

Abstract
Monoclonal antibodies can modulate cancer cell signal transduction and recruit antitumor immune effector mechanisms – including antibody-dependent cellular cytotoxicity (ADCC). Although several clinically effective antibodies can promote ADCC, therapeutic resistance is common. We hypothesized that oncogenic signaling networks within tumor cells affect their sensitivity to ADCC. We developed a screening platform and targeted 60 genes derived from an EGFR gene network using RNA interference (RNAi) in an in vitro ADCC model system. Knockdown of GRB7, PRKCE, and ABL1 enhanced ADCC by primary and secondary screens. ABL1 knockdown also reduced cell proliferation, independent of its ADCC enhancement effects. c-Abl overexpression decreased ADCC sensitivity and rescued the effects of ABL1 knockdown. Imatinib inhibition of c-Abl kinase activity also enhanced ADCC – phenocopying ABL1 knockdown – against several EGFR-expressing head-and-neck squamous cell carcinoma (HNSCC) cell lines by ex vivo primary NK cells. Our findings suggest that combining c-Abl inhibition with ADCC-promoting antibodies, such as cetuximab, could translate into increased therapeutic efficacy of monoclonal antibodies.
Keywords: ADCC, NK cells, EGFR, cetuximab, c-Abl, imatinib, HNSCC
Introduction
Monoclonal antibodies (MAb) have proven useful in the targeted therapy of cancer, due to their specificity, versatility, and efficacy (1). Approved antibody therapies have multiple mechanisms of action including perturbation of growth factor signaling, induction of apoptotic activity, and direct cytotoxicity. Antibodies can also recruit immune effector mechanisms such as antibody-dependent cellular cytotoxicity (ADCC) (2).
ADCC involves the engagement of antibody, bound to the surface of a target cell, by activating Fc receptors (FcR) on immune effector cells. The low-affinity FcγR, FcγRIIIA (FCGR3A), recognizes IgG1 and IgG3 antibody isotypes and is the predominant receptor involved in natural killer (NK) cell-mediated ADCC (3). NK cells are well-studied effectors of ADCC, but other innate immune cells also contribute (1,3,4). The engagement of antibodies and FcRs activates cytotoxic degranulation and cytokine release by NK cells (5). Perforin and granzyme released from cytotoxic granules initiate apoptosis in targeted cells (3,6).
The role of FcRs in modulating antitumor efficacy of antibodies in murine models supports the in vivo relevance of ADCC (7). Higher-affinity FCGR3A polymorphisms have been associated with improved clinical outcomes in antibody therapy of hematological and solid malignancies (8-12). The correlation of patient outcomes with FcR polymorphisms supports the role of ADCC in antibody therapy.
The epidermal growth factor receptor (EGFR) and its family members are frequently altered in cancer. Cetuximab, an anti-EGFR antibody, is approved for treatment of KRAS-wild-type colorectal cancer and head-and-neck squamous cell carcinoma (HNSCC). Cetuximab induces ADCC against both colorectal cancer – independent of KRAS status – and HNSCC cell lines (1,13,14).
Prior studies have focused on various mechanisms of enhancing immune effector cell activity and ADCC (2,15-17). A functional genomics study targeting kinases and phosphatases in myeloma cells assessed for modulation of their sensitivity to NK cell cytotoxicity, independent of ADCC (3,18). However, functional screens targeting oncogenic signaling networks within tumor cells and their resultant sensitivity to ADCC have not been reported.
We describe an RNA interference (RNAi) screen for tumor-based molecular determinants of sensitivity to cetuximab-mediated ADCC. Our screens demonstrate that knockdown of several oncogenic signaling network members – ABL1, GRB7 and PRKCE – modulate sensitivity to ADCC. We confirm that ABL1 knockdown increases tumor cell sensitivity to ADCC, while overexpression of c-Abl reduces ADCC and rescues the effects of knockdown. Imatinib mesylate (Gleevec), a c-Abl kinase inhibitor, also enhances cetuximab-mediated ADCC against several HNSCC cell lines. These results suggest that combining cetuximab and c-Abl inhibition may translate into enhanced ADCC and increase the clinical utility of mAb therapy.
Materials & Methods
Cell lines, primary cells, and culture
A431, A253, FaDu, HNSCC 1483, SCC-4, SCC-9 and SCC-25 cell lines were obtained from the Georgetown Lombardi Tissue Culture Shared Resource (TCSR). The SCC-61 cell line was provided by Igor Astsaturov (Fox Chase Cancer Center, FCCC). The UM-SCC-11a cell line was provided by John Deeken (Georgetown Lombardi Comprehensive Cancer Center). These cell lines were cultured in high-glucose DMEM (HyClone) supplemented with 10% fetal bovine serum (FBS; Omega Scientific) and 2 mM L-glutamine (Gibco). NK92-CD16V cells were provided by Kerry S. Campbell (FCCC) and maintained as previously described (1,3,4,19). Cell lines were confirmed as mycoplasma free and verified by short tandem repeat analysis (TCSR). Frozen primary peripheral blood mononuclear cells (PBMC) from three individual donors (AllCells) were enriched for NK cells (Human NK Cell Enrichment Kit, STEMCELL Technologies) yielding 3.6-6.7% of total PBMCs, maintained in RPMI-1640 with 10% FBS and 2 mM L-glutamine, and stimulated with 500 units/mL recombinant human IL2 (Life Technologies). All cells were cultured at 37°C and 5% CO2.
Antibody-independent natural cytotoxicity and ADCC assays
Target cells were seeded or reverse-transfected in 96-well white-walled, clear bottom tissue culture plates (Corning Costar). Pre-treatments were added as indicated. At the time of assay, four treatments were added: vehicle (growth media); antibody; effector cells; and antibody with effector cells. Antibody was added at concentrations and effector cells were added at effector-to-target ratios (E:T) indicated and incubated for 4 h. CytoTox-Glo (Promega) was used to assess initial and total cytotoxicity signal per manufacturer’s instructions. Specific lysis was determined for antibody-independent natural cytotoxicity (NK92-CD16V cells only) as
and ADCC (effector cells with antibody) as
where tar refers to the plated target cells, eff refers to effector cells, and mAb refers to the monoclonal antibody.
siRNA reverse transfection
All siRNAs, including AllStars Negative Control (siNEG) and Hs Death Control (siDEATH) siRNAs, were from Qiagen. The EGFR (siEGFR) siRNA (EGFR_10) target sequence was TACGAATATTAAACACTTCAA. Supplementary Table 1 and 2 contain the target sequence for siRNAs used in screens. siRNAs working stocks were 1 μM in siRNA Suspension Buffer (Qiagen). Lipofectamine RNAiMAX (Invitrogen) was diluted in OptiMEM and 10 nM siRNA or v/v Suspension Buffer was added. Transfection mixtures were incubated for 10 min, plated, and overlaid with cells.
Screening
Arrayed 96-well siRNA library plates were used with the 60 inner wells containing two pooled siRNAs per gene (Supplementary Table 1). Control siRNAs and treatment control wells were included in the 36 outer wells for quality control and normalization.
A431 cells were reverse transfected with library plate siRNAs into assay plates utilizing fluidic instrumentation. First, 10 μL of diluted Lipofectamine RNAiMAX transfection reagent in OptiMEM (Invitrogen) – 0.3 μL of RNAiMAX per 10 μL of OptiMEM – was plated by Combi-nL (Thermo Scientific). Then, 10 μL of 100 nM siRNAs were aliquoted from library plates into the diluted transfection reagent using a CyBi-Well vario (CyBio) and incubated at room temperature for 10 min. 5,000 A431 cells in 80 μL were overlaid using a WellMate (Thermo Scientific), resulting in 10 nM siRNA. Duplicate plates were reverse transfected for each treatment.
