

Abstract
Adaptive immune responses to antigens released by dying cells play a critical role in the development of autoimmunity, allograft rejection, and spontaneous as well as therapy-induced tumor rejection. Although cell death in these situations is considered sterile, various reports have implicated type I IFNs as drivers of the ensuing adaptive immune response to cell-associated antigens. However, the mechanisms that underpin this type I IFN production are poorly defined. Here we show that dendritic cells (DCs) can uptake and sense nuclear DNA-associated entities released by dying cells to induce type I IFN. Remarkably this molecular pathway requires STING but not TLR or NLR function and results in the activation of IRF3 in a TBK1-dependent manner. DCs are shown to depend on STING function in vivo to efficiently prime IFN-dependent CD8+ T cell responses to tumor antigens. Furthermore, loss of STING activity in DCs impairs the generation of follicular helper T (Tfh) and plasma cells as well as anti-nuclear antibodies in an inducible model of systemic lupus erythematosus (SLE). These findings suggest that the STING pathway could be manipulated to enable the rational design of immunotherapies that enhance or diminish anti-tumor and autoimmune responses, respectively.
Introduction
The immune system carefully balances its response to dead and dying cells in order to maintain homeostasis and prevent the development of autoimmunity. Although uptake and clearance of dying cells is generally considered a tolerogenic process, the existence of immunogenic cell death has been well described (1). Depending on the nature of the cell death, dying cells can emit damage-associated molecular patterns (DAMPs)(2) that act as danger signals and increase a dying cell’s immunogenicity. Many of these DAMPs seem to use the sensing and signaling pathways that are normally associated with for the recognition and elimination of pathogens. Given the importance of immune responses to cell-associated antigens in autoimmunity, allograft rejection, and tumor rejection, the identification of these DAMPs, their cognate sensors, and their pro-inflammatory sequelae have become topics of intense research.
Ample studies have implicated type I IFNs in the development or progression of immune responses to self-antigens in autoimmune diseases such as rheumatoid arthritis (RA), type I diabetes mellitus (T1D), Sjögren’s syndrome and systemic lupus erythematosus (SLE)(3). However, type I IFNs were only recently identified as a crucial mediator in the priming of CD8+ T cells to cell-associated antigens in cancer and cancer treatments. Mice lacking type I IFN sensing—either by genetic IFN receptor (IFNAR) deletion or treatment with blocking Ab—develop more chemically-induced tumors and show poorer rejection of transplanted immunogenic tumors than WT mice, highlighting the requirement for type I IFN in spontaneous tumor rejection (4, 5). Additional studies showed that the spontaneous induction of tumor-specific CD8+ T cells in tumor-bearing mice was predominantly mediated by type I IFN sensing in dendritic cells (DC)(6, 7). A similar role for type I IFN was seen in therapy-induced tumor elimination. Burnette and Kang showed increased intratumoral production of type I IFN upon ablative radiotherapy or chemotherapy (8, 9). The ablative effect of the therapy was associated with enhanced (cross) priming capacity of tumor-infiltrating DCs and could be abolished by eliminating IFNAR from the hematopoietic compartment. Our previous work, using tumor cell therapy in vaccination and therapeutic settings, showed a comparable dependency of type I IFN in the induction of protective anti-tumor CD8+ T cell responses (10–12).
While the general immunostimulatory effects of type I IFN on DCs are well studied, little is known on the cellular source of type I IFN, the type I IFN-inducing ligand, and the receptor/signaling pathways involved in its induction upon the sensing and clearance of dying cells. Our previous work indicated that DCs can produce type I IFN upon phagocytosis of dying cells (10–12). Importantly, DCs from MyD88−/−/TRIFlps/lps double deficient mice showed normal type I IFN production upon phagocytosis of dying cells and type I IFN-dependent CD8+T cell priming to tumor-cell vaccines was comparable in WT and MyD88−/−/TRIFlps/lps mice, indicating that the type I IFN induction requires an unidentified TLR-independent sensing pathway (11).
Using various murine cancer and tumor cell vaccination models and in vitro approaches, we show that the type I IFN production upon sensing of dying cells is not only TLR-independent, but also RLR-independent and requires stimulator of IFN genes (STING)-IRF3 mediated sensing of apoptotic cell-derived nuclear DNA structures by DCs. The ensuing type I IFN production enhances DC functionality in an autocrine manner, resulting in the increased clonal expansion, poly-functionality, and memory formation of tumor-specific CD8+T cells. Importantly, the role of the STING/IRF3/IFNAR nexus was not limited to CD8+ T cell priming or tumor models; elimination of STING or IFNAR significantly impacted the development of CD4+T follicular helper cells, plasma cells, and anti-nuclear antibodies in an inducible model of SLE. Collectively, our results demonstrate that STING/IRF3 sensing of nuclear DNA-derived structures by DCs broadly drives the priming of adaptive immune responses to dying cells.
Materials and methods
Mice, cells lines, and peptides
Mice were maintained under specific pathogen-free conditions in accordance with guidelines by the Association for Assessment and Accreditation of Laboratory Animal Care International. C57BL/6J, B6.PL-Thy1a/CyJ (B6/CD90.1), B6.SJL.Ptpcra (B6/CD45.1), CD11c-DTR mice and B6(C)-H2-Ab1bm12/KhEgJ mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and H2-Kb−/− from Harlan (Indianapolis, IN). IFNAR−/−, MyD88−/−/TRIFlps/lps, CD11c-DTR-IFNAR−/−, and Act-mOVA/H2-Kb−/− mice were bred in our facility. IRF3−/− and IRF7−/− mice were a gift from Dr. K. Fitzgerald (U. Massachusetts, MA), STING−/− mice a gift form Dr. R. Vance (Berkeley, CA), and IPS-1−/− mice a gift from Dr. J. Tschopp (UNIL, Lausanne). Mixed bone marrow chimeric mice were generated using donors on different congenic backgrounds in 1:1 ratios. All BM chimeric mice were rested for 12 weeks before experiments were started.
B16/F10, B16F10-OVA (B16-OVA), EL-4mOVA, MEC.B7.SigOVA, Tap sufficient and deficient MEFs expressing the human adenovirus type 5 early region 1 (Ad5E1-TAKO, I1.2), and ISRE-L929 IFN reporter cells have been described before (13–16). Peptides OVA257–264 (SIINFEKL), E1B192–200 (VNIRNCCYI), TRP-2180-188 (SVYDFFVWL) and LCMV GP33–41 (KAVYNFATC), were obtained from A&A Laboratories (San Diego, CA).
Cell isolations
T cells, B cells, Mph and DCs were sorted by flow cytometry using markers TCRβ, CD4, CD8, CD11c, CD11b, CD19, CD45R, MHC II as described before (12). Purity of sorted cells was generally >98% and viability was >97% as determined by 7-AAD staining.
Erythrocytes, normoblasts, reticulocytes and red blood cells were generated using a mouse adapted protocol for the long-term ex vivo erythroid differentiation culture protocol described by Giarratana et al (17) and Konstantinidis et al (18). Briefly, low-density bone marrow cells were cultured in erythroblast growth medium (StemPro-34 with 2.6% StemPro-34 supplement; Invitrogen), 20% BIT 9500 (StemCell Technologies), 900 ng/mL ferrous sulfate, 90 ng/mL ferrous nitrate, 10−6M hydrocortisone, penicillin/streptomycin, L-glutamine), in 3 subsequent phases. For the proliferative phase (days 1–5) cells were expanded with 100 ng/mL SCF, 5 ng/mL IL-3, and 2 IU/mL human erythropoietin (Amgen). In the differentiation step (days 6–7), the cells were supplemented with only erythropoietin in fibronectin coated plates. For enucleation (day 8–9) cells were grown without cytokines. Cells were sorted based on size gating and nucleotide staining using different combinations of Syto-16, Draq5, Mitotracker Red and MitotrackerGreen (Invitrogen).
