

Abstract
Recent evidence has shown that an increase in CD4+CD25+FoxP3+ regulatory T (Treg) cells may contribute to stroke-induced immunosuppression. However, the molecular mechanisms that underlie this increase in Treg cells remain unclear. Here, we used a transient middle cerebral artery occlusion model in mice and specific pathway inhibitors to demonstrate that stroke activates the sympathetic nervous system, which was abolished by 6-OHDA. The consequent activation of β2-adrenergic receptor (AR) signaling increased prostaglandin E2 (PGE2) level in bone marrow. β2-AR antagonist prevented the upregulation of PGE2. PGE2, which acts on prostaglandin E receptor subtype 4 (EP4), upregulated the expression of receptor activator for NF-κB ligand (RANKL) in CD4+ T cells and mediated the increase in Treg cells in bone marrow Treatment of MCAO mice with RANKL antagonist OPG inhibited the increase in percent of bone marrow Treg cells. PGE2 also elevated the expression of indoleamine 2,3 dioxygenase in CD11C+ dendritic cells and promoted the development of functional Treg cells. The effect was neutralized by treatment with indomethacin. Concurrently, stroke reduced production of stromal cell-derived factor-1 (SDF-1) via β3-AR signals in bone marrow but increased the expression of C-X-C chemokine receptor (CXCR) 4 in Treg and other bone marrow cells. Treatment of MCAO mice with β3-AR antagonist SR-59230A reduced the percent of Treg cells in peripheral blood after stroke. The disruption of the CXCR4–SDF-1 axis may facilitate mobilization of Treg cells and other CXCR4+ cells into peripheral blood. This mechanism could account for the increase in Treg cells, hematopoietic stem cells, and progenitor cells in peripheral blood after stroke. We conclude that cerebral ischemia can increase bone marrow CD4+CD25+FoxP3+ regulatory T cells via signals from the sympathetic nervous system.
Keywords: Bone marrow, Cerebral ischemia, Immunosuppression, RANKL, SDF-1, SNS, Treg cells
1. Introduction
Accumulating evidence suggests that regulatory T cells are key immunomodulators after ischemic stroke and may contribute to post-stroke immunosuppression and infectious complications, such as pneumonia (Chamorro et al., 2007; Dirnagl et al., 2007; Liesz et al., 2009; Meisel et al., 2005; Offner et al., 2006; Prass et al., 2003). However, few studies have investigated the cellular and molecular mechanisms of ischemic stroke-induced immunosuppression.
It has recently become clear that peripheral tolerance and immune homeostasis are largely maintained by immunosuppressive regulatory T cells, such as CD4+CD25+FoxP3+ regulatory T (Treg) cells (Wing and Sakaguchi, 2010). Treg cells exert immune-modulating effects by either direct contact with the suppressed cell or release of immunosuppressive cytokines, such as transforming growth factor (TGF)-β, interleukin (IL)-10, and IL-35 (Sakaguchi et al., 2008; Wing and Sakaguchi, 2010). Evidence from clinical trials and from preclinical studies that used the middle cerebral artery occlusion (MCAO) model showed that stroke causes marked elevations in the number of Treg cells in peripheral blood and spleen (Offner et al., 2006; Yan et al., 2009). Treg cells decrease T cell activation and reduce production of interferon-γ (γ-IFN), one of the most important factors for preventing bacterial infections (Liesz et al., 2009; Liu et al., 2011; Mahic et al., 2006; Offner et al., 2006). Therefore, Treg cells are thought to be strongly associated with stroke-induced immunosuppression (Offner et al., 2006; Offner et al., 2009). However, the cellular and molecular mechanisms that underlie the stroke-induced increase in Treg cells are largely unknown.
Treg cells comprise at least two subpopulations: inducible Treg (iTreg) cells and natural Treg (nTreg) cells (Sakaguchi et al., 2008; Wing and Sakaguchi, 2010). nTreg cells are produced in the thymus and released into peripheral blood. iTreg cells are induced in the periphery from naive T cells, mainly CD4+CD25- T cells (Sakaguchi et al., 2008; Wing and Sakaguchi, 2010). Cyclooxygenase (COX)-2 and its product prostaglandin (PG) E2 play important roles in mediating the generation of iTreg cells in the ultraviolet-irradiated mouse and tumor models (Mahic et al., 2006; Sharma et al., 2005; Soontrapa et al., 2011). In the ultraviolet irradiation model, PGE2 acts on prostaglandin E receptor subtype 4 (EP4), leading to elevated levels of receptor activator for NF-κB ligand (RANKL) in the epidermis (Loser et al., 2006; Soontrapa et al., 2011). RANKL and its receptor, RANK, upregulate CD205 expression in dendritic cells (DCs) (Loser et al., 2006). CD205+ DCs directly utilize endogenous TGF-β to induce the differentiation of CD4+CD25- into CD4+CD25+FoxP3+ cells (Yamazaki et al., 2008). However, it is well known that RANKL is produced mainly by bone marrow cells, including osteoblasts, stromal cells, and activated T cells (especially for CD4+ lymphocytes), in response to immune stimulation (Vernal et al., 2006). EP4 is also mainly expressed in lymphocytes (Tilley et al., 2001). Interestingly, bone marrow increases the production of PGE2 in response to lipopolysaccharide-induced brain inflammation via β2-adrengenic receptor (AR) signaling (Inoue et al., 2003). Moreover, stroke significantly increases the number of bone marrow CD4+ T cells (Denes et al., 2010). Therefore, we asked whether bone marrow can generate iTreg via PGE2-EP4-RANKL signaling after stroke.
Treg cells express CXCR4 receptor and are retained in bone marrow by the CXCR4-SDF-1 axis (Zou et al., 2004a). Under homeostatic conditions, cyclical signals from the sympathetic nervous system (SNS) act via β3-ARs to reduce bone marrow SDF-1 and maintain low levels of CXCR4+ hemopoietic stem/progenitor cells (HSPCs) in peripheral blood (Méndez-Ferrer et al., 2008). It is well known that cerebral infarct increases the number of HSPCs and Treg cells in peripheral blood (Chang et al., 2011a; Paczkowska et al., 2005). However, it is still unclear whether ischemic stroke enhances the mobilization of CXCR4+ cells such as Treg cells and HSPCs from bone marrow into peripheral blood via SNS signaling.
Although studies have shown that stroke might mediate the increase in Treg cells, and bone marrow can be a priming site for immune response and generation of Treg cells (Feuerer et al 2003; Sakaguchi et al., 2008), no study has examined the mechanisms involved. We hypothesize that stroke induces activation of the SNS, which further activates specific signaling pathways downstream and mediates the generation of Treg cells. In the current study, we focused on the molecular regulation of bone marrow Treg generation and its role in immunosuppression after stroke. We found that stroke-induced activation of the SNS mediated the increase in bone marrow Treg cells and enhanced the mobilization of Treg cells via β2-AR and β3-AR signaling pathways, respectively, and that the Treg cells aggravated bacterial loads in lung.
2. Materials and methods
2.1. Animals
Male C57BL/6 mice (25–30 g, 12–14 weeks old; Animal Experimental Center of Zhengzhou University) were used for the study. Animals had free access to food and water and were maintained under temperature-, humidity-, and light-controlled conditions. All animal procedures were performed under an appropriate Home Office License and adhered to regulations as specified in the Animals (Scientific Procedures) Act (1986).
2.2. Surgical procedures
C57BL/6 mice were subjected to transient MCAO as described previously but with some modification (Jin et al., 2013). In brief, mice were anesthetized with an intraperitoneal injection of 4% chloral hydrate. Rectal temperature was maintained at 37±0.6°C throughout the surgical procedure by means of a feedback-regulated water-heating system. The right common carotid artery, external carotid artery, and internal carotid artery were exposed through a midline neck incision. A 6.0 monofilament nylon suture with silicone-coated tip was introduced into the origin of the external carotid artery and advanced through the internal carotid artery to block the origin of the middle cerebral artery. The monofilament was left in place for 45 min until reperfusion. Successful MCAO was defined as ≥80% decrease in cerebral blood flow confirmed by laser-Doppler flowmetry (moorVMS-LDF, Moor Instruments Ltd., UK). In sham-operated animals, the filament was advanced along the internal carotid artery until the origin of the middle cerebral artery and was withdrawn immediately.
2.3. Bone marrow and peripheral blood cell analysis
Bone marrow and peripheral blood mononuclear cells were collected as previously described (Wang et al., 2013). The mice were sacrificed after a sublethal intraperitoneal injection of 4% chloral hydrate. Then, their femurs and tibias were aseptically dissected and both ends cut. Bone marrow was flushed out with 500 μL of phosphate-buffered saline (PBS). Blood samples were collected into heparinized tubes and centrifuged. The supernatant was frozen for later measurement of SDF-1 and PGE2 by enzyme-linked immunosorbent assay (ELISA). The bone marrow and peripheral blood cells were depleted of red cells by hypotonic lysis, resuspended in PBS, and subjected to density-gradient centrifugation (260g, 26 min) in 1.083 g/mL Histopaque 1.083 (Sigma-Aldrich, St. Louis, MO, USA). The mononuclear cell layer was collected from the gradient interface and washed with DMEM F12 in three consecutive steps of 6-min centrifugation and resuspension.