At 48 h, the reverse-transfected cells were treated with: cetuximab; NK92-CD16V effector cells; or cetuximab and NK92-CD16V effector cells. Cetuximab (ImClone/Eli Lilly) was utilized at 1 μg/mL. 20,000 NK92-CD16V effector cells were added to ~1:1 E:T. Treatments were added to duplicate plates using the Combi-nL.
Cytotoxicity values were normalized per plate by the within-plate median of the vehicle-treated negative control siRNA cytotoxicity. ADCC-specific lysis and fold-changes were calculated relative to negative controls. Differential cytotoxicity was
ANOVA was conducted with Dunnett’s multiple comparison correction. Secondary screens are described in the Supplementary Methods.
Quantitative reverse transcription PCR
Reverse-transfected cells were collected at 48 h and processed using Fast SYBR Green Cells-to-Ct Kit (Ambion) per manufacturer’s instructions. QuantiTect Primer Assays (Qiagen) were used (Supplementary Table 3). Quantitative PCR was conducted on a 7900HT in 96-well MicroAmp plates (Applied Biosystems). Relative quantitation was assessed by the comparative Ct (2-ΔΔCt) method calibrated to GAPDH (GAPDH_1_SG) level.
Real-time cell assay (RTCA)
The xCELLigence RTCA with 96-well E-plates (Roche) was utilized per manufacturer’s instructions. E-plates with media or reverse transfection components were equilibrated in an RTCA system at 37°C in 5% CO2. Background measurements were obtained, cells were added, and the assay was started. Cell index was measured every 10 min. Treatments were added by pausing, aliquoting, and restarting the assay.
Viability and proliferation assays
CellTiter-Blue (Promega) assays were conducted in 96-well format per manufacturer’s instructions.
c-Abl overexpression and rescue
Wild type c-Abl in the pCEFL-AU5 plasmid was kindly provided by Anna Riegel (Georgetown Lombardi). Cells were seeded overnight to 50-60% confluence. Forward transfection of c-Abl plasmid and/or 10 nM siRNA was conducted using Lipofectamine 2000 (Invitrogen) per manufacturer’s instructions.
Kinase inhibitors and treatments
Imatinib mesylate (Selleck Chemicals) and dasatinib, ponatinib, and nilotinib (kindly provided by John Deeken) were solubilized in DMSO. Cells seeded overnight were treated with inhibitors diluted in growth media for 48 h. Vehicle treatment (DMSO) was used at the highest equivalent v/v.
Western blotting
Cells were lysed in RIPA buffer with EDTA (Boston BioProducts) supplemented with protease (Roche) and phosphatase inhibitors (Pierce). Cleared lysate concentrations were obtained by DC Protein Assay (BioRad). Lysates were subjected to SDS-PAGE and transferred to nitrocellulose membranes (GE Healthcare). Western blots were conducted using: rabbit anti-Abl (Cell Signaling Technologies [CST], #2862); rabbit anti-pCrkL (CST #3181); mouse anti-CrkL (CST #3182); rabbit anti-EGFR (Abcam, Clone EP38Y); and mouse anti-β-actin (Sigma, Clone AC-74). Goat anti-rabbit or donkey anti-mouse IgG HRP-conjugated secondary antibodies (GE Healthcare) were used with chemiluminescence substrates (Pierce). Densitometry was assessed by ImageJ (5,20).
Data analysis
Data analysis was conducted in R with the reshape and multcomp packages (3,6,21-23). Multiple comparison test correction was used to obtain adjusted p-values by the single-step method (7,23). Otherwise, two-tailed t-tests were conducted. Statistical significance was obtained when p<0.05.
Results
Development of a functional genomics screen for ADCC
Several methods used to assess ADCC, such as chromium-release and flow-based apoptosis detection assays, are low-throughput and not easily scaled for screening. Therefore, we developed a high-throughput screening platform for assessing the effect of RNAi on tumor cell sensitivity to ADCC. The platform utilized siRNAs arrayed in 96-well format for reverse transfection of adherent target cells. Our model system included: A431 squamous cell carcinoma cells, which overexpress EGFR; cetuximab, an IgG1 anti-EGFR antibody capable of engaging FcRs on NK cells; and NK92-CD16V effector cells, an NK-like cell expressing an FCGR3A (CD16) variant that induces ADCC (8-12,19).
We adapted a luminescence-based cytotoxicity assay for screening. This assay utilized protease activity released from dying cells, exhibited greater sensitivity with similar background versus an LDH release assay (data not shown), and provided a two-step cytotoxicity measurement from each well. The assay was capable of assessing cytotoxicity across a range of both effector-to-target ratios (E:T) and cetuximab concentrations (Fig. 1A). Specific lysis, a measurement of NK cell-mediated antibody-independent natural cytotoxicity and ADCC, was readily derived (Fig. 1B). Antibody-independent natural cytotoxicity was low (0-15%) across the range of E:T (1:1-4:1, Fig. 1B & Supplementary Fig. 1A).
Figure 1.
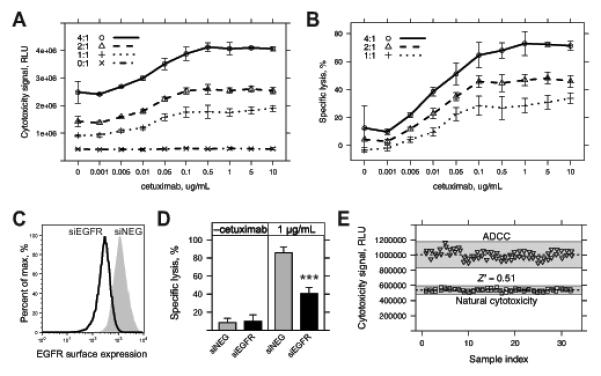
Development and characterization of a functional genomics screening assay for assessment of ADCC. A, A431 cells were seeded overnight in 96-well plates and then treated with vehicle (media), cetuximab only, NK92-CD16V only, and combined cetuximab and NK92-CD16V treatments. Cytotoxicity signal (relative luminescence units, RLU) was assessed 4 h later across a range of concentrations of cetuximab (0-10 μg/mL) and effector-to-target ratios (E:T, 0:1-4:1). B, Specific lysis was calculated from the cytotoxicity signal values in A by subtraction of initial A431 cytotoxicity and NK92-CD16V spontaneous cytotoxicity from combined treatment-induced cytotoxicity, before normalization by total A431 cytotoxicity (see Materials & Methods for details). C, A431 cells were reverse transfected with 10 nM EGFR siRNA (siEGFR) or negative control siRNA (siNEG) and collected 48 h later. Cells were stained with cetuximab and anti-human IgG(H+L)-PE-conjugated secondary antibody to assess EGFR surface expression or with secondary antibody alone. Secondary Ab only staining was similar to unstained controls (not shown) for all transfected cell lines. D, A431 cells were reverse transfected as in C and assessed for ADCC 48 h later. Specific lysis was assessed using a 4:1 E:T using NK92-CD16V effector cells in the absence (–cetuximab) or presence of 1 μg/mL cetuximab. Specific lysis of A431 cells by NK92-CD16V cells following knockdown of EGFR (siEGFR, black bars) was compared to negative control siRNA (siNEG, gray bars) in the absence and presence of cetuximab. ***, p<0.001 from two-tailed t-test. E, A431 cells seeded overnight in 96-well plates were treated with 1:1 E:T of NK92-CD16V cells in the absence (“natural cytotoxicity”, squares) or presence (“ADCC”, triangles) of 1 μg/mL cetuximab. Cytotoxicity was assessed 4 h later. Z’-factor, which measures the suitability of an assay for screening, was assessed between the ADCC vs. natural cytotoxicity treatments. Dashed lines represent the mean of sample replicates and shaded area represents ±3 standard deviations (s.d.) from the mean. The Z’-factor incorporates both sample mean and ±3 s.d. For all panels, error bars represent s.d. of at least three independent experiments (n=3).