DC cytokine production, phagocytosis and T cell activation
Flow cytometry purified DCs were cultured in a 1:3 ratio with irradiated cells (1500–3000 Rad). Parallel experiments were performed with UV irradiation (120–240mJ/cm2), Fas-cross linking (1–20ug/ml), or etoposide treatment (6hr, 1–10 μM) (11). When enucleated cells were compared to nucleated cells, all cells were gamma-irradiated and treated with thrombospondin-1 to upregulate phosphatidylserine on the membrane (19). After 20 hr type I IFN in the supernatant was determined by ISRE-L929 reporter assay that has detection limit of 0.3U/ml (13). For phagosomal acidification studies, diphenyleneiodium (DPI, 10uM), ConB (1 nM), or chloroquine (50uM) were added at the start of the culture (12, 20). Parallel cultures using CpG and LPS were used as controls.
In vitro and ex vivo type I IFN analyses were performed by quantitative real-time PCR using SYBR Green and primers for β-actin (fw, TTGCTGACAGGATGCAGAAG; rev, GTACTTGCGCTCAGGAGGAG) and pan IFN-α (fw, TCTGATGCAGCAGGTGGG; rev AGGGCTCTCCAGACTTCTGCTCTG). Samples were treated with DNAse to eliminate genomic DNA contaminations. Gene expression was analyzed using the relative standard curve method and was normalized to Gapdh and β-actin expression.
For phagocytosis studies, DCs were incubated with CellTrace Violet-labeled irradiated splenocytes (Molecular Probes). After 3 and 16hr, DCs were stained with Abs to CD11c, CD11b, CD8α, nuclear dye Draq5, and fixable live/dead staining and analyzed by ImageStream (Amnis, Seattle, WA). At least 10,000 live events were acquired and the number and size of phagocytosed particles were determined using the spot counting and spot size features after tight masking on the brightfield image to exclude membrane-associated extracellular particles for each condition as shown before (12, 21).
To determine T cell activating capacity, DCs were incubated with irradiated OVA-Kb−/− cells for 4 hr after which CFSE-labeled purified OT-1-CD45.1 CD8+ T cells were added as described before (11). OT-1 cell proliferation and survival were determined after 70 hr by analysis of CFSE dilution together with staining for CD8α, Vα2, CD45.1 and 7-AAD.
DC signaling studies
Immune-coprecipitations: purified DCs were incubated with irradiated IRF3- or STING-deficient splenocytes. At different time points DCs were sorted, lysed, precipitated using agarose-bound antibodies to STING (3337, Cell Signaling) or IRF3 (D83B9, Cell Signaling) and probed for p-IRF3 (4D4G, Cell Signaling), TBK-1 (72B587, Novus Biologicals), p-TBK1 (D52C2, Cell Signaling), STAT6 (9362, Cell Signaling), and IPS1 (77275, Novus Biologicals).
In parallel, CellTrace Violet-labeled irradiated IRF3−/− splenocytes were cultured with WT and STING−/− DCs and analyzed by flow cytometry and AmnisImagestream upon surface staining with CD11c-PacificBlue and intracellular staining with Draq5 and p-IRF3 (anti-pS386, 4D4G) combined with anti-Rabbit IgGAlexafluor 488. Correlations between intensity of phagocytosed material and p-IRF3 localization in the nucleus were determined using Imagestream by gating on CD11c+ single cells followed by tight masking on the cells to discriminate internalized from bound particles followed by tight masking on the nucleus to determine colocalization of the p-IRF3 staining in the nucleus (12).
In vivo models
Immunizations
Mice were injected s.c. or i.p. with irradiated cells (OVA-Kb−/− splenocytes, 106–107; B16-OVA, B16/F10 and 5E1-TAKO, 1–5×106). Alternatively, mice received i.v. 2×105 purified DCs that had been pulsed with peptide or exposed to irradiated cells in vitro (10–12, 21). For DC depletion studies CD11c-DTR mice and mixed bone marrow chimeric mice were treated 24 hr before and 24 hr after immunization with 4ng/g diphtheria toxin i.p.
Tumor models
For the cyroablation model mice were subcutaneously injected with 4×105 B16-OVA or B16/F10 in PBS. Ten days later tumors (7–10 mm) were cryoablated using Verucca-Freeze armed with a 6 mm probe (Brymill Cryogenic Systems) (22) for 3×25-second cycles. To test long-term tumor protection, the mice were challenged with 3×104 B16-OVA or B16/F10 40 days after the ablation of the primary tumor and monitored for tumor growth. In the EL-4-mOVA challenge model, mice were immunized with 107 irradiated OVA-Kb−/− splenocytes as described above and 40 days later challenged with 106 EL-4-mOVA cells s.c.
SLE model (23)
Mice of indicated genetic backgrounds received 3×106 B6(C)-H2-Ab1bm12/KhEgJ (bm12) splenocytes i.p.. Splenic B and T cell composition B was analyzed by flow every 2 weeks. Serum levels of anti-dsDNA IgG, IgG1 and IgG2a were determined by ELISA as previously described (24).
T cell analysis
Unless stated differently, analysis were performed 7 days after immunization. CD8+ T cells were enumerated in spleens and draining lymph nodes using OVA257-264-Kb tetramers (Beckman Coulter) or E1B192–200-Dbdecamers (Immudex) together with staining for CD8α, CD44 and 7-AAD. In parallel cytokine production and polyfunctionality was determined directly ex vivo by intracellular cytokine staining after a 5 hr stimulation with cognate peptide in the presence of brefeldin A as described before (11, 16). Samples were collected on a LSRII flow cytometer with Diva software (BD Pharmingen), and data were analyzed with FlowJo software (Tree Star). In B16/F10 studies, responses to TRP2180-188 were determined by ELISPOT directly ex vivo.
Capacity for secondary expansion in vitro was determined by stimulating the splenocytes or purified CD8+ T cells on irradiated MEC.B7.SigOVA (OVA-specific) or I1.2 cells (E1B specific) for 6 days and dividing the absolute number of antigen-specific CD8+ T cells at the beginning of the culture by the absolute number of antigen-specific CD8+ T cells at the end of the culture as described before (16, 25). In vivo secondary expansion was determined by re-injecting the mice with a 10-fold higher number of irradiated cells than used during immunization. Four days after the secondary challenge the frequency antigen-specific CD8+ T cells was compared to non-challenged immunized mice (16).
Statistical analyses
Data were analyzed using Prism software (GraphPad Software, Inc.). Unless stated otherwise, the data are expressed as means ± SEM. Survival responses were analyzed by Kaplan-Meier using a log-rank test. All other data were evaluated using a two-way ANOVA followed by a Dunnett’s test. A p value of <0.05 was considered statistically significant.