2.4. Flow cytometry
The collected mononuclear cells were resuspended in PBS supplemented with 0.2% bovine serum albumin and 0.1% sodium azide (FACS buffer), incubated for 10 min with anti-CD4-FITC (Miltenyi Biotec, Bergisch-Gladbach, Germany) or anti-CD11C-PE (Miltenyi Biotec), and stained for 30 min at 4°C with one or more of the following antibodies: allophycocyanin (APC)–conjugated anti-CD25 (Miltenyi Biotec), phycoerythrin (PE)–conjugated anti-CXCR4 (Miltenyi Biotec), PE–conjugated anti-RANKL (BD Biosciences, USA). For intracellular IDO and FoxP3 staining, we used a BD Cytofix/Cytoperm Kit (BD Pharmingen, San Diego, CA). Next, for IDO detection, we used rabbit anti-mouse IDO (1:200, Santa Cruz Biotechnology, Santa Cruz, CA, USA) for the primary antibody and goat anti-mouse-APC–conjugated antibody (1:200) for the secondary antibody. For FoxP3 detection, APC–conjugated anti-FoxP3 (Miltenyi Biotec) was used directly. Isotype-matched antibodies and unstained cells were used as negative controls. Data were collected on a FACScan 4-color, 2-laser flow cytometer (BD Biosciences and Cytek Development, Fremont, CA) with CellQuest software (BD Biosciences) and were analyzed with the FlowJo software package (TreeStar, Ashland, OR, USA).
2.5. Western blot analysis
Bone marrow cell extracts were prepared in ice-cold RIPA lysis buffer containing 1× PMSF and 1× phosphatase inhibitor cocktail (Beyotime, China). After centrifugation, the supernatant of each sample was collected, and the protein concentration was determined in triplicate using the protein assay kit (Bio-Rad, USA). Equal amounts of protein (20-50μg) per lane were separated on 4–12% SDS-polyacrylamide gels under reducing conditions. Proteins were transferred to polyvinylidene difluoride membrane (Millipore, Billerica, MA, USA). Membranes were blocked with 10% nonfat milk in PBS–0.1% Tween 20 for 1 h at room temperature. Next, membranes were incubated with one of the following primary antibodies: polyclonal rabbit anti-CXCR4 (1:400, ProteinTech Group, USA), polyclonal rabbit anti-COX-2 (1:600, ProteinTech Group), polyclonal rabbit anti-SDF-1 (1:600, Santa Cruz), polyclonal rabbit anti-RANKL (1:600, Santa Cruz), polyclonal rabbit anti-tyrosine hydroxylase (TH; 1:600, Santa Cruz), polyclonal rabbit anti-IDO (1:600, Santa Cruz). These antibodies were diluted in PBS/0.1% Tween 20 (PBST) containing 1% bovine serum albumin at 4°C. Loading volumes were normalized against anti-GAPDH peroxidase (1:1,000, Zhixian Biotech, Hangzhou, China). Membranes were washed with PBST and incubated with horseradish peroxidase-conjugated secondary antibody (goat anti-rabbit–horseradish peroxidase, Sangon Biotech, Shanghai, China) diluted 1:800 in 6% nonfat milk/PBST for 2 h at room temperature. Protein bands were visualized by enhanced chemiluminescence ECL kit (Beyotime, China) and detected on a ChemiScope 3400 Mini (Clinx Science Instruments Co., Ltd, China). Target protein levels were normalized against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels and expressed as relative fold changes compared to the naïve group.
2.6. ELISA
We measured the concentrations of secreted SDF-1 and PGE2 in the bone marrow supernatants (100 μL per sample) and of γ-IFN in plasma in triplicate using the appropriate ELISA kits (R&D systems, USA) (Petit et al., 2002).
2.7. Drug administration
All drugs were injected intraperitoneally (i.p.), intravenously (i.v.) or subcutaneously. Control animals were injected with the corresponding vehicle at the same time. RU486 (glucocorticoid receptor inhibitor; Sigma-Aldrich) was dissolved in ethanol/sesame oil solution and administered 24 h, 5 h, and immediately before MCAO (30 mg/kg body weight, i.p.) (Prass et al., 2003). 6-hydroxydopamine HBr (6-OHDA; Sigma-Aldrich) was dissolved in sterile 0.01% ascorbic acid/saline and injected 3 days before MCAO (200 mg/kg body weight, i.p.) (Prass et al., 2003). Phentolamine (α-adrenergic receptor blocker; Sigma-Aldrich) was dissolved in saline and administered 6 h before MCAO (0.2 mg/kg body weight, i.v.) (Hagendorff et al., 1998). Propranolol (nonselective β-adrenergic receptor inhibitor; Sigma-Aldrich) was dissolved in 0.9% saline and administered 8 h, 4 h, and immediately before MCAO and 4, 8, 24, and 48 h after MCAO (30 mg/kg body weight, i.p.) (Prass et al., 2003). Butoxamine (selective β2-adrenergic receptor antagonist; Sigma-Aldrich) was dissolved in PBS and injected at 12 h and immediately before MCAO and then 1 day after MCAO (25 mg/kg body weight, i.p.) (Lucin et al., 2009). SR59230A (selective β3-adrenergic receptor antagonist; Sigma-Aldrich) was dissolved in PBS and administered twice on the day before MCAO (5 mg/kg body weight, i.p.) (Méndez-Ferrer et al., 2008).
Indomethacin (selective COX-2 inhibitor; Sigma-Aldrich) was dissolved in drinking water and administered based on daily water consumption to reach a target of 2.5 mg/kg per day (Soontrapa et al., 2011). Treatment of mice with indomethacin began 3 days before MCAO. L-161,982 (selective EP4 antagonist; Sigma-Aldrich) was dissolved in PBS and administered once daily for 4 days before MCAO (10 mg per mouse, i.p.) (Soontrapa et al., 2011). Osteoprotegerin/Fc (OPG/Fc) chimera from mouse (RANKL inhibitor; Sigma-Aldrich) was injected subcutaneously at a dose of 10 mg/kg, three times per week beginning 7 days before MCAO (Weinstein et al., 2011).
2.8 Bacteriological Analysis
The mice were anesthetized and then washed with 70% ethanol under sterile conditions. The lungs were removed after thoracotomy and homogenized. For determination of colony-forming units, 100 μL of tissue homogenate was serially diluted, plated onto blood agar plates (Merck), and incubated at 37°C for 24 h. An investigator blinded to experimental conditions counted the bacterial colonies.
2.9. Statistical analysis
Data are presented as mean ± SD. Differences among multiple groups were assessed with 1-way ANOVA followed by the Bonferroni post hoc test for multiple comparisons. Differences were considered significant at P<0.05. All data were analyzed with SPSS Statistics 13.00.
3. Results
3.1. Ischemic stroke increases the level of PGE2 and Treg cells but reduces the production of SDF-1 in bone marrow by signaling from activated SNS
The SNS innervates bone marrow and can mediate profound changes in bone marrow during conditions of stress (Dutta et al., 2012; Lucin et al., 2009). To determine the influence of stroke on bone marrow, we first assessed the dynamic activity of the SNS. Western blot showed that the level of tyrosine hydroxylase (TH, the rate-limiting enzyme that determines the production of norepinephrine [NE] in sympathetic fibers) (Dutta et al., 2012) was significantly increased at 6 h after stroke (n=6/time point, 6 h, days 1, 3: P<0.05; Fig. 1A, B), indicating activation of the SNS. Expression had returned to baseline on day 7. The CXCR4-SDF-1 axis plays a key role in retaining bone marrow cells (Petit et al., 2002). The disruption of this axis leads to migration of bone marrow cells to the periphery (Petit et al., 2002). To test the influence of stroke on the CXCR4–SDF-1 axis, we first examined the concentration of SDF-1 in bone marrow. Western blot results showed that bone marrow SDF-1 was significantly lower in MCAO mice than in sham-operated mice at 6 h after ischemic stroke, reached the lowest mean value on day 3, and persisted at low values for at least 7 days (n=6/time point, 6 h, days 1, 3, 7: P<0.05; Fig. 1A, C). ELISA results confirmed those of Western blot (Fig. 1F). Western blot also showed that stroke significantly increased the level of CXCR4 in total bone marrow on days 1 and 3 compared to that in sham-operated mice (n=6/time point, days 1, 3: P<0.05; Fig. 1A, D). CXCR4 was restored to the baseline level on day 7 after stroke. PGE2 is an important immunomodulator and can be secreted by bone marrow cells (Porter et al., 2013). Expression of COX-2, the rate-limiting enzyme for synthesis of PGE2, was significantly higher in bone marrow of MCAO mice than in that of sham-operated mice at 6 h, peaked at day 1, and remained elevated for at least 7 days after stroke (n=6/time point, 6 h, days 1, 3, 7: P<0.05; Fig. 1A, E). Quantitative analysis of PGE2 by ELISA further supported the results observed in the Western blot (Fig. 1G).
Fig. 1.
