607. [Google Scholar]
- 33.Lambrechts D, Lenz HJ, de Haas S, Carmeliet P, Scherer SJ. Markers of response for the antiangiogenic agent bevacizumab. J Clin Oncol. 2013;31:1219–30. doi: 10.1200/JCO.2012.46.2762. [DOI] [PubMed] [Google Scholar]
- 34.Maru D, Venook AP, Ellis LM. Predictive biomarkers for bevacizumab: are we there yet? Clin Cancer Res. 2013;19:2824–7. doi: 10.1158/1078-0432.CCR-12-3409. [DOI] [PubMed] [Google Scholar]
- 35.Engelman JA, Chen L, Tan XH, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant KRAS G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–6. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Janku F, Wheler JJ, Westin SN, Moulder SL, Naing A, Tsimberidou AM, et al. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J Clin Oncol. 2012;30:777–82. doi: 10.1200/JCO.2011.36.1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yunokawa M, Koizumi F, Kitamura Y, Katanasaka Y, Okamoto N, Kodaira M, et al. Efficacy of everolimus, a novel mTOR inhibitor, against basal-like triple-negative breast cancer cells. Cancer Sci. 2012;103:1665–71. doi: 10.1111/j.1349-7006.2012.02359.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McAuliffe PF, Meric-Bernstam F, Mills GB, Gonzalez-Angulo AM. Deciphering the role of PI3K/AKT/mTOR pathway in breast cancer biology and pathogenesis. Clin Breast Cancer. 2010;10(Suppl 3):S59–65. doi: 10.3816/CBC.2010.s.013. [DOI] [PubMed] [Google Scholar]
- 39.Singh JC, Volm M, Novik Y, Speyer JL, Adams S, Omene CO, et al. Efficacy of RAD001/carboplatin in triple-negative metastatic breast cancer: a phase II study. J Clin Oncol. 2012;30 suppl 27; abstr 108. [Google Scholar]
- 40.Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68:6084–91. doi: 10.1158/0008-5472.CAN-07-6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hennessy BT, Gonzalez-Angulo AM, Stemke-Hale K, Gilcrease MZ, Krishnamurthy S, Lee JS, et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009;69:4116–24. doi: 10.1158/0008-5472.CAN-08-3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.