Results
Protective anti-tumor immunity requires type I IFN sensing by DCs
We and others recently showed that type I IFN sensing is critical for the induction of protective anti-tumor responses in various tumor models in both vaccination and therapeutic settings (6–8, 22). Cryoablation of B16-OVA tumors in WT mice resulted in effective priming of OVA257-264-specific CD8+T cells that provided protective immunity upon subsequent B16-OVA challenge. In contrast, IFNAR−/− mice failed to induce adequate OVA257-264-specific CD8+ T responses and succumbed upon tumor rechallenge (figure 1a/b). Similarly, immunization with gamma-irradiated OVA-expressing Kb−/− splenocytes (OVA-Kb−/−) induced a significantly more robust OVA257-264-specific CD8+ T cell response in WT mice than in IFNAR−/− mice (figure 1. c/d). The OVA257-264-specific CD8+ T cells from WT mice but not IFNAR−/− mice underwent expansion upon secondary encounter with antigen in vitro and in vivo and protected mice from EL-4-mOVA challenge in vivo (figure 1e, figure S1a). Importantly, this response was not restricted to OVA or the selected pathways of cell death. Similar results were found when the parental line B16/F10 was used and the response to self-antigen TRP-2 was probed (figure S1b/c). In addition, direct immunization with gamma-irradiated, UV-irradiated, Fas-crosslinked, or etoposide treated cells showed significantly decreased CD8+ T cell responses in IFNAR−/− mice, illustrating a central role for type I IFN in CD8+ T cell priming to cell-associated antigens in various scenarios of cell death (figure 2).
Figure 1. Protective CD8+ T cell induction requires IFNAR on DCs.

A. WT and IFNAR−/− mice were s.c. injected with B16-OVA tumor cells and palpable tumors were cryoablated 10 days later. Cryoablated and naïve mice were s.c. challenged with B16-OVA 40 days later and survival was monitored (n=8–10/group). B. OVA257-264-specific CD8+T cell frequency in the spleen 7 days after cryoablation. C. Mice were immunized with irradiated OVA-Kb−/− or Kb−/− splenocytes and 40 days later s.c. challenged with EL-4-mOVA cells s.c. D. Frequency of splenic OVA257-264-specific CD8+ T cells 7 days after immunization as determined by intracellular cytokine staining for IFNγ following peptide restimulation and by OVA257-264-Kb-tetramer staining. E. Fold expansion of OVA257-264-specific CD8+T cells upon stimulation with OVA257-264-expressing cells in vitro. Data of representative experiments (out of 3–5) are shown (mean± s.e.m, n=8–10/group).
Figure 2. Impaired CD8+ T cell priming in IFNAR−/− mice is independent of the method of cell death or cell type.

Frequency of splenic antigen-specific CD8+ T cells in WT and IFNAR−/− mice 7 days after s.c. immunization with actmOVA-Kb−/− splenocytes (which were gamma irradiated (1500 Rad), UV irradiated (120 mJ/cm2) or treated with FAS cross-linking antibody (Jo-1, 20ug/ml; 4 hr, 37C)), 5E1-TAKO cells (3000rad, 240 mJ/cm2, or treated with Etoposide (10μM)), B16-OVA or B16/F10 cells (3000 rad or 240 mJ/cm2). Antigen-specific T cell frequencies were determined by intracellular cytokine staining upon incubation with OVA257-264, E1B192-200, TRP-2180-188, or control peptide GP33-41 (white bar, control peptide; black bar, specific peptide). Data in all experiments are expressed as mean± s.e.m with n=5.
As nearly all cells express IFNAR, we first used bone marrow (BM) chimeras to identify which cells required type I IFN sensing in the CD8+ T cell response to dying cells. BM chimeric mice (WT→IFNAR−/−, WT→WT, IFNAR−/−→WT, IFNAR−/−→IFNAR−/−) demonstrated that type I IFN needed to be sensed by the hematopoietic compartment (figure 3a/b). Additional studies with WT/IFNAR−/−→WT mixed BM chimeric mice indicated that the diminished CD8+ T cell response could not be attributed to the lack of IFNAR on the CD8+ T cells as IFNAR−/− CD8+ T cells only showed a marginal decrease in clonal expansion (figure 3c). Given the important role of DCs in cross-presentation of cell-associated antigens, we next examined the role of type I IFN sensing on DCs. Mixed BM chimeras were generated in which IFNAR−/− recipients received a combination of WT-CD11c-DTR and IFNAR−/−/CD45.1 BM or IFNAR−/−-CD11c-DTR and WT/CD45.1 BM. In this model the administration of DT yielded animals that contained either WT or IFNAR−/− DCs with an otherwise comparable composition of hematopoietic cells. Depletion of WT DCs significantly decreased the number and frequency of antigen-specific CD8+ T cells. In contrast, depletion of IFNAR−/− DCs had no effect on the number or frequency of antigen specific CD8+ T cells (figure 3d). Similar results were observed when purified WT or IFNAR−/− DCs were pulsed with irradiated cells and transferred into WT and IFNAR−/− recipients, illustrating the critical role of type I IFN sensing by DCs in CD8+ T cell priming to cell-associated antigens (figure 3e).
Figure 3. Selective requirement for type I IFN sensing in the DCs.

A. Bone-marrow chimeric mice (WT→IFNAR−/−, WT→WT, IFNAR−/−→WT, IFNAR−/−→IFNAR−/−) were immunized i.p. with irradiated 5E1-TAKO cells and the frequency of E1B192-200-specific CD8+ T cells in the spleens was determined 7 days later (white bar, control peptide; black bar, E1B192-200 peptide). B. The effect on memory formation using these chimeras was determined by measuring antigen-specific T cell expansion ex vivo. Secondary expansion of E1B192-200-specific CD8+ T cells was calculated by dividing the absolute number of E1B192-200-specific CD8+ T cells at the end of a6 day culture by the absolute number at the start of the culture. C. WT/CD90.1 mice were irradiated and reconstituted with WT, IFNAR−/− or a 1:1 ratio of WT (CD45.1):IFNAR−/−(CD45.2) bone-marrow. Mice were i.p. immunized with irradiated 5E1-TAKO cells and the frequency of splenic E1B192-200-specific CD8+ T cells in each graft was determined 7 days later. D. WTCD11cDTR:IFNAR−/− and WT:IFNAR−/−CD11cDTR mixed bone-marrow chimeras were treated with DT or vehicle and immunized with irradiated 5E1-TAKO cells. The frequency of E1B192-200-specific CD8+ T cells in the WT compartment was determined 7 days later. E. Freshly isolated WT and IFNAR−/− DCs were incubated with irradiated 5E1-TAKO cells, sorted and i.v. injected into WT and IFNAR−/− mice. The frequency of splenic E1B192-200-specific CD8+ T cells was determined 7 days later. Data of representative experiments (out of 3–5) are shown (mean± s.e.m, n=5/group).
DCs predominate the type I IFN production elicited by dying cells
Besides sensing type I IFNs in the context of CD8+ T cell priming to dying cells, we reasoned that DCs may also be the main producers of type I IFN, thereby establishing a positive IFN-feedback loop. In vitro studies using purified cell populations indicated that type I IFN was produced by DCs upon incubation with dying cells, whereas only nominal IFN production was observed in cultures depleted of DCs (figure 4a/b/c)(11, 12). The in vitro data were supported by our observation that IFNα mRNA was readily detectable in the draining LN of control-treated but not DT-treated CD11c-DTR mice upon subcutaneous immunization with irradiated cells (figure 4d).
Figure 4. Type I IFN production by DCs in vitro and in vivo.