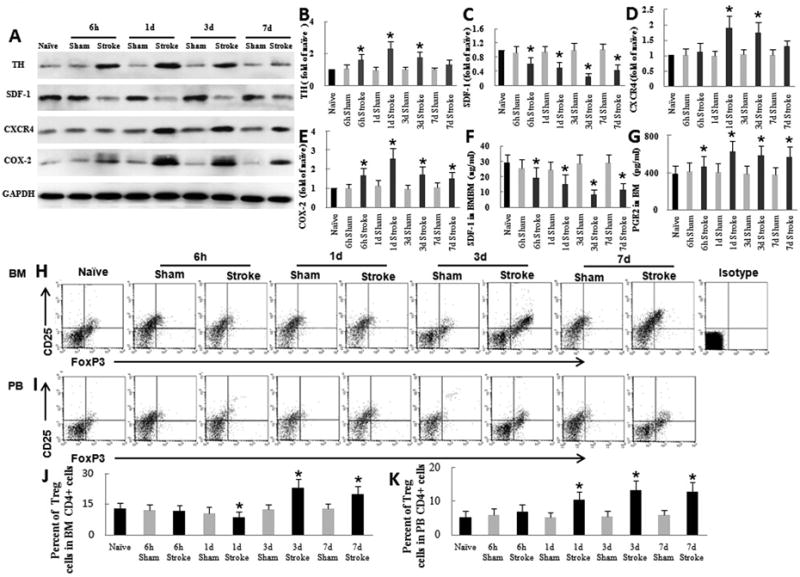
Ischemic stroke induces changes in bone marrow by activating the sympathetic nervous system. (A) Western blot analysis of tyrosine hydroxylase (TH), SDF-1, CXCR4, and COX-2 in bone marrow (BM) after stroke. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a loading control. (B–E) Quantification of Western blot band densities. (F) ELISA analysis of bone marrow SDF-1 showed that SDF-1 was significantly decreased beginning 6 h after stroke. (G) ELISA analysis of bone marrow PGE2 showed that PGE2 was significantly increased beginning at 6 h after stroke. (H-I) Flow cytometric quantification of Treg cells in BM and peripheral blood (PB). All plots are gated on CD4+ cells. (J–K) Quantification of flow cytometry results. (J) The percent of Treg cells in BM after ischemic stroke was modestly reduced on day 1 but significantly increased on day 3. (K) The percent of Treg cells in peripheral blood after ischemic stroke was significantly increased on day 1 and remained high through day 7. *P<0.05 vs. corresponding sham group, n=6 per group.
Bone marrow contains various immune cells, including CD4+ T cells and Treg cells; 1-2% of bone marrow mononuclear cells are CD4+T cells (Price and Cerny, 1999). In naïve mice Treg cells accounted for 13.2 ± 2.8% of all CD4+ T cells in bone marrow and 5.2 ± 1.8% of all CD4+ T cells in peripheral blood (n=6, P<0.05; Fig. 1H–K), consistent with previously published data (Chang et al., 2011; Offner et al., 2006; Yan et al., 2009). Sham-operated mice exhibited a slight decrease in Treg cells in bone marrow within 3 days, but the percent of Treg cells was not significantly different from that of naïve mice (n=6/time point, days 1, 3, 7: P>0.05; Fig. 1 H, J). However, stroke disrupted the homeostasis and caused a modest reduction in the percent of Treg cells on day 1 (n=6; P<0.05) and a significant elevation in the fraction of CD4+ T cells that were Treg cells in bone marrow on days 3 and 7 (n=6/time point, days 3, 7: P<0.05; Fig. 1H, J). The percent of Treg cells in peripheral blood also was significantly elevated on days 1, 3, and 7 after stroke (n=6/time point, days 1, 3, 7: P<0.05; Fig. 1I, K), consistent with recent data from stroke patients and experimental animals (Chang et al., 2011; Yan et al., 2009).
Experimental stroke induces systemic immune changes via the hypothalamic-pituitary axis (HPA) and the SNS (Chamorro et al., 2012). 6-OHDA, which ablates SNS innervations, and the glucocorticoid receptor inhibitor RU486 were used to confirm whether the HPA axis mediates the changes in bone marrow after stroke. RU486 had no effect on the levels of SDF-1 (Fig. 2A-C), CXCR4 (Fig. 2D, E), COX-2 (Fig. 2D, F), PGE2 (Fig. 2G), or Treg cells (Fig. 2H-K) in bone marrow after stroke (n=6, P>0.05). However, 6-OHDA significantly increased the level of SDF-1 (Fig. 2A-C) and reduced the production of CXCR4 (Fig. 2D-E), COX-2 (Fig. 2D, F), PGE2 (Fig. 2G), and Treg cells (Fig. 2H-K) in bone marrow (n=6; P<0.05). The results suggest that the changes in bone marrow are mainly mediated by the SNS rather than the HPA.
Fig. 2.

Ischemic stroke induces changes in bone marrow by activating the sympathetic nervous system (SNS), but not the hypothalamic-pituitary axis (HPA). Mice underwent middle cerebral artery occlusion (MCAO) and blockade of the SNS (6-OHDA) or HPA (RU486). One group of MCAO mice and one group of sham-operated mice were treated with ethanol/sesame oil solution (the vehicle for RU486) to rule out any effect. (A) Western blot analysis of SDF-1 in bone marrow. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a loading control. (B) Quantification showed that 6-OHDA, but not RU486, significantly increased the SDF-1 level in bone marrow on day 3. (C) ELISA analysis of SDF-1 in bone marrow (BM). (D) Western blot analysis of CXCR4 and COX-2 in bone marrow of mice that underwent MCAO and blockade of SNS or HPA. (E–F) Quantification showed that 6-OHDA, but not RU486, significantly decreased CXCR4 and COX-2 levels in bone marrow on day 1 compared with those in the vehicle-treated group. (G) ELISA analysis showed that 6-OHDA, but not RU486, significantly decreased the PGE2 level in bone marrow on day 1 compared with that in the vehicle-treated group. (H-I) Flow cytometry analysis of Treg cells in bone marrow (H) and peripheral blood (PB; I). (J-K) Quantification shows that 6-OHDA, but not RU486, significantly decreased the percent of Treg cells in bone marrow (J) and increased the percent of Treg cells in peripheral blood (K). *P<0.05 vs. 1-day PBS group, n=6 per group; #P<0.05 vs. 3-day PBS group, n=6 per group.
3.2. SNS signals regulate Treg cells and PGE2 via β2-AR and SDF-1 production through β3-AR
The results described above indicate that stroke causes changes in bone marrow through SNS signaling. However, NE released from the SNS can act on α-and β-ARs. Therefore, we examined which AR subtype mediates the increase in percent of Treg cells and expression of COX-2. We first used phentolamine to block α-ARs. Treatment of mice with phentolamine had no effect on COX-2 expression or the percent of Treg cells, (n=6, P>0.05; Fig. 3A, C, G, H). In contrast, treatment of mice with the nonselective β-AR antagonist propranolol markedly reduced bone marrow COX-2 expression and the percent of Treg cells compared to that in vehicle-treated mice (n=6, P<0.05; Fig. 3A, C, G, H). Previous studies have shown that SDF-1 level in bone marrow is controlled by β-AR signaling under homeostasis (Méndez-Ferrer et al., 2008). Therefore, we used butoxamine (selective β2-AR antagonist) and SR59230A (selective β3-AR antagonist) to identify which receptor subtype primarily mediates these changes. Treatment of MCAO mice with butoxamine reduced the production of COX-2 and the percent of Treg cells in bone marrow but did not affect SDF-1 level (n=6, P<0.05; Fig. 3A-D, G, H). Conversely, SR59230A inhibited the reduction in bone marrow SDF-1 but had no effect on COX-2 or Treg cells (n=6, P<0.05; Fig. 3A-D, G, H). Quantitative analysis of PGE-2 and SDF-1 by ELISA supported the results observed on Western blot (Fig. 3E, F). Taken together, these results suggest that the changes in PGE2 and percent of Treg cells are mediated by β2-AR signaling and that the change in SDF-1 levels is mediated by β3-AR.
Fig. 3.
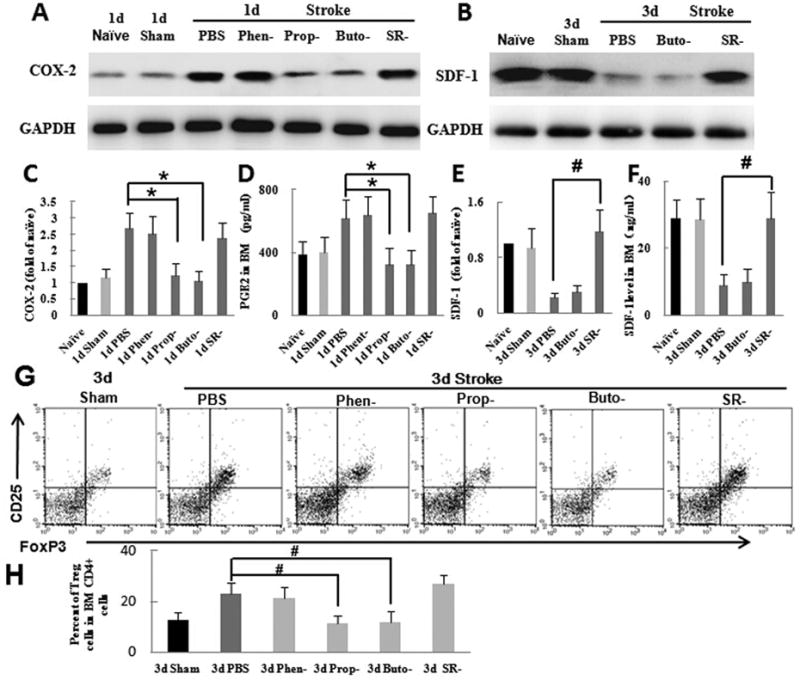
After ischemic stroke, the activated sympathetic nervous system upregulates COX-2/PGE2 by β2-adrenergic receptor (AR) signaling and downregulates SDF-1 by β3-AR signaling in bone marrow. Mice were pretreated with α-AR antagonist (phentolamine), nonselective β-AR antagonist (propranolol), selective β2-AR antagonist (butoxamine), or selective β3-AR antagonist (SR59230A) before undergoing transient middle cerebral artery occlusion (MCAO). All inhibitors were all dissolved in PBS; a control group was treated with PBS only. (A-B) Western blot analysis of COX-2 (A) and SDF-1 (B) in bone marrow. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a loading control. (C) Quantification showed that pretreatment with α-AR and β3-AR antagonists had no effect on COX-2 expression in bone marrow after ischemic stroke but that β-AR and β2-AR antagonists significantly decreased COX-2 expression compared with that in the vehicle-treated group. (D) Quantification showed that pretreatment with β3-AR antagonist significantly increased SDF-1 expression compared with that in the vehicle-treated group. (E) ELISA analysis showed that β-AR and β2-AR antagonists significantly decreased PGE2 level in bone marrow (BM) on day 1 after MCAO compared with that in the vehicle-treated group. (F) ELISA analysis showed that the β3-AR antagonist significantly increased SDF-1 level in bone marrow on day 3 after MCAO compared with that in the vehicle-treated group. (G) Flow cytometry analysis of Treg cells in bone marrow (BM) from mice treated with PBS, phentolamine, propranolol, butoxamine or SR59230A. (H) Quantification showed that propranolol and butoxamine significantly reduced the percent of Treg cells compared to that in the control group. *P<0.05 vs. 1-day PBS group, n=6 per group; #P<0.05 vs. 3-day PBS group, n=6 per group.