We assessed two additional antibodies to verify that ADCC was specific to antigen-bound antibody engagement of NK cells. Rituximab (anti-CD20) does not bind A431 target cells due to lack of antigen expression and panitumumab (anti-EGFR) binds A431 cells, but has limited capacity to engage NK92-CD16V FcγR. Neither rituximab (Supplementary Figs. 1B & C) nor panitumumab (Supplementary Figs. 1D & E) promoted ADCC.
Next, we identified conditions for siRNA reverse transfection wherein modulation of ADCC could be detected. Surface expression of EGFR has been correlated with magnitude of cetuximab-mediated ADCC (24). We confirmed that siRNA knockdown of EGFR significantly reduced EGFR surface expression (Fig. 1C) and ADCC (Fig. 1D).
Because we aimed to identify genes whose knockdown enhanced sensitivity to ADCC, we sought assay conditions with modest levels of ADCC. At an E:T of 1:1 and using 1 μg/mL of cetuximab, ADCC-specific lysis plateaued at ~30% (Fig. 1B). We anticipated that siRNA knockdown could enhance this level of ADCC, outside of the antibody concentration-dependent range of ADCC (Fig. 1B, 1-100 ng/μL). Z’-factor, a measure of dynamic range and signal-to-noise, was assessed to determine robustness for screening (25). Under our prescribed conditions, a Z’-factor ≥ 0.5 indicated that assay conditions were suitable for screening (Fig. 1E, Z’-factor=0.51).
Screening for sensitizers of ADCC
In a prior screening study of an EGFR-centered siRNA library targeting 638 genes, knockdown of 61 genes enhanced the effectiveness of EGFR-targeted therapeutics, independent of ADCC (26). We assessed whether knockdown of any of those 61 genes could also enhance cetuximab-mediated ADCC, aiming to find gene targets wherein two antitumor effects – synthetic lethality and ADCC sensitivity – would be enhanced. An arrayed library of pooled siRNAs was prepared, with the exception of one target gene, RPL10P16, because it was re-annotated as a pseudogene (Supplementary Table 1).
In two primary screens, A431 cells were reverse transfected with two pooled siRNAs per target gene. Two days later, three treatments were added to the transfected cells: cetuximab; NK92-CD16V cells; and the combination of cetuximab and NK92-CD16V cells (Supplementary Fig. 2A). Cytotoxicity was assessed after 4 h and normalized by per plate controls. ADCC sensitivity was analyzed by both specific lysis and differential cytotoxicity (see Materials & Methods). ADCC-specific lysis was determined for the combined cetuximab and NK92-CD16V treatment relative to the cetuximab only treatment.
Knockdown of 22 genes significantly enhanced ADCC by either specific lysis (Fig. 2A) and/or differential cytotoxicity (Supplementary Fig. 2B) with a multiple-comparison adjusted p<0.05. We prioritized the selection of gene hits for secondary screens based on enhancement of ADCC, first including those that significantly enhanced differential cytotoxicity (Supplementary Fig. 2B): RASA3 and GRB7. The top five additional genes whose knockdown significantly enhanced specific lysis were included (Fig. 2A): PRKCE, EPHA5, PLSCR1, RET, and ABL1. These seven genes – PRKCE, EPHA5, PLSCR1, RASA3, RET, ABL1, and GRB7 – were promoted to secondary screens.
Figure 2.
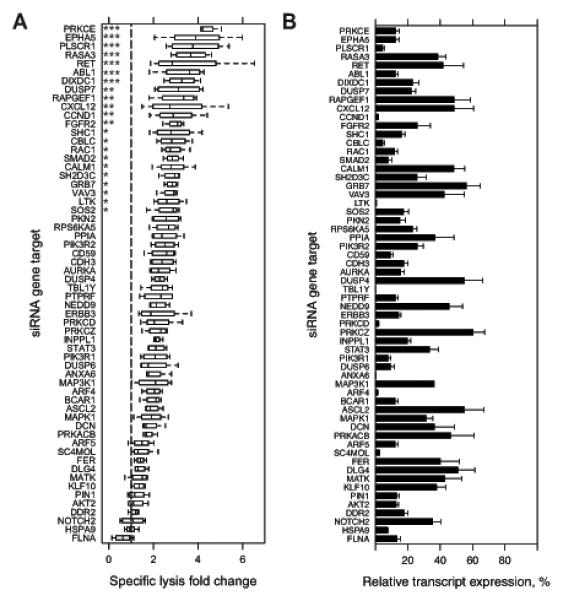
Primary siRNA screen targeting A431 cells for genes whose knockdown enhances ADCC. A, Two primary screens were conducted in which an arrayed library of two pooled siRNA at 10 nM each per 60 targeted genes were reverse transfected in A431 cells in 96-well plates. At 48 h post-transfection, three treatments were added: 1 μg/mL cetuximab alone; 20,000 NK92-CD16V cells alone; and the combination of cetuximab and NK92-CD16V cells. All transfections and treatments were conducted in duplicate for each primary screen. Specific lysis of the combined cetuximab and NK92-CD16V cell treatments was calculated for each replicate siRNA gene target. Specific lysis fold-change relative to a negative control siRNA (dashed vertical line, normalized to 1) was calculated for each replicate in each screen. Specific lysis fold-change values are represented as boxplots of two independent measurements in two primary screens (n=4). Statistical significance was assessed by ANOVA followed by Dunnett’s multiple comparison test correction for each siRNA gene target versus the negative control siRNA. *, p<0.05; **, p<0.01; and ***, p<0.001. B, A431 cells were reverse transfected as described in A, RNA isolated 48 h later, cDNA generated, and real-time quantitative reverse-transcription PCR (q-RT-PCR) was conducted to assess knockdown of each targeted genes. Percent relative expression was calculated compared to the negative control siRNA transfection and calibrated by GAPDH expression. Percent relative expression for each siRNA target genes are ordered as in A. *, p<0.05; and **, p<0.01. Error bars represent s.d. of the mean from three independent experiments (n=3).
Two secondary screens were conducted using an arrayed library of four individual unpooled siRNAs per hit (Supplementary Table 2) to minimize individual siRNA off-target effects. The criteria for validation of hits were: at least two of four individual siRNAs assessed must corroborate primary screen results; and, knockdown of the target gene must be confirmed. Aside from using unpooled siRNAs and including EGFR knockdown as an additional control, secondary screens were conducted and analyzed similarly to primary screens (Supplementary Fig. 2C & D). These secondary screens identified three of the primary hits – GRB7, PRKCE, and ABL1 – whose knockdown by at least two independent siRNAs significantly enhanced ADCC (Supplementary Fig. 2C & D and Table 1, adjusted p<0.05). Knockdown of these three genes, along with all screened genes, was confirmed at the transcript level by real-time quantitative reverse-transcription PCR (qRT-PCR) (Fig. 2B). Relative expression of transcripts after knockdown ranged from 1-60% and did not correlate with ADCC-specific lysis (Supplementary Fig. 2E).
Table 1.
Validated gene targets whose knockdown by at least two independent siRNAs significantly enhanced ADCC by specific lysis (Fig. 2A) and/or differential cytotoxicity (Supplementary Fig. 2B) in secondary screens. siRNA target sequences can be found in Supplementary Table 2.
| Gene target | Specific lysis, p<0.05 | Differential cytotoxicity, p<0.05 |
|---|---|---|
| GRB7 | siGRB7_7, siGRB7_3 | siGRB7_7, siGRB7_3 |
| PRKCE | siPRKCE_5, siPRKCE_2 | siPRKCE_5 |
| ABL1 | siABL1_9 (“siABL1 #1”)*, | – |
| siABL1_10 (“siABL1 #2”)* |
,used in subsequent characterization studies (Fig. 3C)
Characterization of the effects of ABL1 knockdown on proliferation and sensitivity to ADCC
We focused on ABL1 as several clinically useful inhibitors target c-Abl kinase activity. After identifying ABL1 as a putative modulator of ADCC, we further characterized the effects of its knockdown. A real-time cell assay (RTCA) system was used to assess cell index, a surrogate measurement of adhesion, viability, and proliferation. By cell index, NK cell and ADCC responses were detectable across a broad range of E:T (0.5:1-4:1) and cetuximab (0.01-1 μg/mL) concentrations (Supplementary Fig. 3).