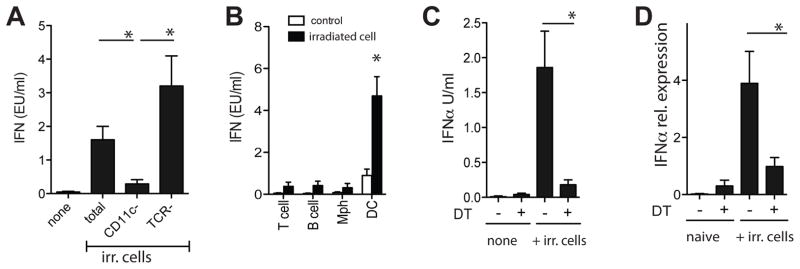
Total and subset-depleted splenocytes (A) or purified cell populations (B) from spleens of naïve WT mice were cultured with irradiated OVA-Kb−/− splenocytes and type I IFN in the supernatant was determined 20 hr later (black bars, irradiated cells; white bars, no cells). Data are expressed as mean± s.e.m with n=4. C. Splenocytes fromCD11c-DTR mice (treated with PBS or DT 24 hr prior) were isolated and cultured with irradiated OVA-Kb−/− splenocytes. Type I IFN in the supernatant was determined 20 hr later. D. CD11c-DTR mice were treated with vehicle or DT and 24 hr later s.c. injected with irradiated cells. Type I IFNα mRNA in draining lymph nodes was assessed 6–8 hr later. Representative data of one experiment (of 3–4) are shown (mean± s.e.m., n=5).
The STING/IRF3 pathway drives type I IFN in response to cell-associated antigen
Induction of type I IFNs is generally associated with innate sensing of pathogenic danger signals. To identify which innate sensing pathways facilitated the type I IFN production, primary DCs from WT, MyD88−/−/Triflps/lps, IRF3−/−, IRF7−/−, IPS1−/− (MAVS/Cardif), and STING−/− mice deficient were tested for their type I IFN production upon culture with dying cells. Type I IFN production was similar in WT, MyD88−/−/Triflps/lps, IPS1−/−, and IRF7−/− DCs indicating that the IFN induction was TLR-, RIG-I- and Mda5-independent. In contrast, significant reductions in type I IFN production were seen with DCs deficient in STING and the transcription factor IRF3 even though their subset compositions, maturation status and phagocytic capacity were similar to WT DCs (figure 5a/b and not shown). Immunoprecipitation and western blotting studies indicated rapid IRF3 phosphorylation in WT DCs and a significant reduction and delay in IRF3 phosphorylation in STING−/− DCs upon exposure to dying cells (figure 5c/d). To determine whether the magnitude of the nuclear p-IRF3 signal was associated with the frequency and size of phagocytosed particles, WT and STING−/− DCs were incubated with CellTrace Violet (VT)-labeled, irradiated, IRF3−/− splenocytes and analyzed by conventional flow cytometry and imaging cytometry. WT DCs showed a strong correlation between nuclear p-IRF3 intensity and the amount of phagocytosed material. Although STING−/− DCs displayed similar uptake of VT material as WT DCs, they exhibited significantly reduced nuclear p-IRF3 staining, consistent with their limited IFN production (figure 5e/f, figure S2a). Using UV-irradiation, Fas-crosslinking, or etoposide to induce cell death yielded similar outcomes, indicating that the STING/IRF3 pathway was the dominant IFN-inducing pathway in various forms of sterile cell death (figure S2b).
Figure 5. Type I IFN induction requires the STING/IRF3 pathway.

A. Type I IFN production by purified DCs from indicated strains upon 20hr culture with irradiated splenocytes. B. Purified DCs from indicated strains were cultured at 4°C (white bars) and 37°C (black bars) with CellTrace Violet-labeled irradiated splenocytes. Uptake of CellTrace Violet materials was determined 6 hr later by flow cytometry. Data is expressed as percentage of DCs that contain CellTrace Violet. Representative data of one experiment (of 3–4) are shown (mean± s.e.m., n=4). C/D. p-IRF3 kinetics in WT and STING−/− DCs upon incubation with irradiated IRF3−/− splenocytes. E. ImageStream images of p-IRF3 nuclear translocation in WT DCs 8 hr after incubation with VT-labeled irradiated IRF3−/− cells. F. ImageStream analysis of nuclear p-IRF3 intensity and intensity of cytosolic phagocytosed material in WT and STING−/− DCs. Tight masking on the cytosol was done to discriminate between bound and internalized cellular material. At least 3 experiments were performed with 800–1000 analyzed cells/condition.
Nuclear DNA-derived structures induce type I IFN production by DCs
We next set out to determine the ligand upstream of STING that was responsible for inducing type I IFN. STING facilitates immune responses to various nucleotide structures, including cytosolic dsDNA, and in some cases dsRNA (26–28). As dsRNA sensing utilizes the RIG-I/IPS-1 pathway and IPS1−/− DCs have normal type I IFN production, it is likely that the IFN-inducing species released by dying cells is a DNA- and not an RNA-based entity. Indeed, addition of DNAses, but not RNAses to WT DC/irradiated cell co-cultures significantly reduced type I IFN production without affecting uptake of cellular material or responses to non-nucleic TLR ligands (figure 6a, figure S2c/d). To more rigorously address the role of DNA complexes in the type I IFN induction, we exploited the process of erythropoiesis where red blood cell (RBC) precursors sequentially lose their nuclei, mitochondria and ribosomes. Timed RBC cultures were sorted, irradiated and treated with thrombospondin-1 (irr/TSP) to induce comparable phosphatidylserine expression on the membrane and facilitate an “apoptotic phenotype” in the non-nucleated cells (figure 6b). ImageStream analysis showed comparable uptake of the irr/TSP cell subsets by DCs (figure 6c). Nucleated irr/TSP erythroblasts readily induced type I IFN in DCs while enucleated reticulocytes and RBC failed to do so (figure 6d). These data indicate that the IFN-inducing species is nuclear DNA-derived.
Figure 6. IFN induction by a nuclear DNA-derived structure.

A. Type I IFN production by WT DCs upon culture with irradiated splenocytes in the presence or absence of RNAses and DNAses. B. Cells at different stages of erythropoiesis were sorted based on nucleotide content, irradiated and incubated with thrombospondin-1. Flowcytometric analysis of nuclear content (Draq5) and Annexin-V levels in indicated cell types before (red line) and after (blue line) irradiation and thrombospondin-1 treatment. C. Purified WT DCs were cultured with CellTrace Violet-labeled irradiated/TPS1 treated cells. Uptake of CellTrace Violet material was determined 6 hr later by flow cytometry. Data is expressed as percentage of DCs that contain CellTrace Violet D. Type I IFN production by WT DCs upon culture with VT-labeled irradiated/Thrombospondin-1 treated cells at different stages of erythropoiesis. Representative data of one experiment (of 3) are shown (mean± s.e.m, n=5).