3.3. The PGE2-EP4 signaling pathway plays a key role in mediating the increase in bone marrow Treg cells after stroke
Our data indicate that stroke induces the increase in PGE2 production and the percent of Treg cells. To test the relationship between PGE2 and Treg cells in bone marrow, we treated mice with the selective COX-2 inhibitor indomethacin. Indomethacin-treated mice exhibited significantly fewer Treg cells in bone marrow than did vehicle-treated mice (n=6, P<0.05; Fig. 4A, B). To verify whether EP4 receptor signaling plays a role in regulating the percent of Treg cells, we administered the EP4-selective antagonist L-161,982 to MCAO mice. Notably, injection of L-161,982 significantly prevented the increase in Treg cells after stroke (n=6, P<0.05; Figure 4A, B). Under normal conditions, CD11C+ DCs express little IDO. It has been shown that the upregulation of IDO in CD11C+ DCs promotes functional development of Treg cells but does not affect the generation of Treg cells (van der Marel et al., 2007). Flow cytometry results showed that IDO production in CD11C+ DCs was significantly upregulated at 6 h after stroke, peaked on day 3, and persisted at high levels for at least 7 days (Figure 4C, D). Treatment of mice with indomethacin for 3 days inhibited the expression of IDO (Figure 4C, D), indicating that PGE2 mediates this upregulation of IDO in CD11C+ DCs after stroke. However, L-161-982 did not alter IDO expression in CD11+ DCs, suggesting that EP4 signaling does not mediate the increase in IDO expression. Taken together, our results show that PGE2 promotes the generation of bone marrow Treg cells via EP4 signaling and that the upregulation of IDO may contribute to the immunosuppressive function of Treg cells.
Fig. 4.
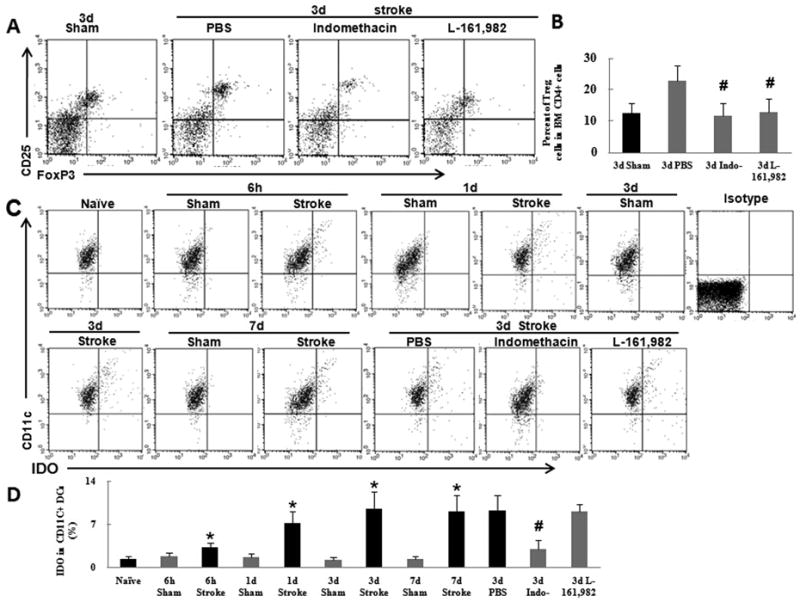
Treg cells are upregulated by the PGE2–EP4 signaling pathway in bone marrow after ischemic stroke. (A) Flow cytometry analysis of Treg cells in bone marrow (BM) from mice treated with vehicle, indomethacin, or L-161,982. (B) Quantification showed that indomethacin and L-161,982 significantly reduced the percent of Treg cells compared to that in the control group. (C) Flow cytometry analysis of IDO production in CD11C+ cells from mice that underwent ischemic stroke. (D) Quantification showed that IDO level was significantly increased in CD11C+ dendritic cells at 6 h after stroke and remained high for 3 days. This effect was abolished by treatment with indomethacin. *P<0.05 vs. corresponding sham group, n=6 per group; #P<0.05 vs. 3-day PBS group, n=6 per group.
3.4. PGE2-EP4 signaling mediates the increase in percent of Treg cells via RANKL signaling in bone marrow CD4+ T cells after stroke
Soontrapa et al. (Soontrapa et al., 2011) recently reported that PGE2–EP4 signaling mediates the ultraviolet-induced increase in number of Treg cells through upregulation of RANKL expression in the epidermis. To verify whether PGE2–EP4–RANKL signaling also mediates the increase in bone marrow Treg cells, we first investigated RANKL production in the bone marrow. Western blot results showed that the bone marrow RANKL expression was strongly upregulated at 6 h after stroke compared to that in sham-operated mice, peaked on day 1, and persisted at high levels for at least 7 days (n=6/time point, 6 h, days 1, 3, 7: P<0.05; Fig. 5A, B). To verify the effect of PGE2–EP4 signaling on RANKL production, we treated MCAO mice with indomethacin or L-161,982. Blockade of PGE2–EP4 signaling markedly inhibited the production of RANKL in bone marrow (n=6, P<0.05; Fig. 5C, D). It is well known that RANKL is mainly expressed by lymphocytes, especially CD4+ T cells in bone marrow in response to immunostimulation (Kong et al., 1999; Loser et al., 2006). Therefore, we used flow cytometry to test RANKL production in CD4+ T cells from MCAO mice. We found that RANKL production in CD4+ T cells was significantly elevated at 6 h, peaked on day 1, and remained high for at least 7 days after stroke (n=6/time point, 6 h, days 1, 3, 7: P<0.05; Fig. 5E, G). These data suggested that stroke activated CD4+ T cells and elevated RANKL level in bone marrow through PGE2–EP4 signaling. To further determine the relationship between increased RANKL and Treg cells in bone marrow, we pretreated MCAO mice with the RANKL antagonist OPG/Fc. Flow cytometry showed that OPG-treated mice had significantly fewer Treg cells in bone marrow than did vehicle-treated mice on day 3 after MCAO (n=6/time point, days 3, 7: P<0.05; Fig. 5F, H). Taken together, these findings suggest that the PGE2–EP4–RANKL pathway can mediate the increase in Treg cells in bone marrow, consistent with its reported role in the ultraviolet model (Soontrapa et al., 2011).
Fig. 5.
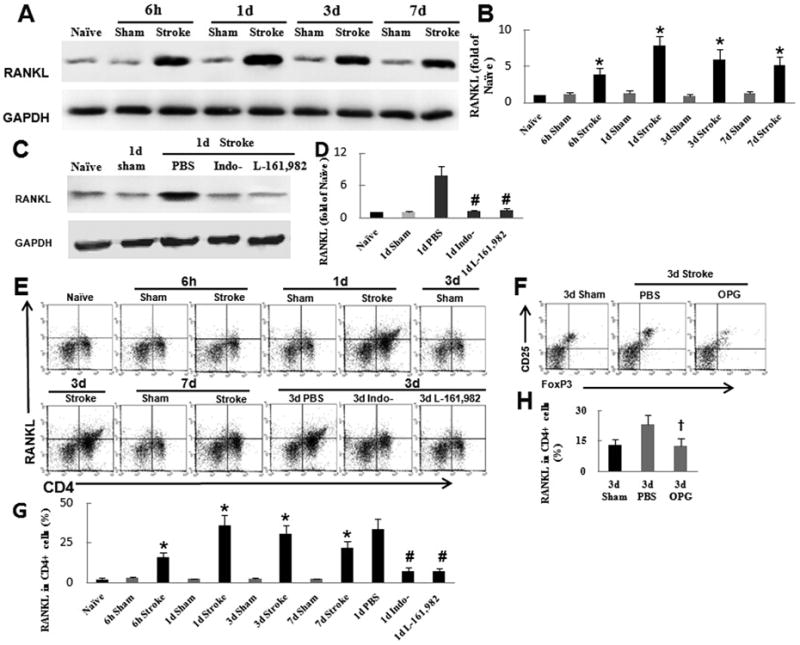
Activation of the PGE2–EP4 signaling pathway increases the percent of Treg cells in bone marrow of mice after ischemic stroke via RANKL signaling in CD4+ T cells. (A) Western blot analysis of RANKL in bone marrow from mice after ischemic stroke. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a loading control. (B) Quantification showed that ischemic stroke significantly increased RANKL expression compared with that in sham-operated mice. (C) Western blot analysis of RANKL in bone marrow from mice that were pretreated with indomethacin or L-161,982 before undergoing ischemic stroke. (D) Quantification showed that the inhibition of COX-2 with indomethacin and of EP4 with L-161,982 significantly reduced RANKL expression. (E) Flow cytometry analysis of RANKL production in CD4+ cells after stroke. (F) Flow cytometry analysis of Treg cells in the bone marrow of mice pretreated with RANKL antagonist osteoprotegerin (OPG) before ischemic stroke. (G) Quantification showed that stroke significantly increased RANKL production in CD4+ T cells. This effect was abolished by treatment with indomethacin or L-161,982. (H) Quantification showed that inhibition of RANKL significantly reduced the percent of Treg cells in bone marrow (BM) after ischemic stroke in mice.*P<0.05 vs. corresponding sham group, n=6 per group; #P<0.05 vs. 1-day PBS group,n=6 per group; †P<0.05 vs. 3-day PBS group, n=6 per group.