We first assessed the effects of siRNA gene knockdown alone by RTCA. Reverse transfection of ABL1 siRNA in A431 cells reduced cell index relative to control siRNA (Fig. 3A, top left). Viability assays across a comparable time series demonstrated similar effects of ABL1 knockdown on proliferation (Fig. 3B). Seven days post-transfection, A431 cells recovered from the effects of transient ABL1 knockdown by cell index (Fig. 3A, top left) and viability (Fig. 3B) assessments.
Figure 3.
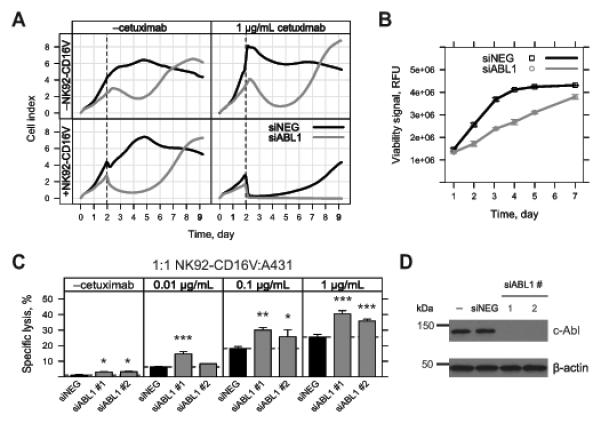
Characterizing the effect of ABL1 knockdown on proliferation, viability, and sensitivity to ADCC in A431 cells. A, A431 cells were reverse transfected with 10 nM ABL1 (siABL1, gray) or negative control siRNA (siNEG, black) in a 96-well real-time cell assay (RTCA) plate. Cell index was measured by RTCA every 10 min over the entire time course of the experiment (9 days). Cell index was derived from the impedance of adhered cells in the individual wells of the RTCA plate. Following reverse transfection, treatments were added at 48 h (dashed line) as follows: top left, vehicle (media); top right, 1 μg/mL cetuximab; bottom left, 20,000 NK92-CD16V effector cells; bottom right, 1 μg/mL cetuximab and 20,000 NK92-CD16V effector cells. Each cell index line represents the mean of three independent assessments (n=3) from one representative of three experiments. B, A431 cells were reverse transfected 10 nM ABL1 siRNA (siABL1, gray) or negative control siRNA (siNEG, black) in 96-well format. Fluorometric viability assays were conducted at the indicated time points. Each viability measurement represents the mean of three independent measurements (n=3) from one representative of four experiments. C, A431 cells were reverse transfected in six-well plates with 10 nM ABL1 siRNA (siABL1 #1 or #2, gray bars) or negative control siRNA (siNEG, black bars). siABL1 #1 is the same ABL1 siRNA used in A. Reverse transfected cells were collected at 48 h and re-plated at 20,000 viable cells/well in a 96-well plate. Following a brief incubation, the following treatments were added: vehicle (media); cetuximab (0.01, 0.1 and 1 μg/mL); 20,000 NK92-CD16V cells; and 20,000 NK92-CD16V cells and cetuximab (0.01, 0.1 and 1 μg/mL). Cytotoxicity was assessed 4 h later and specific lysis by NK92-CD16V cells was determined in the absence or presence of cetuximab. Each ABL1 siRNA was compared to the negative control siRNA within each sub-panel. *, p<0.05; **, p<0.01; and ***, p<0.001 from two-tailed t-tests. Results are from three independent experiments (n=3). D, A431 cells were reverse transfected without siRNA (–) or with 10 nM negative control (siNEG) or ABL1 siRNA (siABL1 #1 or #2 as used in C). Cell lysates were collected, applied to SDS-PAGE, and transferred to a membrane, and blotted for c-Abl. c-Abl expression was undetectable for the two ABL1 siRNAs transfections, although faint bands were visible upon overexposure (data not shown). The same membrane was blotted for β-actin as a loading control. Results are representative of two experiments. For panels B and C, error bars represent s.d. of the mean.
We also used RTCA to assess the effects of ABL1 knockdown on ADCC. As evident in Figure 3, cetuximab (top right) or NK92-CD16V cell (bottom left) modestly enhanced the effects of ABL1 knockdown alone (top left). However, the combined ADCC treatment dramatically reduced cell index in ABL1 knockdown cells (Fig. 3A, bottom right). By the end of the assay, the negative control siRNA transfected cells had recovered from the ADCC treatment, whereas ABL1 knockdown cells were at background (Fig. 3A, bottom right). Endpoint crystal violet staining corroborated the RTCA results (data not shown).
Because ABL1 knockdown reduced the proliferation of A431 cells, it was plausible that specific lysis was enhanced due to differences in relative E:T at the time of ADCC assay. ABL1 knockdown resulted in 50-60% reduction of A431 cells at 48 h, when assay treatments were added (Fig. 3A, top left; and 3B). Because NK92-CD16V cells were added at the same density to A431 cells in the negative control and ABL1 knockdown conditions, relative E:T was approximately two-fold higher in the latter, which alone could account for an increase in ADCC-specific lysis (Fig. 1B).
To account for these effects of ABL1 knockdown, we utilized a modified ADCC assay. Negative control and ABL1 knockdown A431 cells were collected 48 h post-transfection, re-plated at equivalent density, and assayed. With these modifications and using two independent siRNAs, ABL1 knockdown significantly enhanced ADCC, with a minor effect on cetuximab-independent NK cell natural cytotoxicity (Fig. 3C). ABL1 knockdown by the two independent siRNAs resulted in c-Abl levels undetectable by Western blot (Fig. 3D).
EGFR expression is correlated with magnitude of ADCC, which we (Fig. 1C & D) and others have demonstrated (24). Enhanced ADCC following ABL1 knockdown could be due to increased EGFR surface expression, as c-Abl activity has been shown to regulate EGFR endocytosis (27). Therefore, EGFR surface expression was assessed by flow cytometry following ABL1 knockdown. In A431 cells reverse transfected with ABL1 siRNA, EGFR surface expression was unchanged compared to controls (Supplementary Fig. 4A & B).
c-Abl expression and sensitivity to ADCC
Knockdown of c-Abl by siRNA enhanced the sensitivity of A431 cells to ADCC, independent of its effects on proliferation. We further investigated whether the level of c-Abl could regulate ADCC responsiveness. A wild type c-Abl plasmid expression construct was forward transfected into A431 cells and ADCC sensitivity was assessed. Because c-Abl overexpression has been shown to disrupt the cell cycle (28), minimal quantities (1-50 ng) of the plasmid were utilized in short-term (24 h) transfections. Increasing quantities of transfected c-Abl plasmid were associated with concomitant decreases in ADCC sensitivity, without altering antibody-independent natural cytotoxicity (Fig. 4A). We confirmed that c-Abl expression was proportional to the amount of plasmid transfected by Western blot (Fig. 4B).
Figure 4.