DC-intrinsic STING regulates CD8+ T cell responses to dying cells
Given the importance of STING-mediated DNA sensing in the type I IFN production, we next assessed the relative contribution of STING in the priming of CD8+ T cells to dying cell-associated antigens. WT, MyD88−/−/Triflps/lps, IPS−/−, and IRF7−/− mice showed comparable CD8+ T cell priming as determined by tetramer staining, intracellular cytokine staining, and capacity for secondary expansion upon immunization with irradiated 5E1-TAKO cells (figure 7a/b). In contrast, IFNAR−/−, IRF3−/− and STING−/− mice showed significantly reduced CD8+ T cell priming (figure 7c/d). Moreover, the antigen-specific IFNAR−/−, IRF3−/− and STING−/− CD8+ T cells displayed less cytokine polyfunctionality and impaired capacity for secondary expansion (figure 7e). The defect in CD8+ T priming was fully DC-regulated as similar results on CD8+ T cell clonal burst, secondary expansion and cytokine polyfunctionality were seen when purified IFNAR−/−, IRF3−/− and STING−/− DCs were exposed to irradiated cells in vitro and transferred into WT recipients. (figure7f/g/h). The priming defect of the IFNAR−/−, IRF3−/−, and STING−/− DCs could not be attributed to a decrease in overall DC functionality as peptide-pulsed IFNAR−/−, IRF3−/−, and STING−/− DCs induced comparable CD8+ T cell responses as WT DCs upon transfer in WT recipients (figure 7i). Together, these data demonstrate the requirement for STING, IRF3, and IFNAR in DCs in the cross-priming of CD8+ T cells to cell-associated antigens.
Figure 7. STING regulates the CD8+ T cells responses to dying cells in vivo.

A. Mice of indicated strains were immunized with irradiated 5E1-TAKO cells and the frequency of splenic E1B192-200-specific CD8+ T cells was determined 7 days later (white bar, control peptide; black bar, E1B192-200 peptide). B. Fold expansion of E1B192-200-specific CD8+ T cells from indicated mouse strains upon culture with E1B192-200-expressing feeder cells in vitro. C. E1B192-200-specific CD8+ T cell frequency in indicated mouse strains 7 days after immunization with irradiated 5E1-TAKO cells. D. Secondary expansion of E1B192-200-specific CD8+ T cells in vitro. E. Ex vivo polyfunctionality of E1B192-200-specific CD8+ T cells from fig C as determined by flow cytometry. F. Frequency of E1B192-200-specific CD8+ T cells in WT mice 7 days after transfer of indicated DCs pulsed with irradiated 5E1-TAKO cells in vitro. G. Secondary expansion of E1B192-200-specific CD8+ T cells primed by indicated DCs. H. Ex vivo polyfunctionality of E1B192-200-specific CD8+ T cells primed by indicated DCs. I. DCs were purified from WT, IFNAR−/− and STING−/− mice and exposed to irradiated 5E1-TAKO cells or pulsed with 1uM E1B192-200 peptide. DCs were repurified and 2 × 105 were injected into WT recipients. Seven days later the frequency of splenic E1B192-200-specific CD8+ T cells was determined (white bar, control peptide; black bar, E1B192-200 peptide). Representative data of one experiment (of 3–4) are shown (mean± s.e.m., n=5–7).
STING regulates CD4+ T cell and B cell responses in the bm12 SLE model
To assess whether STING has a broader role in the priming of adaptive immune responses to dying cells, we assessed the induction of CD4+ T cell and B cell responses in the bm12-cGVHD model where H-2b B6 hosts develop lupus-like disease upon transfer of B6.C-H2bm12 CD4+ T cells (29). Both IFNAR−/− and STING−/− mice developed considerably less activated CD4+ T cells and T follicular helper cells than WT recipients upon transfer of bm12 CD4+ T cells (figure 8a/b). Moreover, both IFNAR−/− and STING−/− mice developed considerably less activated B cells, plasma cells and pathogenic anti-dsDNA IgG2a antibodies than WT recipients (figure 8a/c/d). Given that the transferred bm12-CD4+ T cells were IFNAR- and STING-sufficient, these data further support the role for antigen presenting cell-intrinsic STING and IFNAR in the induction of adaptive immune responses to dying cell-derived antigens.
Figure 8. STING regulates CD4+ T cell and B cell responses in the bm12 SLE model.

A. Flow cytometric analysis of Tfh (gated on CD4+ T cells) and plasma cells (total spleen) in spleens of WT, STING−/− and IFNAR−/− mice 14 days after i.p. transfer of live bm12 splenocytes. B. Number of splenic Tfh cells and activated CD4+ T cells in the spleen of indicated mouse strains. C. Number of plasma cells and activated B cells in the spleen of indicated mouse strains. Representative data of one experiment (of 3–5) are shown. D. Anti-dsDNA IgG1 and IgG2a levels in indicated mouse strains (mean± s.e.m, n=10–22).
Discussion
Type I IFNs have been implicated as the upstream events precipitating autoimmune disease and a prerequisite for effective anti-tumor radiotherapy. Here we identify DC sensing of cell-derived nuclear DNA entities via the STING/IRF3 pathway as a key component in the early type I IFN response to dying cells.
Dying cells can emit a plethora of structurally distinct DAMPs and it is likely that the molecular pathways involved in the sensing of these DAMPs are equally diverse. While many DAMPs can contribute to the final adaptive immune response to cell-associated antigens, our data identified type I IFN as the dominant pro-inflammatory factor and STING/IRF3 signaling as the principle pathway in the initiation of the immune responses to cell-associated antigens in our tumor models as well as the autoimmune responses in our SLE model. Although all nucleated cells can sense type I IFN and type I IFN has been shown to directly act on T cells, our data indicate that the early STING/IRF3-mediated type I IFN predominantly acts on DCs. Transfer of IFNAR−/− DCs (pulsed with irradiated cells) into WT recipients resulted in similar deficiencies in CD8+ T cell expansion, functionality and memory formation as the direct immunization of IFNAR−/− mice. Moreover, immunization of WT/IFNAR−/− mixed bone marrow chimeric mice did not show any significant difference in CD8+ T cell clonal expansion or polyfunctionality between the WT and IFNAR−/− grafts. Importantly, transfer of STING−/− or IRF3−/− DCs into WT recipients resulted in similar defects in CD8+ T cell responses as the transfer of IFNAR−/− DCs. Likewise, STING−/− recipients showed identical reduction in Tfh cell and plasma cell formation as IFNAR−/− recipients in our SLE model. Together with the observation that STING−/− and IRF3−/− DC have significantly reduced type I IFN induction upon phagocytosis of dying cells, these data implicate that the STING/IRF3 pathway is the critical component in the type I IFN-dependent T cell priming to cell-associated antigens.
It has been suggested that different types of death may induce different DAMPs. We observed comparable type I IFN induction and STING/IRF3 engagement in DCs upon phagocytosis of cells treated with gamma irradiation, UV irradiation, Fas-crosslinking antibody, or etoposide, suggesting that these different types of cell death generated a similar nuclear DNA-derived DAMP (figure S2b, and not shown). It is also likely that similar nuclear DNA-associated DAMPs become available in vivo as type I IFN induction and type I IFN-dependent CD8+ T cell priming was readily observed in WT and MyD88/Trif deficient mice, but significantly reduced in STING−/− and IRF3−/− mice upon administration of dying cells (gamma, UV, or FAS-treated) or upon in vivo tumor cryo-ablation (11, 22).