3.5. SNS decreases bone marrow SDF-1 level and enhances the mobilization of Treg cells from bone marrow to peripheral blood after ischemic stroke
Previous work has suggested that the CXCR4–SDF-1 axis plays an important role in controlling the migration of Treg cells. We used flow cytometry to further investigate changes of CXCR4 in Treg cells after stroke. The expression of CXCR4 in Treg cells was significantly increased on days 1 and 3 after MCAO (n=6, P<0.05; Fig. 6A, B). Pretreatment with 6-OHDA, but not RU486, significantly reduced the expression of CXCR4 on CD4+FoxP3+ Treg cells (n=6, P<0.05; Fig. 6A, B), suggesting that the SNS mediates the increase in expression of CXCR4 in Treg cells. Pretreatment of MCAO mice with SR59230A (selective β3-AR antagonist) had no effect on the percent of Treg cells in bone marrow but decreased the percent of Treg cells in peripheral blood (Fig. 6C), suggesting that decreased bone marrow SDF-1 enhances the mobilization of Treg cells to peripheral blood after stroke.
Fig. 6.
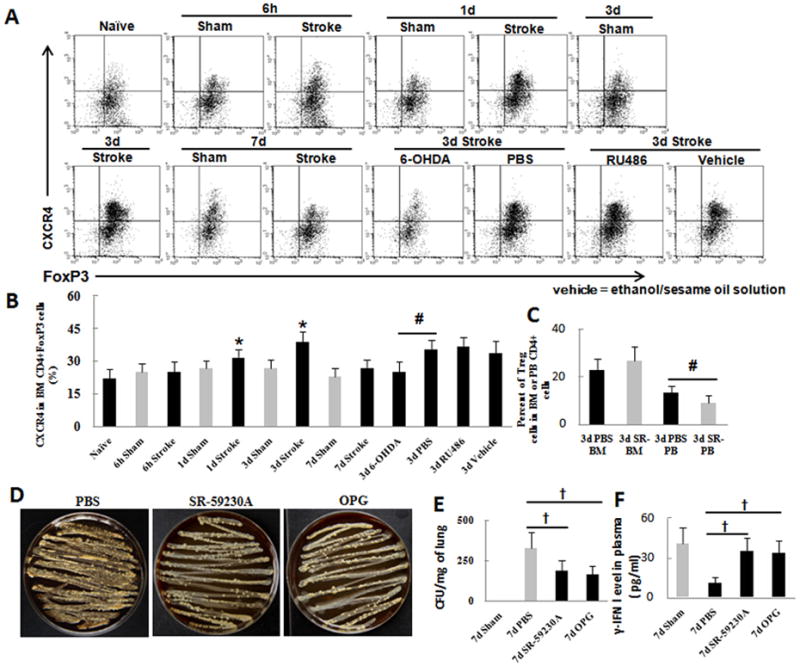
The CXCR4–SDF-1 axis contributes to the mobilization of Treg cells from bone marrow to peripheral blood after ischemic stroke and increased bacterial loads in lung. (A) Flow cytometry analysis of CXCR4 production on Treg cells of mice pretreated with RU486 or 6-OHDA before ischemic stroke. (B) Quantification showed that ischemic stroke significantly increased CXCR4-postive Treg cells in the bone marrow on day 3. Pretreatment of mice with 6-OHDA, but not RU486, significantly reduced the expression of CXCR4 on CD4+FoxP3+ Treg cells. (C) Flow cytometry analysis showed that pretreatment of mice with SR59230A before ischemic stroke had no effect on the percent of bone marrow (BM) Treg cells but decreased the percent of Treg cells in peripheral blood (PB). (D) Analysis of bacterial load in lungs of MCAO mice treated with PBS, β3-AR blocker, or RANKL inhibitor. (E) Quantification showed that treatment of MCAO mice with β3-AR or RANKL inhibitor significantly reduced bacterial load in lung on day 7 after stroke. (F) ELISA analysis showed that the β3-AR or RANKL antagonist significantly reduced γ-IFN level in plasma on day 7 after MCAO compared with that in the vehicle-treated group. *P<0.05 vs. corresponding sham group, n=6 per group; #P<0.05 vs. 3-day PBS group, n=6 per group, †P<0.05 vs. 7-day PBS group, n=6 per group.
3.6 Bone marrow Treg cells contribute to immunosuppression after stroke
The mortality of MCAO mice reaches a peak of 57% on day 6 after stroke, and the main cause of death is considered to be pulmonary infection (Meisel et al., 2004). In addition, 6-OHDA or β2-AR blockade is able to reduce the bacterial load in lung. To determine whether Treg cell contribute to immunosuppression in the delayed phase after stroke, we evaluated the bacterial load in lung of MCAO mice on day 7 after stroke by using β3-AR inhibitor SR-59230A and RANKL inhibitor OPG. Analysis of lung homogenate showed that SR-59230A or OPG-treated mice had significantly lower bacterial loads than did vehicle-treated mice, suggesting that bone marrow Treg cells may contribute to immunosuppression after stroke (Fig. 6D, E). The concentration of γ-IFN in plasma significantly increased in SR-59230A and OPG-treated mice compared to that in vehicle-treated mice on day 7 after stroke (Fig. 6F).
4. Discussion
In this study, we show that stroke induces activation of the SNS. The resulting elevation in TH leads to increased production of PGE2 via β2-AR signaling. Increased PGE2 acts directly on EP4 to mediate upregulation of RANKL, which plays an important role in regulating the percent of Treg cells in bone marrow after stroke (Fig. 5F). Although it has already been reported that bone marrow might be a priming site for generating iTreg cells, to our knowledge, this study is the first to demonstrate the molecular mechanisms that underlie this process after stroke. We found that, simultaneously, stroke leads to a significant decrease in the concentration of SDF-1 via β3-AR signaling and upregulates the expression of CXCR4 in Treg cells and other bone marrow cells. The reduction in bone marrow SDF-1 facilitates the mobilization of CXCR4+ Treg cells and HSPCs from bone marrow into peripheral blood, accounting for the increase in these cells in peripheral blood after stroke.
Bone marrow is innervated by the SNS but is also part of the lymphocyte recirculation network and a priming site for T cells in response to blood-borne antigens (Feuerer et al., 2003). Approximately 8% to 20% of bone marrow mononuclear cells are lymphocytes (Schirrmacher et al., 2003). Studies in bone metabolism have confirmed that the anti-osteogenic function of leptin, which is secreted from adipocytes, is mediated by the SNS through β2-AR signals, suggesting that the SNS can directly control the biologic function of osteoblasts (Takeda et al., 2002). Mesenchymal stem cells (MSCs) are also innervated by the SNS (Méndez-Ferrer et al., 2008). MSCs and osteoblasts have the capacity to synthesize and secrete PGE2. We found that stroke increased the levels of both COX-2 and PGE2. Treatment with 6-OHDA reduced the level of PGE2, suggesting that activation of the SNS can promote the secretion of PGE2 in bone marrow. Previous studies have reported that β2 is the only β-AR expressed in osteoblasts, but MSCs express both β2- and β3-ARs, and osteoblasts derive from MSCs (Méndez-Ferrer et al., 2008). Therefore, both β2- and β3-AR signaling can affect the production of PGE2. Treatment of mice with a selective β2-AR inhibitor, but not a β3-AR inhibitor, markedly reduced the level of COX-2/PGE2 in bone marrow, confirming that the SNS directly enhances synthesis and release of PGE2 via β2-AR signaling. Prostaglandins have been reported to stimulate expression of COX-2, suggesting that COX-2/PGE2 expression in vivo is regulated by a positive feedback mechanism (Obermajer et al., 2011). In our study, the main mediator of TH returned to normal values on day 7 after stroke; however the production of COX-2/PGE2 persisted at high levels for at least 7 days after stroke. The positive feedback mechanism may be responsible for these results.
Treg cells are composed of iTreg and nTreg cells. iTreg cells play a vital role in mediating immune tolerance or immunosuppression. Previous reports revealed that iTreg cells, which come from CD4+CD25- T cells, can be generated in the periphery by prolonged or repeated antigenic and cytokine stimulation, including PGE2 (Chen et al., 2008; Loser et al., 2006; Obermajer et al., 2011; Sakaguchi et al., 2008; Sharma et al., 2005; Soontrapa et al., 2011; Yamazaki et al., 2008). PGE2 is the most abundant prostanoid found in primates. Recent studies have confirmed that PGE2 has strong immunomodulatory effects on the immune system (Braun et al., 2005; de Visser et al., 2006; Mahic et al., 2006; Sharma et al., 2005). PGE2 inhibits T cell proliferation through EP2 receptor signals in a concentration-dependent manner, but raises the level of IDO in DCs in the presence of TNF-α (Braun et al., 2005). When unstimulated CD4+CD25- T cells are cultured for 2 days in the presence of PGE2, the expression of FoxP3 increases 3- to 5-fold in a concentration-dependent manner (Mahic et al., 2006). In our studies, the level of PGE2 in bone marrow increased rapidly beginning at 6 h after stroke. However, at 24 h after stroke, the percent of Treg cells in bone marrow was modestly decreased while the percent of Treg cells in peripheral blood was significantly increased. These results suggest that the total percent of Treg cells is unlikely to increase. Therefore, PGE2 probably does not mediate the increase in Treg cells in bone marrow directly.