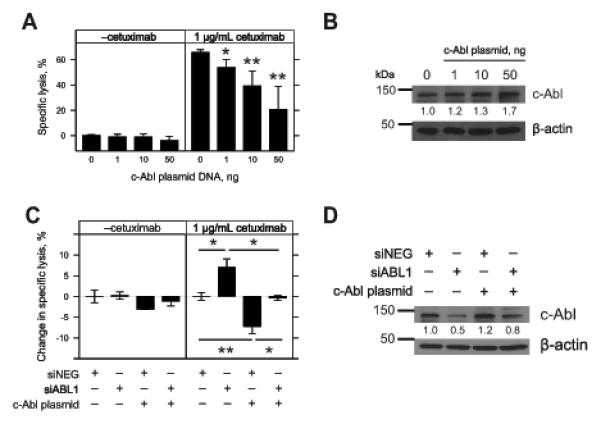
Modulating c-Abl expression and sensitivity to ADCC in A431 cells. A, A431 cells were seeded overnight in 96-well plates and forward transfected with increasing quantities (1, 10, and 50 ng) of wild type c-Abl plasmid and compared to a mock transfection (0). After 24 h, the following treatments were added: vehicle (media); 1 μg/mL cetuximab; 20,000 NK92-CD16V cells; and 20,000 NK92-CD16V cells and 1 μg/mL cetuximab. Cytotoxicity was assessed 4 h later and specific lysis by NK92-CD16V cells was determined in the absence or presence of cetuximab. Each quantity of transfected plasmid was compared to the mock transfection control. Results are from three independent experiments (n=3). B, Cell lysates were collected from A431 cells following forward transfection of the wild type c-Abl plasmid as described in A. Western blot of c-Abl and β-actin was performed. Densitometry was conducted and c-Abl levels were normalized to β-actin levels and compared relative to the mock transfection control. Results are from one representative of three experiments. C, A431 cells were seeded overnight in 96-well plates and forward transfected with combinations of 10 nM negative control siRNA (siNEG), 10 nM ABL1 siRNA (siABL1), and 1 ng wild type c-Abl plasmid c-Abl. After 24 h, specific lysis was assessed (as described in A). Change in specific lysis was calculated by subtracting the negative control siRNA specific lysis within each sub-panel. Change in specific lysis was compared to the negative control siRNA within each sub-panel. Results are from three independent experiments (n=3). D, Cell lysates were collected following forward transfection of the negative control siRNA (siNEG), ABL1 siRNA (siABL1), and 1 ng wild type c-Abl plasmid as described in C. Western blot of c-Abl and β-actin was performed. Densitometry was conducted and c-Abl levels were normalized to β-actin and compared to negative control siRNA. Results are depicted for one representative of two experiments. For panels A and C, error bars represent s.d. of the mean. *, p<0.05 and **, p<0.01 by two-tailed t-test.
We hypothesized that c-Abl expression could rescue the effects of ABL1 knockdown. We selected an ABL1 siRNA that targets the 3’-untranslated region (UTR) of endogenous c-Abl transcript (siABL1 #1 from Fig. 3D and Supplementary Table 2), but not the c-Abl plasmid, which contains only the ABL1 coding sequence. In short-term forward transfections, siRNA knockdown of ABL1 enhanced ADCC (Fig. 4C) while reducing c-Abl expression by ~50% (Fig. 4D). c-Abl plasmid co-transfected with a negative control siRNA reduced ADCC sensitivity (Fig. 4C) with a modest increase in c-Abl expression of ~20% (Fig. 4D). However, co-transfection of c-Abl plasmid with ABL1 siRNA abrogated enhancement of ADCC (Fig. 4C). Western blotting revealed that c-Abl plasmid co-transfection partially rescued endogenous c-Abl knockdown by ABL1 siRNA to ~80% of the endogenous c-Abl levels (Fig. 4D).
Inhibition of c-Abl kinase activity and sensitivity to ADCC
c-Abl is a ubiquitously expressed protein with pleiotropic roles in growth factor response, cell cycle, DNA damage, apoptosis, and cytoskeletal dynamics reflected in its functional domains (29). The N-terminus of c-Abl contains regulatory Src Homology 3 (SH3) and SH2 domains and a kinase catalytic core, much like c-Src. To dissect the role of c-Abl on modulation of ADCC, we utilized imatinib to inhibit c-Abl tyrosine kinase activity. Imatinib was the first approved kinase inhibitor of the fusion gene product, Bcr-Abl, found in chronic myelogenous leukemia (CML). Beyond inhibiting the constitutive kinase activity of the fused c-Abl kinase domain, imatinib also inhibits endogenous c-Abl, platelet-derived growth factor receptors (PDGFRs), and c-Kit (30).
We pre-treated A431 cells with imatinib to assess the effects of c-Abl kinase inhibition on ADCC sensitivity. Just prior to assessment, we replaced the imatinib treatments with fresh growth media to focus on target cell effects and limit potential off-target effects on NK92-CD16V effector function (31). Pre-treatment of A431 cells with imatinib for 48 h significantly enhanced ADCC (Fig. 5A, 10 μM imatinib with 0.1 or 1 μg/mL cetuximab and NK92-CD16V cells). Higher doses of imatinib resulted in significant direct cytotoxicity to A431 cells independent of ADCC (data not shown). Imatinib pre-treatment of A431 cells also significantly enhanced ex vivo IL2-stimulated human NK cell-mediated ADCC (Fig. 5B).
Figure 5.
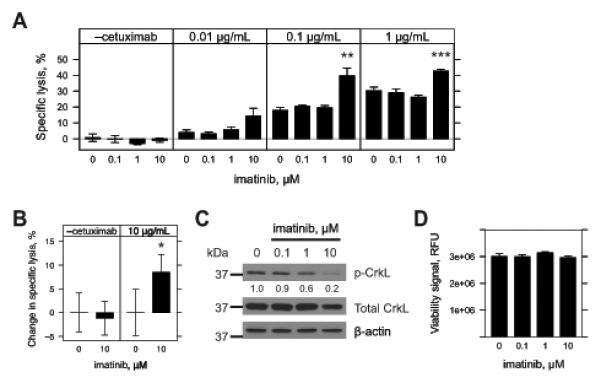
Inhibition of c-Abl kinase activity by imatinib in A431 cells and sensitivity to ADCC. A & B, A431 cells were seeded overnight in 96-well plates and treated with vehicle (0, DMSO) or imatinib (0.1, 1 or 10 μM) for 48 h. Treatments were aspirated and replaced with fresh growth media. Effector cells were added in the absence or presence of cetuximab (0.01, 0.1, and 1 μg/mL). Cytotoxicity was assessed 4 h later and specific lysis by effector cells was determined. For A, 20,000 NK92-CD16V effector cells were used. Results represent three independent experiments (n=3). For B, 50,000 IL-2 negatively-selected, IL2 stimulated NK effector cells were used. Percent-change in specific lysis was quantified to account for varying levels of donor specific lysis against target cells. Results represent one of two independent experiments using three independent donors (n=3). For A & B, imatinib pre-treatment was compared to vehicle within each sub-panel. *, p<0.05; **, p<0.01; and ***, p<0.001 by two-tailed t-test. C, A431 cells were seeded in six-well plates overnight and treated with vehicle (0, DMSO) or imatinib (0.1, 1 or 10 μM) and cell lysates collected 48 h later. Western blots were conducted and membranes blotted for phospho-CrkL (p-CrkL), stripped and re-blotted for both total CrkL and then β-actin. Densitometry was conducted and p-CrkL levels were normalized to total CrkL levels and then compared to vehicle treatment. Results are for one representative of two experiments. D, A431 cells were seeded overnight in 96-well plates and treated with vehicle (0, DMSO) or imatinib (0.1, 1 or 10 μM) for 48 h. Viability was assessed by fluorometric assay. No statistically significant differences were found between treatments by t-test. Results are from six independent experiments (n=6). For panels A, B & C, error bars represent s.d. of the mean.
We next assessed phospho-CrkL (p-CrkL), an endogenous surrogate for c-Abl kinase activity (32). A concentration-dependent reduction in p-CrkL vs. total CrkL was observed by Western blot, with the most reduction at 10 μM imatinib (Fig. 5C, 20% of vehicle). Because ABL1 knockdown inhibited proliferation of A431 cells (Fig. 3B), we assessed the effect of imatinib on proliferation. Imatinib pre-exposure did not affect the proliferation of A431 cells over 48 h (Fig. 5D). Similar to ABL1 knockdown, 10 μM imatinib did not affect EGFR surface expression (Supplementary Fig. 4C & D).