Importantly, the crucial role for type I IFN in the priming of protective adaptive anti-tumor responses is not restricted to situations where massive tumor cell death occurs, as is the case for radiotherapy, chemotherapy, and cryoablative tumor therapies. Recent publications indicate that spontaneous and limited tumor cell death in tumor-bearing mice also resulted in type I IFN-dependent protective immune responses. Fuertes and Diamond showed spontaneous anti-tumor CD8+ T cell induction and tumor rejection in tumor-bearing mice that was critically dependent on type I IFN sensing by cross-priming DCs (6, 7). In this light, it is interesting to notice that STING−/− mice—like IFNAR−/− mice—develop significantly more lung metastases than WT mice upon i.v. injection of low numbers of untreated B16 melanoma cells, illustrating a role for the STING/IFNAR nexus in the antitumor response when cell death is limited (not depicted).
STING can facilitate innate responses to cytosolic bacterial cyclic dinucleotides (c-di-GMP)(30, 31), dsDNA and in some cases cytosolic dsRNA (26–28). Our data strongly suggest that the main type I IFN-inducing ligand is a nuclear DNA species. STING-mediated dsRNA sensing requires RNA with 5′-triphosphate groups in combination with the IPS-1/RIG-I pathway. The absence of IPS-1 recruitment to STING in WT DCs upon phagocytosis of cellular materials as well as the normal type I IFN production and CD8+ T cell responses in IPS1−/− mice, effectively argue against a role for dsRNA sensing in the STING/IFN phenotype. Our hypothesis that the type I IFN inducing ligand is a DNA structure is strongly supported by our in vitro data that show significantly reduced type I IFN induction upon addition of DNAses to the dying cell/DC co-culture. Moreover, the use of enucleated cells dramatically reduced type I IFN production and cross-priming by WT DCs in vitro. Importantly, the latter experiment also suggested that mitochondrial or ribosomal nucleotide structures had no notable role in the type I IFN production as reticulocytes—enucleated but still containing mitochondria and ribosomes—failed to induce type I IFN.
Recent studies indicate direct binding of c-di-GMP and cyclic-GMP-AMP (cGAMP) to STING but have not provided evidence for direct dsDNA-STING interactions (32, 33) suggesting the involvement of upstream DNA sensors. Over the last few years several of candidate sensors have been identified, including cyclic GMP-AMP synthase (cGAS), which has been shown to signal through STING via the production of cGAMP (34–37). DExD/H-box protein family member DDX41 and IFI16 (p204) have also been implicated as possible DNA sensors acting through STING, but their precise molecular interactions have not been fully elucidated (38, 39). At present these candidate DNA sensors are studied in in vitro systems where the DNA is directly delivered into the cytosol via transfection, transduction or infection pathways. However, in order for the phagocytosed DNA-derived structures to be sensed by the STING pathway, either its key components should be recruited to the phagosome or the DNA-derived species should escape into the cytosol. Although STING can translocate from the ER to the Golgi and autophagosome-like compartments, we and others did not observe STING in phagosomes or phagolysosomes (not shown)(40–44). However, phagosomal DNA sensing via STING could still be possible by phagosomal recruitment of p204 that has reported migratory capacity or cGAS that produces the highly mobile secondary messenger cGAMP (34, 36, 39). On the other hand, phagosomal escape is a well reported process in cross-presentation where protein structures escape into the cytosol to be processed for presentation in MHC class I (20, 45). The exact mechanism by which proteins escape into the cytosol is not known but it is strongly associated with alkalinization of the phagosome and prevention of phagosomal acidification (20, 45, 46). Consistent with the latter possibility, STING-mediated type I IFN production was strongly associated with inhibition of phagosomal acidification (45); in vitro treatments of DCs with agents that accelerated phagosomal acidification decreased type I IFN production while alkalinization or the delay of endosomal acidification significantly enhanced type I IFN production (figure S3). Moreover, we found that DC populations that have the greatest capacity for cross-presentation and slowest phagosomal acidification rate also produced the most type I IFN upon phagocytosis of dying cells (data not shown and (11, 12, 47)).
While the exact mechanism by which the nuclear DNA-derived structure activates the STING pathway needs further elucidation, our data strongly demonstrate its potent role in anti-tumor immunity and autoimmunity. Our observations are in line with the findings that mice lacking DNAse II, responsible for degradation of phagocytosed DNA, die of TLR-independent pro-inflammatory cytokine production unless the mice are crossed to the IFNAR−/− or STING−/− background (48–51). Intriguingly, gain of function mutations in STING were recently associated with vasculopathy with onset in infancy (SAVI), a syndrome characterized by a severe cutaneous vasculopathy leading to extensive tissue loss and structural damage, with neonatal-onset systemic inflammation (52). Moreover, the deletion of TREX1 –DNAse III, a DNA exonuclease with functions in DNA degradation– was shown to trigger type I IFN-associated inflammatory responses in a cGAS-dependent manner (53, 54). Furthermore, mutations in TREX1 have been associated with a variety of (multi-organ) inflammatory syndromes, including SLE, Sögrens Syndrome, chilblain lupus, retinal vasculopathy, and Aicardi-Goutieres syndrome (53–60).
Together our data indicate that sensing of nuclear DNA-derived structures via the STING/IRF3 pathway in DCs is responsible for the early type I IFN induction upon phagocytosis of dying cells. This early type I IFN directly acts on the DCs and endows them with greater capacity to activate adaptive immune responses to cell-associated antigens. Given that type I IFNs have been implicated as the upstream events precipitating autoimmune disease and a prerequisite for effective anti-tumor therapy further characterization of the STING pathway as sensor of dying cell-derived DNA could enable the rational design of new therapies to enhance anti-tumor responses and interfere with the development and progression of autoimmune diseases.
Supplementary Material
Acknowledgments
We are grateful to Dr. H. Singh for comments and discussion on the manuscript; Dr. T. Kalfa for providing reagents and advice for the in vitro erythropoiesis experiments; A. White and M. DeLay for support with the AmnisImagestream; and the Research Flow Cytometry Core at Cincinnati Children’s Hospital Medical Center.