Studies in bone metabolism have shown that RANKL and RANK are important regulators of osteoclasts (Ashcroft et al., 2003; Weinstein et al., 2011). RANKL expression can be upregulated by bone-resorbing factors, including PGE2 (Hofbauer et al., 2000). In the immune system, RANKL is highly expressed in activated T cells (Loser et al., 2006; Vernal et al., 2006), and RANKL cognate receptor RANK is expressed on DCs (Loser et al., 2006; Soontrapa et al., 2011). In the ultraviolet irradiation model, PGE2 increased expression of RANKL by acting through EP4 on keratinocytes (Soontrapa et al., 2011). RANKL–RANK signaling subsequently mediates the increase in Treg cells in the periphery by increasing CD205 expression in DCs (Yamazaki et al., 2008). It is clear that stroke can induce activation of lymphocytes and an increase in the number of CD4+ T cells in bone marrow (Chamorro et al., 2012; Denes et al., 2010; Meisel et al., 2005). Additionally, RANKL production can be upregulated in CD4+ T cells via exogenous stimulation in vitro and in vivo (Kong et al., 1999). In our study, we first verified that stroke significantly elevated the level of RANKL, which increased the percent of Treg cells. Treatment of MCAO mice with COX-2 inhibitor or EP4 antagonist significantly reduced RANKL production, suggesting that PGE2-EP4 signaling mediated the increase in bone marrow RANKL. Flow cytometry analysis further revealed that CD4+ T cells are a primary source for RANKL in bone marrow after stoke. Together with our finding that stroke increases the production of PGE2 in bone marrow via β2-AR signals, our results also show that the SNS can mediate the increase in Treg cells in bone marrow via PGE2-EP4 signaling on CD4+ T cells.
IDO, which is the rate-limiting enzyme in the kynurenine pathway, is mainly present in macrophages and DCs (van der Marel et al., 2007). During the progress of iTreg development, the IDO pathway is essential for plasmacytoid DC-driven Treg generation from CD4+CD25- T cells (Chen et al., 2008). Blockade of IDO does not affect FoxP3 expression but inhibits the development of functional Treg cells (van der Marel et al., 2007). In our studies, SNS increased the level of IDO via PGE2 signaling, suggesting that iTreg generation from the bone marrow microenvironment is functional and can induce immune tolerance in the periphery.
Under homeostatic conditions, circadian oscillations of SDF-1 in bone marrow are controlled by the SNS (Méndez-Ferrer et al., 2008). Granulocyte-colony stimulating factor induces stem cell mobilization by decreasing bone marrow SDF-1 and upregulating CXCR4 through activation of SNS (Petit et al., 2002), suggesting that disruption of the CXCR4–SDF-1 axis has profound influence on bone marrow. We found that acute stroke constitutively activates SNS and leads to elevated expression of TH and NE, which acts on the β3-AR to inhibit MSC secretion of SDF-1. The resulting reduction in SDF-1 in bone marrow promotes mobilization of CXCR4+ cells, including Treg cells, to peripheral blood. Therefore, disruption in the CXCR4–SDF-1 equilibrium may be responsible for the alterations in percent of Treg cells in bone marrow and peripheral blood on day 1 after stroke and for the increase in percent of Treg cells in peripheral blood on day 3 after stroke. A recent study showed that the SNS negatively regulates the percent of Treg cells in the spleen. 6-OHDA treatment significantly reduced the concentration of NE and increased the percent of Treg cells in spleen through inhibition of apoptosis (Wirth et al., 2014). However, we and other groups found that stroke-induced SNS activity not only elevates NE level but also increases the percent of Treg cells in bone marrow and peripheral blood (Chang et al., 2011). In fact, the two findings are not contradictory. An ex vivo experiment showed that NE induces the apoptosis of Treg cells in a dose-dependent manner via β2-AR signaling; however, a concentration of NE less than 10-4 M did not increase apoptosis of CD4+CD25+Treg cells (Wirth et al., 2014). Under steady state conditions, the concentration of NE in spleen (∼100 ng/g) is significantly higher than that in bone marrow (1-3 ng/g) and peripheral blood (<1 μM/L) (Bellinger et al., 2008; Chang et al., 2011). 6-OHDA treatment reduced the concentration of NE to less than 17% and led to an increase in the percent of Treg cells in spleen (Wirth et al., 2014), suggesting that an in vivo NE concentration less than 17 ng/g does not induce apoptosis of Treg cells. In our research and that of others, stroke led to a 2-fold increase of NE in bone marrow and peripheral blood, but the concentration of NE remained less than that in spleen after 6-OHDA treatment (Chang et al., 2011). Therefore, the NE concentration after stroke does not increase apoptosis of Treg cells. In addition, treatment of mice after stroke with cocaine- and amphetamine-regulated transcript downregulated the NE level and simultaneously reduced the percent of Treg cells in peripheral blood (Chang et al., 2011), suggesting that increased NE may be responsible for the increase in percent of Treg cells after stroke, further supporting our findings.
Stroke can cause immunodepression syndrome, which is characterized by the apoptosis and inactivation of lymphocytes and a shift from T helper cell (Th)1 toTh2 cytokine production (Meisel et al., 2004; Meisel et al., 2005; Offner et al., 2006; Prass et al., 2003; Wirth et al., 2014). It has been shown that an approximately 90% reduction in spleen and thymus cell numbers in MCAO mice leads to significant atrophy of spleen and thymus (Offner et al., 2006). The lost components are mainly immune cells, including B cells, T cells, and natural kill cells (Offner et al., 2006). The loss and deactivation of lymphocytes may contribute to decreased production of γ-IFN (Chamorro et al., 2012; Wirth et al., 2014), which results in increased mortality from bacteremia and/or pneumonia (Offner et al., 2006; Prass et al., 2003; Venet et al., 2009). A recent emerging concept of immunodepression after stroke is the increase in Treg cells (Chang et al., 2011; Offner et al., 2006; Offner et al., 2009; Yan et al., 2009). Studies have shown that Treg cells have an immunosuppressive function in cancer, autoimmunity, allergy, trauma, and infectious diseases, such as pneumonia and sepsis, through cell-to-cell contact and/or secretion of immunosuppressive cytokines, including IL-10 (Dziennis et al., 2011; MacConmara et al., 2006; Nascimento et al., 2010; Venet et al., 2009; Wing and Sakaguchi, 2010; Zhang et al., 2010). Manifestations of systemic immunodepression are not unique to stroke, as they can also occur after traumatic injury, severe burns, or brain surgery (Chamorro et al., 2012). These injuries all seem to affect the innate and adaptive immune responses profoundly. Treg cells promote increased susceptibility to subsequent bacterial infections by reducing production of γ-IFN in these disorders (MacConmara et al., 2006; Venet et al., 2009). However, there is no direct evidence to show the relationship between exogenous Treg and immunosuppression after stroke. We found that inhibiting the increase in bone marrow Treg cells led to reductions in bacterial load in lung and increases in plasma level of γ-IFN. Together with previous findings, we conclude that exogenous Treg cells contribute to stroke-induced immunosuppression by inhibiting the secretion of γ-IFN. Although Liesz et al. showed that depletion of endogenous Treg cells with anti-CD25 in MCAO mice led to significant downregulation of IL-10 level and upregulation of γ-IFN in peripheral blood after stroke (Liesz et al., 2009b; Liesz et al., 2013), the effect of Treg cells on the peripheral immune system has not been examined. We are the first to confirm the role of Treg cells in immunosuppression after stroke.
A recent study showed that transfer of Treg cells collected from inguinal and axillary lymph nodes and spleens of MCAO mice could not increase the bacterial loads on day 3 after stroke (Li et al., 2013). The source of Treg cells and time point used may account for the discrepancy First, Treg cells derived from bone marrow are more suppressive than other Treg cells in thymus and spleen because they express higher levels of FoxP3 and CD25 (Zou et al., 2004). Second, CD4+ CD25+ T cells are not a homogeneous population; they contain varying numbers of regulatory and non-regulatory T cells (Yagi et al., 2004). Transfer of naïve lymphocytes or splenocytes into MCAO mice can inhibit the reduction of plasma γ-IFN and prevent pneumonia (Prass et al., 2003). Third, the effect on the innate and adaptive immune system of deleting or increasing endogenoucs Treg cells does not become evident until 3 days after stroke, indicating that endogenous Treg cells may primarily be involved in immune response during the delayed phase of stroke (Chamorro et al., 2012; Liesz et al., 2009; Liesz et al., 2013).
The major limitation of our study is the use of pharmacological inhibitors in animals. Although we paid great attention to the potential side effects of these inhibitors, we cannot exclude potential systemic effects of these agents beyond their impact on immune cells in vivo.
In summary, our study reveals novel mechanistic insights into the generation of bone marrow Treg cells and mobilization of CXCR4+ cells from the bone marrow to the circulation after stroke. We show that stroke-induced activation of the SNS mediates the generation of bone marrow Treg cells by β2-AR-EP4-RANKL signaling. Additionally, we found that bone marrow SDF-1 degradation by β3-AR signaling and upregulation of CXCR4 in bone marrow cells facilitate the mobilization of Treg cells and other CXCR4+ cells, including HSPCs, to peripheral blood. These changes could account for the increase in Treg cells and HSPCs in peripheral blood after stroke. Manipulation of these interactions could lead to new perspectives on the development of improved clinical protocols for stroke-induced immunosuppression.
Fig. 7.