We assessed additional tyrosine kinase inhibitors including dasatinib, nilotinib, and ponatinib that have been developed to treat Bcr-Abl-positive cancers. These inhibitors have been shown to inhibit c-Abl as well as several other kinases (33). Dasatinib (Sprycel) is a second-generation dual Src/Abl tyrosine kinase inhibitor with a pan-tyrosine kinase inhibitor profile (33). Dasatinib pre-treatment of A431 cells demonstrated concentration-dependent inhibition and complete abrogation of ADCC (Supplementary Fig. 5A, 100 μM).
Nilotinib (Tasigna) is a second-generation Abl inhibitor with a kinase profile similar to imatinib (33). Nilotinib pre-treatment enhanced ADCC in a concentration-dependent manner with a modest, but significant enhancement at the highest concentration assessed (Supplementary Fig. 5B, 1 μM). Ponatinib (AP24534) is a third-generation c-Abl inhibitor with dual Src/Abl tyrosine kinase activity like dasatinib, but with the capacity to inhibit Bcr-Abl mutations associated with resistance (34). Ponatinib significantly enhanced ADCC at sub-nanomolar concentrations (Supplementary Fig. 5C, 0.01 and 0.1 nM), but inhibited ADCC of A431 cells at higher concentrations (Supplementary Fig. 5C, 10 and 100 nM). Ponatinib also had significant, but inconsistent effects on antibody-independent natural cytotoxicity (Supplementary Fig. 5C, 0.1 and 100 nM).
As c-Src and Src family kinases (SFK) are important for NK cell activity, we hypothesized that inhibition of ADCC with dasatinib and ponatinib was due to c-Src inhibition of NK cell function (35,36). Even with pre-treatment and replacement prior to ADCC assessment, abrogation of ADCC still occurred (Supplementary Fig. 5A & C). However, it was plausible that inhibition of c-Src or other SFKs in targeted cells protected them from ADCC. To clarify the role of c-Src on target cell susceptibility to ADCC, we assessed the effects of SRC knockdown in A431 cells. SRC knockdown in A431 cells did not change their sensitivity to ADCC (Supplementary Fig. 5D).
Imatinib inhibition of c-Abl kinase activity in EGFR-expressing HNSCC lines and sensitivity to ADCC
Cetuximab-mediated ADCC has been reported against HNSCC cell lines and is a purported mechanism of therapeutic activity (14,37). To examine the translational potential of imatinib on cetuximab-mediated ADCC, we assessed a panel of eight HNSCC cell lines: A253, FaDu, HNSCC 1483, SCC-4, SCC-9, SCC-25, SCC-61, and UM-SCC-11a.
Using our NK92-CD16V model system, imatinib pre-treatment of A253, FaDu, HNSCC 1483 and UM-SCC-11a cells significantly enhanced cetuximab-mediated ADCC (Fig. 6A). Imatinib also significantly enhanced antibody-independent natural cytotoxicity of HNSCC 1483 and UM-SCC-11a (Fig. 6A). SCC-9 and SCC-25 demonstrated no change and SCC-4 and SCC-61 cell lines had significantly reduced ADCC after imatinib treatment (Supplementary Fig. 6A). Using ex vivo IL2-stimulated NK cells, we found that imatinib pre-treatment enhanced ADCC of A253, FaDu, HNSCC 1483, and UM-SCC-11a cells (Fig. 6B). As was demonstrated with NK92-CD16V cells (Fig. 6A), imatinib pre-treatment also enhanced natural cytotoxicity of UM-SCC-11a cells by ex vivo NK cells (Fig. 6B). In the cell lines where imatinib enhanced ADCC, we assessed whether imatinib also affected target cell viability. Imatinib treatment was associated with significant reduction in UM-SCC-11a viability, whereas A253, FaDu and HNSCC 1483 cells were unaffected (Fig. 6C).
Figure 6.
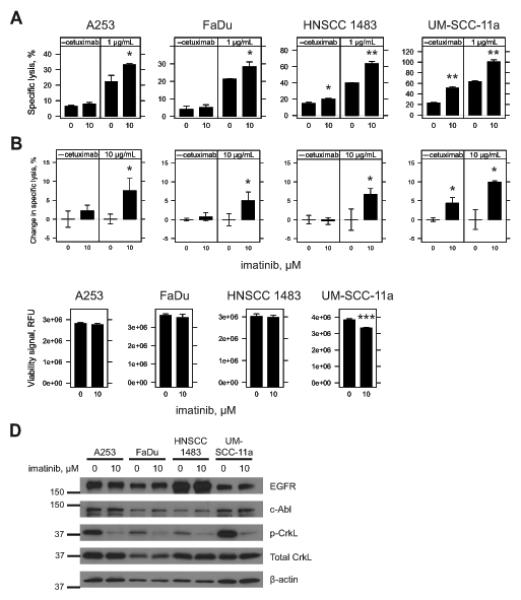
Inhibition of c-Abl kinase activity by imatinib and sensitivity of HNSCC cell lines to ADCC. A & B, A253, FaDu, HNSCC 1483 and UM-SCC-11a cells were seeded overnight in 96-well plates and treated for 48 h with vehicle (0, DMSO) or imatinib (10 μM). Treatments were aspirated and replaced with fresh growth media just prior to addition of effector cells in the absence or presence of cetuximab. Cytotoxicity was assessed 4 h later and specific lysis was determined. For A, 40,000 NK92-CD16V cells were used in the absence or presence of 1 μg/mL cetuximab. For B, 50,000 IL2 negatively-selected, IL2 stimulated NK effector cells were used in the absence or presence of 10 μg/mL cetuximab. Percent change in specific lysis was quantified to account for varying levels of donor specific lysis against target cells. Results represent one of two independent experiments using three independent donors (n=3). C, A253, FaDu, HNSCC 1483 and UM-SCC-11a cells were seeded overnight in 96-well plates and treated for 48 h with vehicle (0, DMSO) or imatinib (10 μM). Viability was assessed by fluorometric assay. For A, B and C, imatinib pre-treatment was compared to vehicle control within each sub-panel. *, p<0.05; **, p<0.01; and ***, p<0.001 by two-tailed t-test. Results are from three independent experiments (n=3) for each cell line. Error bars represent s.d. of the mean. D, A253, FaDu, HNSCC 1483 and UM-SCC-11a cells were seeded in six-well plates overnight and treated for 48 h with vehicle (0, DMSO) or imatinib (10 μM). Cell lysates were collected and Western blots were conducted. c-Abl was blotted before stripping and re-blotting for EGFR. Phospho-CrkL (p-CrkL) was blotted for before re-blotting for both total CrkL and then β-actin as a loading control. Densitometry was conducted and relative expression was assessed within each cell line for vehicle and imatinib treatments (Supplementary Fig. 6C). Results are representative of two independent experiments.
We next assessed imatinib-treated A253, FaDu, HNSCC 1483, and UM-SCC-11a cell lines for expression of EGFR, c-Abl or total CrkL (Fig. 6D & Supplementary Fig. 6B). EGFR expression was evident and varied, with the most expression in HNSCC 1483 cells. Imatinib treatment of the FaDu cell line was associated with a modest increase in EGFR (1.3-fold), c-Abl (1.4-fold), and CrkL (1.2-fold) expression compared to vehicle-treated cells (Supplementary Fig. 6B). HNSCC 1483 cells had a modest increase in c-Abl (1.2-fold) expression with imatinib treatment (Supplementary Fig. 6B). For A253 and UM-SCC-11a, imatinib did not result in apparent changes in expression of EGFR, c-Abl or total CrkL. For all cell lines, imatinib treatment – regardless of changes in c-Abl expression – correlated with reduced CrkL phosphorylation (Fig. 6D & Supplementary Fig. 6B, p-CrkL vs. total CrkL ≤ 0.3-fold).