This work was supported by the National Institutes of Health via National Cancer Institute grant CA138617 (E.M.J.) and National Institute of Diabetes and Digestive and Kidney Diseases grant DK090978 (J.K./E.M.J.) and the Charlotte Schmidlapp Award (E.M.J.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nature reviews Immunology. 2009;9:353–363. doi: 10.1038/nri2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 3.Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (alpha/beta) in immunity and autoimmunity. Annual review of immunology. 2005;23:307–336. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- 4.Dunn GP, Bruce AT, Sheehan KC, Shankaran V, Uppaluri R, Bui JD, Diamond MS, Koebel CM, Arthur C, White JM, Schreiber RD. A critical function for type I interferons in cancer immunoediting. Nature immunology. 2005;6:722–729. doi: 10.1038/ni1213. [DOI] [PubMed] [Google Scholar]
- 5.Swann JB, Hayakawa Y, Zerafa N, Sheehan KC, Scott B, Schreiber RD, Hertzog P, Smyth MJ. Type I IFN contributes to NK cell homeostasis, activation, and antitumor function. J Immunol. 2007;178:7540–7549. doi: 10.4049/jimmunol.178.12.7540. [DOI] [PubMed] [Google Scholar]
- 6.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, Murphy KM, Schreiber RD. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. The Journal of experimental medicine. 2011;208:1989–2003. doi: 10.1084/jem.20101158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, Gajewski TF. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. The Journal of experimental medicine. 2011;208:2005–2016. doi: 10.1084/jem.20101159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burnette BC, Liang H, Lee Y, Chlewicki L, Khodarev NN, Weichselbaum RR, Fu YX, Auh SL. The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer research. 2011;71:2488–2496. doi: 10.1158/0008-5472.CAN-10-2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang TH, Mao CP, Lee SY, Chen A, Lee JH, Kim TW, Alvarez RD, Roden RB, Pardoll D, Hung CF, Wu TC. Chemotherapy acts as an adjuvant to convert the tumor microenvironment into a highly permissive state for vaccination-induced antitumor immunity. Cancer research. 2013;73:2493–2504. doi: 10.1158/0008-5472.CAN-12-4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hennies CM, Reboulet RA, Garcia Z, Nierkens S, Wolkers MC, Janssen EM. Selective expansion of merocytic dendritic cells and CD8DCs confers anti-tumour effect of Fms-like tyrosine kinase 3-ligand treatment in vivo. Clinical and experimental immunology. 2011;163:381–391. doi: 10.1111/j.1365-2249.2010.04305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Janssen E, Tabeta K, Barnes MJ, Rutschmann S, McBride S, Bahjat KS, Schoenberger SP, Theofilopoulos AN, Beutler B, Hoebe K. Efficient T cell activation via a Toll-Interleukin 1 Receptor-independent pathway. Immunity. 2006;24:787–799. doi: 10.1016/j.immuni.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 12.Reboulet RA, Hennies CM, Garcia Z, Nierkens S, Janssen EM. Prolonged antigen storage endows merocytic dendritic cells with enhanced capacity to prime anti-tumor responses in tumor-bearing mice. J Immunol. 2010;185:3337–3347. doi: 10.4049/jimmunol.1001619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang Z, Georgel P, Du X, Shamel L, Sovath S, Mudd S, Huber M, Kalis C, Keck S, Galanos C, Freudenberg M, Beutler B. CD14 is required for MyD88-independent LPS signaling. Nature immunology. 2005;6:565–570. doi: 10.1038/ni1207. [DOI] [PubMed] [Google Scholar]
- 14.Bellone M, Cantarella D, Castiglioni P, Crosti MC, Ronchetti A, Moro M, Garancini MP, Casorati G, Dellabona P. Relevance of the tumor antigen in the validation of three vaccination strategies for melanoma. J Immunol. 2000;165:2651–2656. doi: 10.4049/jimmunol.165.5.2651. [DOI] [PubMed] [Google Scholar]
- 15.van Stipdonk MJ, Lemmens EE, Schoenberger SP. Naive CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nature immunology. 2001;2:423–429. doi: 10.1038/87730. [DOI] [PubMed] [Google Scholar]
- 16.Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421:852–856. doi: 10.1038/nature01441. [DOI] [PubMed] [Google Scholar]
- 17.Garderet L, Kobari L, Mazurier C, De Witte C, Giarratana MC, Perot C, Gorin NC, Lapillonne H, Douay L. Unimpaired terminal erythroid differentiation and preserved enucleation capacity in myelodysplastic 5q(del) clones: a single cell study. Haematologica. 2010;95:398–405. doi: 10.3324/haematol.2009.012773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Konstantinidis DG, Pushkaran S, Johnson JF, Cancelas JA, Manganaris S, Harris CE, Williams DA, Zheng Y, Kalfa TA. Signaling and cytoskeletal requirements in erythroblast enucleation. Blood. 2012;119:6118–6127. doi: 10.1182/blood-2011-09-379263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Head DJ, Lee ZE, Swallah MM, Avent ND. Ligation of CD47 mediates phosphatidylserine expression on erythrocytes and a concomitant loss of viability in vitro. British journal of haematology. 2005;130:788–790. doi: 10.1111/j.1365-2141.2005.05668.x. [DOI] [PubMed] [Google Scholar]
- 20.Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, Lennon-Dumenil AM, Seabra MC, Raposo G, Amigorena S. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell. 2006;126:205–218. doi: 10.1016/j.cell.2006.05.035. [DOI] [PubMed] [Google Scholar]
- 21.Katz JD, Ondr JK, Opoka RJ, Garcia Z, Janssen EM. Cutting edge: merocytic dendritic cells break T cell tolerance to beta cell antigens in non obese diabetic mouse diabetes. J Immunol. 2010;185:1999–2003. doi: 10.4049/jimmunol.1001398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nierkens S, den Brok MH, Garcia Z, Togher S, Wagenaars J, Wassink M, Boon L, Ruers TJ, Figdor CG, Schoenberger SP, Adema GJ, Janssen EM. Immune adjuvant efficacy of CpG oligonucleotide in cancer treatment is founded specifically upon TLR9 function in plasmacytoid dendritic cells. Cancer research. 2011;71:6428–6437. doi: 10.1158/0008-5472.CAN-11-2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morris SC, Cheek RL, Cohen PL, Eisenberg RA. Allotype-specific immunoregulation of autoantibody production by host B cells in chronic graft-versus host disease. J Immunol. 1990;144:916–922. [PubMed] [Google Scholar]
- 24.Seavey MM, Lu LD, Stump KL. Animal models of systemic lupus erythematosus (SLE) and ex vivo assay design for drug discovery. In: Enna SJ, editor. Current protocols in pharmacology/editorial board. Unit 5. Chapter 5. 2011. p. 60. [DOI] [PubMed] [Google Scholar]
- 25.Janssen EM, Droin NM, Lemmens EE, Pinkoski MJ, Bensinger SJ, Ehst BD, Griffith TS, Green DR, Schoenberger SP. CD4+ T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nature. 2005;434:88–93. doi: 10.1038/nature03337. [DOI] [PubMed] [Google Scholar]
- 26.Burdette DL, Vance RE. STING and the innate immune response to nucleic acids in the cytosol. Nature immunology. 2013;14:19–26. doi: 10.1038/ni.2491. [DOI] [PubMed] [Google Scholar]
- 27.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barber GN. Innate immune DNA sensing pathways: STING, AIMII and the regulation of interferon production and inflammatory responses. Current opinion in immunology. 2011;23:10–20. doi: 10.1016/j.coi.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eisenberg R. Mechanisms of systemic autoimmunity in murine models of SLE. Immunologic research. 1998;17:41–47. doi: 10.1007/BF02786429. [DOI] [PubMed] [Google Scholar]