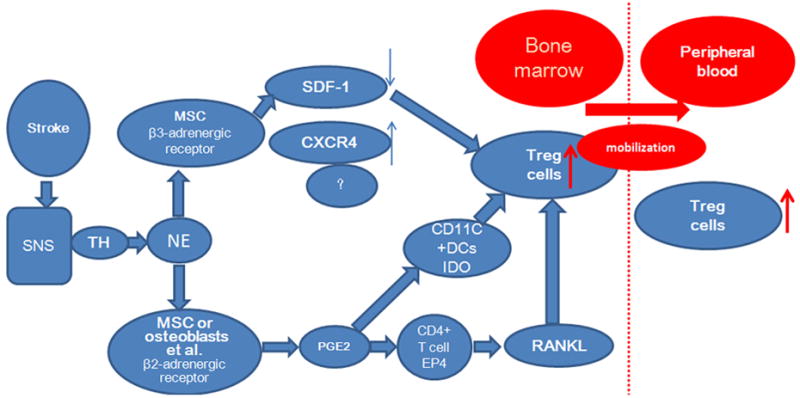
Diagram of the proposed mechanism by which the sympathetic nervous system (SNS) promotes mobilization of T regulatory (Treg) cells from the bone marrow to peripheral blood. CXCR4, C-X-C chemokine receptor; DCs, dendritic cells; IDO, indoleamine 2,3 dioxygenase; MSC, mesenchymal stem cell; NE, norepinephrine; PGE2, prostaglandin E2; RANKL, receptor activator for NF-κB ligand; TH, tyrosine hydroxylase.
Acknowledgments
This work was supported by grants from NSFC (81271284), Medical Science and Technology Research Programs of Henan Province (WKJ2010-2-016), The Overseas Training Program of Henan Province Medical Academic Leaders (2011023), AHA 13GRNT15730001, and NIH (K01AG031926, R01AT007317, R01NS078026). We thank Claire Levine for assistance with this manuscript.
Footnotes
Declaration of Conflicting Interests: No potential conflicts of interest were disclosed.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ashcroft A, Cruickshank S, Croucher P, Perry M, Rollinson S, Lippitt J, Child J, Dunstan C, Felsburg P, Morgan G. Colonic dendritic cells, intestinal inflammation, and T cell-mediated bone destruction are modulated by recombinant osteoprotegerin. Immunity. 2003;19:849–861. doi: 10.1016/s1074-7613(03)00326-1. [DOI] [PubMed] [Google Scholar]
- Bellinger DL, Millar BA, Perez S, Carter J, Wood C, ThyagaRajan S, Molinaro C, Lubahn C, Lorton D. Sympathetic modulation of immunity: relevance to disease. Cellular immunology. 2008;252:27–56. doi: 10.1016/j.cellimm.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun D, Longman RS, Albert ML. A two-step induction of indoleamine 2, 3 dioxygenase (IDO) activity during dendritic-cell maturation. Blood. 2005;106:2375–2381. doi: 10.1182/blood-2005-03-0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamorro A, Meisel A, Planas AM, Urra X, van de Beek D, Veltkamp R. The immunology of acute stroke. Nat Rev Neurol. 2012;8:401–410. doi: 10.1038/nrneurol.2012.98. [DOI] [PubMed] [Google Scholar]
- Chamorro Á, Urra X, Planas AM. Infection after acute ischemic stroke a manifestation of brain-induced immunodepression. Stroke. 2007;38:1097–1103. doi: 10.1161/01.STR.0000258346.68966.9d. [DOI] [PubMed] [Google Scholar]
- Chang L, Chen Y, Li J, Liu Z, Wang Z, Chen J, Cao W, Xu Y. Cocaine-and amphetamine-regulated transcript modulates peripheral immunity and protects against brain injury in experimental stroke. Brain, behavior, and immunity. 2011;25:260–269. doi: 10.1016/j.bbi.2010.09.017. [DOI] [PubMed] [Google Scholar]
- Chen W, Liang X, Peterson AJ, Munn DH, Blazar BR. The indoleamine 2, 3-dioxygenase pathway is essential for human plasmacytoid dendritic cell-induced adaptive T regulatory cell generation. The Journal of Immunology. 2008;181:5396–5404. doi: 10.4049/jimmunol.181.8.5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nature reviews cancer. 2006;6:24–37. doi: 10.1038/nrc1782. [DOI] [PubMed] [Google Scholar]
- Denes A, McColl BW, Leow-Dyke SF, Chapman KZ, Humphreys NE, Grencis RK, Allan SM, Rothwell NJ. Experimental stroke-induced changes in the bone marrow reveal complex regulation of leukocyte responses. Journal of Cerebral Blood Flow & Metabolism. 2010;31:1036–1050. doi: 10.1038/jcbfm.2010.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U, Klehmet J, Braun JS, Harms H, Meisel C, Ziemssen T, Prass K, Meisel A. Stroke-induced immunodepression experimental evidence and clinical relevance. Stroke. 2007;38:770–773. doi: 10.1161/01.STR.0000251441.89665.bc. [DOI] [PubMed] [Google Scholar]
- Dutta P, Courties G, Wei Y, Leuschner F, Gorbatov R, Robbins CS, Iwamoto Y, Thompson B, Carlson AL, Heidt T. Myocardial infarction accelerates atherosclerosis. Nature. 2012;487:325–329. doi: 10.1038/nature11260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziennis S, Akiyoshi K, Subramanian S, Offner H, Hurn PD. Role of dihydrotestosterone in post-stroke peripheral immunosuppression after cerebral ischemia. Brain, behavior, and immunity. 2011;25:685–695. doi: 10.1016/j.bbi.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerer M, Beckhove P, Garbi N, Mahnke Y, Limmer A, Hommel M, Hämmerling GJ, Kyewski B, Hamann A, Umansky V. Bone marrow as a priming site for T-cell responses to blood-borne antigen. Nature medicine. 2003;9:1151–1157. doi: 10.1038/nm914. [DOI] [PubMed] [Google Scholar]
- Hagendorff A, Dettmers C, Danos P, Hümmelgen M, Vahlhaus C, Martin C, Heusch G, Lüderitz B. Cerebral vasoconstriction during sustained ventricular tachycardia induces an ischemic stress response of brain tissue in rats. Journal of molecular and cellular cardiology. 1998;30:2081–2094. doi: 10.1006/jmcc.1998.0772. [DOI] [PubMed] [Google Scholar]
- Hofbauer LC, Khosla S, Dunstan CR, Lacey DL, Boyle WJ, Riggs BL. The roles of osteoprotegerin and osteoprotegerin ligand in the paracrine regulation of bone resorption. Journal of Bone and Mineral Research. 2000;15:2–12. doi: 10.1359/jbmr.2000.15.1.2. [DOI] [PubMed] [Google Scholar]
- Inoue H, Kondo A, Togari A. Activation of the peripheral sympathetic nervous system increased the expression of cyclooxygenase-2 (COX-2) mRNA in mouse calvaria. Neuroscience letters. 2003;338:37–40. doi: 10.1016/s0304-3940(02)01352-6. [DOI] [PubMed] [Google Scholar]
- Jin R, Zhu X, Liu L, Nanda A, Granger DN, Li G. Simvastatin Attenuates Stroke-induced Splenic Atrophy and Lung Susceptibility to Spontaneous Bacterial Infection in Mice. Stroke. 2013;44:1135–1143. doi: 10.1161/STROKEAHA.111.000633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, Capparelli C, Li J, Elliott R, McCabe S. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402:43–47. doi: 10.1038/46303. [DOI] [PubMed] [Google Scholar]
- Li P, Mao L, Zhou G, Leak RK, Sun BL, Chen J, Hu X. Adoptive regulatory T-cell therapy preserves systemic immune homeostasis after cerebral ischemia. Stroke. 2013;44:3509–3515. doi: 10.1161/STROKEAHA.113.002637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S, Giese T, Veltkamp R. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nature medicine. 2009;15:192–199. doi: 10.1038/nm.1927. [DOI] [PubMed] [Google Scholar]
- Liesz A, Zhou W, Na SY, Hämmerling GJ, Garbi N, Karcher S, Mracsko E, Backs J, Rivest S, Veltkamp R. Boosting regulatory T cells limits neuroinflammation in permanent cortical stroke. The Journal of Neuroscience. 2013;33:17350–17362. doi: 10.1523/JNEUROSCI.4901-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wang L, Kikuiri T, Akiyama K, Chen C, Xu X, Yang R, Chen W, Wang S, Shi S. Mesenchymal stem cell-based tissue regeneration is governed by recipient T lymphocytes via IFN-[gamma] and TNF-[alpha] Naturemedicine. 2011;17:1594–1601. doi: 10.1038/nm.2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loser K, Mehling A, Loeser S, Apelt J, Kuhn A, Grabbe S, Schwarz T, Penninger JM, Beissert S. Epidermal RANKL controls regulatory T-cell numbers via activation of dendritic cells. Nature medicine. 2006;12:1372–1379. doi: 10.1038/nm1518. [DOI] [PubMed] [Google Scholar]
- Lucin KM, Sanders VM, Popovich PG. Stress hormones collaborate to induce lymphocyte apoptosis after high level spinal cord injury. Journal of neurochemistry. 2009;110:1409–1421. doi: 10.1111/j.1471-4159.2009.06232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méndez-Ferrer S, Lucas D, Battista M, Frenette PS. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. 2008;452:442–447. doi: 10.1038/nature06685. [DOI] [PubMed] [Google Scholar]
- MacConmara MP, Maung AA, Fujimi S, McKenna AM, Delisle A, Lapchak PH, Rogers S, Lederer JA, Mannick JA. Increased CD4+ CD25+ T regulatory cell activity in trauma patients depresses protective Th1 immunity. Annals of surgery. 2006;244:514. doi: 10.1097/01.sla.0000239031.06906.1f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahic M, Yaqub S, Johansson CC, Taskén K, Aandahl EM. FOXP3+ CD4+ CD25+ adaptive regulatory T cells express cyclooxygenase-2 and suppress effector T cells by a prostaglandin E2-dependent mechanism. The Journal of Immunology. 2006;177:246–254. doi: 10.4049/jimmunol.177.1.246. [DOI] [PubMed] [Google Scholar]