Because imatinib-treated FaDu cells had a slight increase in EGFR expression by Western blot, we assessed EGFR surface expression in A253, FaDu, HNSCC 1483, and UM-SCC-11a cells by flow cytometry. Relative EGFR surface expression across the cell lines was similar to levels detected by Western blot (Supplementary Fig. 6C). Imatinib treatment was not associated with a significant change in EGFR surface expression across all cell lines assessed (Supplementary Fig. 6D).
Discussion
The anti-EGFR mAb cetuximab, which is capable of inducing ADCC, plays an important role in the treatment of colorectal and HNSCC cancers. Although KRAS mutations have been associated with cetuximab resistance, FcγR polymorphisms were independent of KRAS status in predicting clinical outcomes in colorectal cancer and ADCC is independent of KRAS status in vitro (12,13,17). The Cancer Genome Atlas (TCGA) data suggests that KRAS is altered in more than 40% of colorectal cancer cases; however, less than 2% of HNSCC have alterations in KRAS (6 of 302 cases) in which cetuximab resistance also occurs (38,39). Therefore, we hypothesized that other components of EGFR oncogenic signaling networks could modulate ADCC response. We targeted a set of EGFR-related genes, assessing how their knockdown could enhance ADCC sensitivity, in addition to ADCC-independent synthetic lethal responses previously reported (26). Screening revealed and stringently validated three genes – GRB7, PRKCE and ABL1 – whose knockdown enhanced ADCC. Silencing of ABL1 expression was phenocopied by imatinib, suggesting a new approach for augmenting the ADCC-mediated antitumor effects of cetuximab.
Previous studies have defined a role for c-Abl in cell cycle regulation and proliferation (28,40). We also found that c-Abl silencing affected proliferation. However, imatinib inhibition of c-Abl kinase activity did not affect proliferation in most cell lines assessed. Considering that ABL1 knockdown and imatinib treatment enhanced ADCC these results suggest a kinase-independent role of c-Abl in proliferation, but a kinase-dependent role of c-Abl in ADCC sensitization.
We investigated the mechanism of enhanced ADCC by ABL1 knockdown or c-Abl kinase inhibition. Although c-Abl has been shown to modulate EGFR endocytosis, and EGFR surface expression positively correlates with ADCC (24,27), EGFR surface expression was unchanged by ABL1 knockdown or imatinib exposure. c-Abl could regulate apoptosis, which is a critical mechanism of NK cell-mediated cytotoxicity (6,29). We assessed apoptosis sensitivity independent of ADCC following ABL1 knockdown in A431 cells, but did not detect differential caspase-3/7 response (data not shown). Fluctuations in target cell membrane cytoskeletal organization can modify ADCC responsiveness (41). Subtle morphologic changes observed following ABL1 knockdown and imatinib treatment (data not shown) have spurred us to investigate the role of cytoskeletal dynamics in ADCC responsiveness. We speculate that c-Abl, perhaps through actin modulation, may modify how targeted cells engage NK cells at the immune synapse (29,42). Although preliminary flow cytometry studies following ABL1 knockdown in A431 cells have not shown significant differences in conjugation (data not shown), further studies are needed.
Imatinib has complex effects on human NK cells (31,43), and has been shown to indirectly enhance NK cell activity, independent of the effects we demonstrate on tumor cell sensitivity to ADCC. Imatinib inhibition of c-Kit in dendritic cells (DC) has been shown to enhance NK cell activity in vivo (43,44). Two Phase I studies have combined imatinib and IL2 to enhance NK cell antitumor activity (45,46). In our studies, imatinib enhanced ADCC in some but not all HNSCC cell lines assessed, suggesting that imatinib also exerts an NK cell-autonomous effect on tumor cells. Furthermore, our studies of ABL1 knockdown, overexpression, and rescue demonstrate that the effects of imatinib were not attributable to c-Abl inhibition in NK cells. Irrespective of the mechanism by which imatinib promotes ADCC, our findings and the reported effects on NK cell activity warrant further assessment.
While investigating the effects of c-Abl inhibition on tumor cells, we found that two Src/Abl inhibitors, dasatinib and ponatinib, abrogated ADCC, possibly due to the inhibition of c-Src and/or other SFKs, which are critical for NK-cell activation (5,36). Although SRC knockdown in target cells did not affect their sensitivity to ADCC, other SFKs inhibited by dasatinib may modulate ADCC sensitivity (33). Our results corroborate prior studies showing dasatinib inhibition of ex vivo NK cell activity (47,48). Considering all the evidence at hand, inhibition of c-Abl in tumor cells and c-Kit in DCs – without SFK inhibition in NK cells – could enhance antitumor ADCC.
Primary screening studies were facilitated by the use of A431, an ADCC-sensitive squamous carcinoma cell line, and the NK-like cell line, NK92-CD16, to provide a consistent pool of effector and target cells for large-scale ADCC assays. NK92-CD16 cells constitutively express the activating CD16 necessary for ADCC, but also lack inhibitory co-receptors commonly found on NK cells in vivo. Importantly, the findings of our screening studies were confirmed in a panel of HNSCC cell lines, using ex vivo NK cells from multiple donors. It remains to be seen whether imatinib can also enhance ADCC of colorectal cancer cells and, furthermore, if c-Abl kinase activity is a mechanism of resistance to cetuximab or other ADCC-mediating antibody therapies in patients. Future studies may reveal how c-Abl modulates ADCC and identify the molecular determinants that impart the heterogeneity in imatinib responsiveness we observed in our in vitro and ex vivo studies. It is clear that the functions of c-Abl are diverse and still being unraveled (49). Lastly, additional studies examining how the other genes identified in our screens – GRB7 and PRKCE – modulate ADCC have yielded different mechanisms of action – including sensitivity to apoptosis and regulation of EGFR expression (data not shown) – that are being pursued independently.
Our findings justify further examination of the combination of imatinib and an ADCC-promoting antibody. A distinct challenge in the assessment of ADCC in vivo is the lack of well-characterized xenograft-based animal models that can appropriately recapitulate the anticipated therapeutic effects in patients. Although humanized tumor mouse models do exist, they have not been well-characterized to date (50). Instead, we have used this work as a basis for an ongoing Phase I trial combining cetuximab with the c-Abl kinase inhibitor, nilotinib, in patients with HNSCC or colorectal cancer (http://clinicaltrials.gov/show/NCT01871311). We anticipate that this trial – and others combining targeted therapies – will reveal efficacious strategies for enhancing antibody-based immunotherapy in cancer.
Supplementary Material
Acknowledgments
We acknowledge Kerry Campbell, Rishi Surana, and Casey Shuptrine for their critiques and feedback. We thank the Georgetown Lombardi Shared Resources (NCI P30-CA051008) for their expertise. JCM was supported by an MD-PhD NRSA fellowship, NCI F30-CA165474. LMW was supported by NCI R01-CA050633 and P30-CA051008.
Footnotes
Conflict of Interest – LM Weiner has a commercial research grant from Symphogen, serves as a consultant to Merrimack Phramaceuticals, and has received compensation from Novartis and Abbvie. The other authors declare no potential conflicts of interest.