- 30.Yin Q, Tian Y, Kabaleeswaran V, Jiang X, Tu D, Eck MJ, Chen ZJ, Wu H. Cyclic di-GMP sensing via the innate immune signaling protein STING. Molecular cell. 2012;46:735–745. doi: 10.1016/j.molcel.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011;478:515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shu C, Yi G, Watts T, Kao CC, Li P. Structure of STING bound to cyclic di-GMP reveals the mechanism of cyclic dinucleotide recognition by the immune system. Nature structural & molecular biology. 2012;19:722–724. doi: 10.1038/nsmb.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ouyang S, Song X, Wang Y, Ru H, Shaw N, Jiang Y, Niu F, Zhu Y, Qiu W, Parvatiyar K, Li Y, Zhang R, Cheng G, Liu ZJ. Structural analysis of the STING adaptor protein reveals a hydrophobic dimer interface and mode of cyclic di-GMP binding. Immunity. 2012;36:1073–1086. doi: 10.1016/j.immuni.2012.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, Hopfner KP, Ludwig J, Hornung V. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498:380–384. doi: 10.1038/nature12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, Hornung V, Hopfner KP. Structural mechanism of cytosolic DNA sensing by cGAS. Nature. 2013;498:332–337. doi: 10.1038/nature12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang X, Wu J, Du F, Xu H, Sun L, Chen Z, Brautigam CA, Zhang X, Chen ZJ. The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell reports. 2014;6:421–430. doi: 10.1016/j.celrep.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu YJ. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nature immunology. 2011;12:959–965. doi: 10.1038/ni.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, Fitzgerald KA, Paludan SR, Bowie AG. IFI16 is an innate immune sensor for intracellular DNA. Nature immunology. 2010;11:997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Konno H, Konno K, Barber GN. Cyclic Dinucleotides Trigger ULK1 (ATG1) Phosphorylation of STING to Prevent Sustained Innate Immune Signaling. Cell. 2013 doi: 10.1016/j.cell.2013.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saitoh T, Fujita N, Yoshimori T, Akira S. Regulation of dsDNA-induced innate immune responses by membrane trafficking. Autophagy. 2010;6:430–432. doi: 10.4161/auto.6.3.11611. [DOI] [PubMed] [Google Scholar]
- 42.Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell. 2012;150:803–815. doi: 10.1016/j.cell.2012.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McFarlane S, Aitken J, Sutherland JS, Nicholl MJ, Preston VG, Preston CM. Early induction of autophagy in human fibroblasts after infection with human cytomegalovirus or herpes simplex virus 1. Journal of virology. 2011;85:4212–4221. doi: 10.1128/JVI.02435-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, Matsunaga K, Kageyama S, Omori H, Noda T, Yamamoto N, Kawai T, Ishii K, Takeuchi O, Yoshimori T, Akira S. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:20842–20846. doi: 10.1073/pnas.0911267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Amigorena S, Savina A. Intracellular mechanisms of antigen cross presentation in dendritic cells. Current opinion in immunology. 2010;22:109–117. doi: 10.1016/j.coi.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 46.Savina A, Peres A, Cebrian I, Carmo N, Moita C, Hacohen N, Moita LF, Amigorena S. The small GTPase Rac2 controls phagosomal alkalinization and antigen crosspresentation selectively in CD8(+) dendritic cells. Immunity. 2009;30:544–555. doi: 10.1016/j.immuni.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 47.Thacker RI, Janssen EM. Cross-presentation of cell-associated antigens by mouse splenic dendritic cell populations. Frontiers in immunology. 2012;3:41. doi: 10.3389/fimmu.2012.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Okabe Y, Sano T, Nagata S. Regulation of the innate immune response by threonine-phosphatase of Eyes absent. Nature. 2009;460:520–524. doi: 10.1038/nature08138. [DOI] [PubMed] [Google Scholar]
- 49.Kitahara Y, Kawane K, Nagata S. Interferon-induced TRAIL-independent cell death in DNase II−/− embryos. European journal of immunology. 2010;40:2590–2598. doi: 10.1002/eji.201040604. [DOI] [PubMed] [Google Scholar]
- 50.Okabe Y, Kawane K, Akira S, Taniguchi T, Nagata S. Toll-like receptor-independent gene induction program activated by mammalian DNA escaped from apoptotic DNA degradation. The Journal of experimental medicine. 2005;202:1333–1339. doi: 10.1084/jem.20051654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ahn J, Gutman D, Saijo S, Barber GN. STING manifests self DNA-dependent inflammatory disease. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:19386–19391. doi: 10.1073/pnas.1215006109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Montealegre Sanchez GA, Tenbrock K, Wittkowski H, Jones OY, Kuehn HS, Lee CC, DiMattia MA, Cowen EW, Gonzalez B, Palmer I, DiGiovanna JJ, Biancotto A, Kim H, Tsai WL, Trier AM, Huang Y, Stone DL, Hill S, Kim HJ, St Hilaire C, Gurprasad S, Plass N, Chapelle D, Horkayne-Szakaly I, Foell D, Barysenka A, Candotti F, Holland SM, Hughes JD, Mehmet H, Issekutz AC, Raffeld M, McElwee J, Fontana JR, Minniti CP, Moir S, Kastner DL, Gadina M, Steven AC, Wingfield PT, Brooks SR, Rosenzweig SD, Fleisher TA, Deng Z, Boehm M, Paller AS, Goldbach-Mansky R. Activated STING in a vascular and pulmonary syndrome. The New England journal of medicine. 2014;371:507–518. doi: 10.1056/NEJMoa1312625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ablasser A, Hemmerling I, Schmid-Burgk JL, Behrendt R, Roers A, Hornung V. TREX1 deficiency triggers cell-autonomous immunity in a cGAS-dependent manner. J Immunol. 2014;192:5993–5997. doi: 10.4049/jimmunol.1400737. [DOI] [PubMed] [Google Scholar]
- 54.Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134:587–598. doi: 10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K, Robins P, Harvey S, Hollis T, O’Hara A, Herrick AL, Bowden AP, Perrino FW, Lindahl T, Barnes DE, Crow YJ. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. American journal of human genetics. 2007;80:811–815. doi: 10.1086/513443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Namjou B, Kothari PH, Kelly JA, Glenn SB, Ojwang JO, Adler A, Alarcon-Riquelme ME, Gallant CJ, Boackle SA, Criswell LA, Kimberly RP, Brown E, Edberg J, Stevens AM, Jacob CO, Tsao BP, Gilkeson GS, Kamen DL, Merrill JT, Petri M, Goldman RR, Vila LM, Anaya JM, Niewold TB, Martin J, Pons-Estel BA, Sabio JM, Callejas JL, Vyse TJ, Bae SC, Perrino FW, Freedman BI, Scofield RH, Moser KL, Gaffney PM, James JA, Langefeld CD, Kaufman KM, Harley JB, Atkinson JP. Evaluation of the TREX1 gene in a large multi-ancestral lupus cohort. Genes and immunity. 2011;12:270–279. doi: 10.1038/gene.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang YG, Lindahl T, Barnes DE. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell. 2007;131:873–886. doi: 10.1016/j.cell.2007.10.017. [DOI] [PubMed] [Google Scholar]
- 58.Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, de Silva U, Bailey SL, Witte T, Vyse TJ, Kere J, Pfeiffer C, Harvey S, Wong A, Koskenmies S, Hummel O, Rohde K, Schmidt RE, Dominiczak AF, Gahr M, Hollis T, Perrino FW, Lieberman J, Hubner N. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nature genetics. 2007;39:1065–1067. doi: 10.1038/ng2091. [DOI] [PubMed] [Google Scholar]
- 59.Abe J, Nakamura K, Nishikomori R, Kato M, Mitsuiki N, Izawa K, Awaya T, Kawai T, Yasumi T, Toyoshima I, Hasegawa K, Ohshima Y, Hiragi T, Sasahara Y, Suzuki Y, Kikuchi M, Osaka H, Ohya T, Ninomiya S, Fujikawa S, Akasaka M, Iwata N, Kawakita A, Funatsuka M, Shintaku H, Ohara O, Ichinose H, Heike T. A nationwide survey of Aicardi-Goutieres syndrome patients identifies a strong association between dominant TREX1 mutations and chilblain lesions: Japanese cohort study. Rheumatology (Oxford) 2014;53:448–458. doi: 10.1093/rheumatology/ket372. [DOI] [PubMed] [Google Scholar]
- 60.Barizzone N, Monti S, Mellone S, Godi M, Marchini M, Scorza R, Danieli MG, D’Alfonso S. Rare variants in the TREX1 gene and susceptibility to autoimmune diseases. BioMed research international. 2013;2013:471703. doi: 10.1155/2013/471703. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.