- Meisel C, Prass K, Braun J, Victorov I, Wolf T, Megow D, Halle E, Volk HD, Dirnagl U, Meisel A. Preventive antibacterial treatment improves the general medical and neurological outcome in a mouse model of stroke. Stroke. 2004;35:2–6. doi: 10.1161/01.STR.0000109041.89959.4C. [DOI] [PubMed] [Google Scholar]
- Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U. Central nervous system injury-induced immune deficiency syndrome. Nature Reviews Neuroscience. 2005;6:775–786. doi: 10.1038/nrn1765. [DOI] [PubMed] [Google Scholar]
- Nascimento DC, Alves-Filho JC, Sônego F, Fukada SY, Pereira MS, Benjamim C, Zamboni DS, Silva JS, Cunha FQ. Role of regulatory T cells in long-term immune dysfunction associated with severe sepsis. Critical care medicine. 2010;38:1718–1725. doi: 10.1097/CCM.0b013e3181e78ad0. [DOI] [PubMed] [Google Scholar]
- Obermajer N, Muthuswamy R, Lesnock J, Edwards RP, Kalinski P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood. 2011;118:5498–5505. doi: 10.1182/blood-2011-07-365825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offner H, Subramanian S, Parker SM, Wang C, Afentoulis ME, Lewis A, Vandenbark AA, Hurn PD. Splenic atrophy in experimental stroke is accompanied by increased regulatory T cells and circulating macrophages. The Journal of Immunology. 2006;176:6523–6531. doi: 10.4049/jimmunol.176.11.6523. [DOI] [PubMed] [Google Scholar]
- Offner H, Vandenbark A, Hurn P. Effect of experimental stroke on peripheral immunity: CNS ischemia induces profound immunosuppression. Neuroscience. 2009;158:1098–1111. doi: 10.1016/j.neuroscience.2008.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paczkowska E, Larysz B, Rzeuski R, Karbicka A, Jałowiński R, Kornacewicz-Jach Z, Ratajczak M, Machaliński B. Human hematopoietic stem/progenitor‐enriched CD34+ cells are mobilized into peripheral blood during stress related to ischemic stroke or acute myocardial infarction. European journal of haematology. 2005;75:461–467. doi: 10.1111/j.1600-0609.2005.00536.x. [DOI] [PubMed] [Google Scholar]
- Petit I, Szyper-Kravitz M, Nagler A, Lahav M, Peled A, Habler L, Ponomaryov T, Taichman RS, Arenzana-Seisdedos F, Fujii N. G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nature immunology. 2002;3:687–694. doi: 10.1038/ni813. [DOI] [PubMed] [Google Scholar]
- Porter RL, Georger MA, Bromberg O, McGrath KE, Frisch BJ, Becker MW, Calvi LM. Prostaglandin E2 increases hematopoietic stem cell survival and accelerates hematopoietic recovery after radiation injury. Stem Cells. 2013;31:372–383. doi: 10.1002/stem.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prass K, Meisel C, Höflich C, Braun J, Halle E, Wolf T, Ruscher K, Victorov IV, Priller J, Dirnagl U. Stroke-induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1–like immunostimulation. The Journal of experimental medicine. 2003;198:725–736. doi: 10.1084/jem.20021098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price PW, Cerny J. Characterization of CD4+ T cells in mouse bone marrow. I. Increased activated/memory phenotype and altered TCR Vβ repertoire. European journal of immunology. 1999;29:1051–1056. doi: 10.1002/(SICI)1521-4141(199903)29:03<1051::AID-IMMU1051>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Schirrmacher V, Feuerer M, Fournier P, Ahlert T, Umansky V, Beckhove P. T-cell priming in bone marrow: the potential for long-lasting protective anti-tumor immunity. Trends in molecular medicine. 2003;9:526–534. doi: 10.1016/j.molmed.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Sharma S, Yang SC, Zhu L, Reckamp K, Gardner B, Baratelli F, Huang M, Batra RK, Dubinett SM. Tumor Cyclooxygenase-2/Prostaglandin E2–dependent promotion of FoxP3 expression and CD4+ CD25+ T regulatory cell activities in lung cancer. Cancer research. 2005;65:5211–5220. doi: 10.1158/0008-5472.CAN-05-0141. [DOI] [PubMed] [Google Scholar]
- Soontrapa K, Honda T, Sakata D, Yao C, Hirata T, Hori S, Matsuoka T, Kita Y, Shimizu T, Kabashima K. Prostaglandin E2–prostoglandin E receptor subtype 4 (EP4) signaling mediates UV irradiation-induced systemic immunosuppression. Proceedings of the National Academy of Sciences. 2011;108:6668–6673. doi: 10.1073/pnas.1018625108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Elefteriou F, Levasseur R, Liu X, Zhao L, Parker KL, Armstrong D, Ducy P, Karsenty G. Leptin regulates bone formation via the sympathetic nervous system. Cell. 2002;111:305–317. doi: 10.1016/s0092-8674(02)01049-8. [DOI] [PubMed] [Google Scholar]
- Tilley SL, Coffman TM, Koller BH. Mixed messages: modulation of inflammation and immune responses by prostaglandins and thromboxanes. Journal of Clinical Investigation. 2001;108:15–23. doi: 10.1172/JCI13416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Marel AP, Samsom JN, Greuter M, van Berkel LA, O'Toole T, Kraal G, Mebius RE. Blockade of IDO inhibits nasal tolerance induction. J Immunol. 2007;179:894–900. doi: 10.4049/jimmunol.179.2.894. [DOI] [PubMed] [Google Scholar]
- Venet F, Chung CS, Kherouf H, Geeraert A, Malcus C, Poitevin F, Bohé J, Lepape A, Ayala A, Monneret G. Increased circulating regulatory T cells (CD4+ CD25+ CD127−) contribute to lymphocyte anergy in septic shock patients. Intensive care medicine. 2009;35:678–686. doi: 10.1007/s00134-008-1337-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernal R, Dutzan N, Hernández M, Chandía S, Puente J, León R, García L, Valle ID, Silva A, Gamonal J. High expression levels of receptor activator of nuclear factor-kappa B ligand associated with human chronic periodontitis are mainly secreted by CD4+ T lymphocytes. Journal of periodontology. 2006;77:1772–1780. doi: 10.1902/jop.2006.050376. [DOI] [PubMed] [Google Scholar]
- Wang J, Yu L, Jiang C, Chen M, Ou C, Wang J. Bone marrow mononuclear cells exert long-term neuroprotection in a rat model of ischemic stroke by promoting arteriogenesis and angiogenesis. Brain, behavior, and immunity. 2013;34:56–66. doi: 10.1016/j.bbi.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein RS, O'Brien CA, Almeida M, Zhao H, Roberson PK, Jilka RL, Manolagas SC. Osteoprotegerin prevents glucocorticoid-induced osteocyte apoptosis in mice. Endocrinology. 2011;152:3323–3331. doi: 10.1210/en.2011-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol. 2010;11:7–13. doi: 10.1038/ni.1818. [DOI] [PubMed] [Google Scholar]
- Wirth T, Westendorf AM, Bloemker D, Wildmann J, Engler H, Mollerus S, Wadwa M, Schäfer MK, Schedlowski M, del Rey A. The sympathetic nervous system modulates CD4+ FoxP3+ regulatory T cells via noradrenaline-dependent apoptosis in a murine model of lymphoproliferative disease. Brain, behavior, and immunity. 2014;38:100–110. doi: 10.1016/j.bbi.2014.01.007. [DOI] [PubMed] [Google Scholar]
- Yagi H, Nomura T, Nakamura K, Yamazaki S, Kitawaki T, Hori S, Maeda M, Onodera M, Uchiyama T, Fujii S. Crucial role of FOXP3 in the development and function of human CD25+ CD4+ regulatory T cells. International immunology. 2004;16:1643–1656. doi: 10.1093/intimm/dxh165. [DOI] [PubMed] [Google Scholar]
- Yamazaki S, Dudziak D, Heidkamp GF, Fiorese C, Bonito AJ, Inaba K, Nussenzweig MC, Steinman RM. CD8+ CD205+ splenic dendritic cells are specialized to induce Foxp3+ regulatory T cells. The Journal of Immunology. 2008;181:6923–6933. doi: 10.4049/jimmunol.181.10.6923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Greer JM, Etherington K, Cadigan GP, Cavanagh H, Henderson RD, O'Sullivan JD, Pandian JD, Read SJ, McCombe PA. Immune activation in the peripheral blood of patients with acute ischemic stroke. J Neuroimmunol. 2009;206:112–117. doi: 10.1016/j.jneuroim.2008.11.001. [DOI] [PubMed] [Google Scholar]
- Zhang B, Subramanian S, Dziennis S, Jia J, Uchida M, Akiyoshi K, Migliati E, Lewis AD, Vandenbark AA, Offner H. Estradiol and G1 reduce infarct size and improve immunosuppression after experimental stroke. The Journal of Immunology. 2010;184:4087–4094. doi: 10.4049/jimmunol.0902339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Barnett B, Safah H, LaRussa VF, Evdemon-Hogan M, Mottram P, Wei S, David O, Curiel TJ, Zou W. Bone marrow is a reservoir for CD4+ CD25+ regulatory T cells that traffic through CXCL12/CXCR4 signals. Cancer research. 2004;64:8451–8455. doi: 10.1158/0008-5472.CAN-04-1987. [DOI] [PubMed] [Google Scholar]