References
- 1.Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 2010;10:317–27. doi: 10.1038/nri2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weiner LM, Murray JC, Shuptrine CW. Antibody-based immunotherapy of cancer. Cell. 2012;148:1081–4. doi: 10.1016/j.cell.2012.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wallace PK, Howell AL, Fanger MW. Role of Fc gamma receptors in cancer and infectious disease. J Leukoc Biol. 1994;55:816–26. doi: 10.1002/jlb.55.6.816. [DOI] [PubMed] [Google Scholar]
- 4.Chen Z, Freedman MS. CD16+ gammadelta T cells mediate antibody dependent cellular cytotoxicity: potential mechanism in the pathogenesis of multiple sclerosis. Clin Immunol. 2008;128:219–27. doi: 10.1016/j.clim.2008.03.513. [DOI] [PubMed] [Google Scholar]
- 5.Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. 2008;9:495–502. doi: 10.1038/ni1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trapani JA, Smyth MJ. Functional significance of the perforin/granzyme cell death pathway. Nat Rev Immunol. 2002;2:735–47. doi: 10.1038/nri911. [DOI] [PubMed] [Google Scholar]
- 7.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med. 2000;6:443–6. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 8.Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99:754–8. doi: 10.1182/blood.v99.3.754. [DOI] [PubMed] [Google Scholar]
- 9.Weng W-K, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. 2003;21:3940–7. doi: 10.1200/JCO.2003.05.013. [DOI] [PubMed] [Google Scholar]
- 10.Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G, et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol. 2008;26:1789–96. doi: 10.1200/JCO.2007.14.8957. [DOI] [PubMed] [Google Scholar]
- 11.Zhang W, Gordon M, Schultheis AM, Yang DY, Nagashima F, Azuma M, et al. FCGR2A and FCGR3A polymorphisms associated with clinical outcome of epidermal growth factor receptor expressing metastatic colorectal cancer patients treated with single-agent cetuximab. J Clin Oncol. 2007;25:3712–8. doi: 10.1200/JCO.2006.08.8021. [DOI] [PubMed] [Google Scholar]
- 12.Bibeau F, Lopez-Crapez E, Di Fiore F, Thezenas S, Ychou M, Blanchard F, et al. Impact of Fc{gamma}RIIa-Fc{gamma}RIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J Clin Oncol. 2009;27:1122–9. doi: 10.1200/JCO.2008.18.0463. [DOI] [PubMed] [Google Scholar]
- 13.Ashraf SQ, Nicholls AM, Wilding JL, Ntouroupi TG, Mortensen NJ, Bodmer WF. Direct and immune mediated antibody targeting of ERBB receptors in a colorectal cancer cell-line panel. Proc Natl Acad Sci USA. 2012;109:21046–51. doi: 10.1073/pnas.1218750110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.López-Albaitero A, Lee SC, Morgan S, Grandis JR, Gooding WE, Ferrone S, et al. Role of polymorphic Fc gamma receptor IIIa and EGFR expression level in cetuximab mediated, NK cell dependent in vitro cytotoxicity of head and neck squamous cell carcinoma cells. Cancer Immunol Immunother. 2009;58:1853–64. doi: 10.1007/s00262-009-0697-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang X-R, Song A, Bergelson S, Arroll T, Parekh B, May K, et al. Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat Rev Drug Discov. 2011;10:101–11. doi: 10.1038/nrd3365. [DOI] [PubMed] [Google Scholar]
- 16.Kohrt HE, Houot R, Goldstein MJ, Weiskopf K, Alizadeh AA, Brody J, et al. CD137 stimulation enhances the antilymphoma activity of anti-CD20 antibodies. Blood. 2011;117:2423–32. doi: 10.1182/blood-2010-08-301945. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Diaz LA, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537–40. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bellucci R, Nguyen H-N, Martin A, Heinrichs S, Schinzel AC, Hahn WC, et al. Tyrosine kinase pathways modulate tumor susceptibility to natural killer cells. J Clin Invest. 2012;122:2369–83. doi: 10.1172/JCI58457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Binyamin L, Alpaugh RK, Hughes TL, Lutz CT, Campbell KS, Weiner LM. Blocking NK cell inhibitory self-recognition promotes antibody-dependent cellular cytotoxicity in a model of anti-lymphoma therapy. J Immunol. 2008;180:6392–401. doi: 10.4049/jimmunol.180.9.6392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.R Core Team . R: A Language and Environment for Statistical Computing [Internet] Vienna, Austria: 2013. Available from: http://www.R-project.org/ [Google Scholar]
- 22.Wickham H. Reshaping data with the reshape package. J Statistical Software. 2007:21. [Google Scholar]
- 23.Hothorn T, Bretz F, Westfall P. Simultaneous Inference in General Parametric Models. Biometrical J. 2008;50:346–63. doi: 10.1002/bimj.200810425. [DOI] [PubMed] [Google Scholar]
- 24.Derer S, Bauer P, Lohse S, Scheel AH, Berger S, Kellner C, et al. Impact of Epidermal Growth Factor Receptor (EGFR) Cell Surface Expression Levels on Effector Mechanisms of EGFR Antibodies. J Immunol. 2012;189:5230–9. doi: 10.4049/jimmunol.1202037. [DOI] [PubMed] [Google Scholar]
- 25.Zhang J, Chung T, Oldenburg K. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 26.Astsaturov I, Ratushny V, Sukhanova A, Einarson MB, Bagnyukova T, Zhou Y, et al. Synthetic lethal screen of an EGFR-centered network to improve targeted therapies. Sci Signal. 2010;3:ra67. doi: 10.1126/scisignal.2001083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanos B, Pendergast AM. Abl tyrosine kinase regulates endocytosis of the epidermal growth factor receptor. J Biol Chem. 2006;281:32714–23. doi: 10.1074/jbc.M603126200. [DOI] [PubMed] [Google Scholar]
- 28.Sawyers CL, McLaughlin J, Goga A, Havlik M, Witte O. The nuclear tyrosine kinase c-Abl negatively regulates cell growth. Cell. 1994;77:121–31. doi: 10.1016/0092-8674(94)90240-2. [DOI] [PubMed] [Google Scholar]
- 29.Colicelli J. ABL tyrosine kinases: evolution of function, regulation, and specificity. Sci Signal. 2010;3:re6. doi: 10.1126/scisignal.3139re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–6. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 31.Peterson ME, Long EO. Inhibitory receptor signaling via tyrosine phosphorylation of the adaptor Crk. Immunity. 2008;29:578–88. doi: 10.1016/j.immuni.2008.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Srinivasan D, Plattner R. Activation of Abl tyrosine kinases promotes invasion of aggressive breast cancer cells. Cancer Res. 2006;66:5648–55. doi: 10.1158/0008-5472.CAN-06-0734. [DOI] [PubMed] [Google Scholar]
- 33.Rix U, Hantschel O, Dürnberger G, Remsing Rix LL, Planyavsky M, Fernbach NV, et al. Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood. 2007;110:4055–63. doi: 10.1182/blood-2007-07-102061. [DOI] [PubMed] [Google Scholar]
- 34.O'Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16:401–12. doi: 10.1016/j.ccr.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hesslein DGT, Palacios EH, Sun JC, Beilke JN, Watson SR, Weiss A, et al. Differential requirements for CD45 in NK-cell function reveal distinct roles for Syk-family kinases. Blood. 2011;117:3087–95. doi: 10.1182/blood-2010-06-292219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.James AM, Hsu H-T, Dongre P, Uzel G, Mace EM, Banerjee PP, et al. Rapid activation receptor- or IL-2-induced lytic granule convergence in human natural killer cells requires Src, but not downstream signaling. Blood. 2013;121:2627–37. doi: 10.1182/blood-2012-06-437012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taylor RJ, Chan S-L, Wood A, Voskens CJ, Wolf JS, Lin W, et al. FcgammaRIIIa polymorphisms and cetuximab induced cytotoxicity in squamous cell carcinoma of the head and neck. Cancer Immunol Immunother. 2009;58:997–1006. doi: 10.1007/s00262-008-0613-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fertig EJ, Ren Q, Cheng H, Hatakeyama H, Dicker AP, Rodeck U, et al. Gene expression signatures modulated by epidermal growth factor receptor activation and their relationship to cetuximab resistance in head and neck squamous cell carcinoma. BMC Genomics. 2012;13:160. doi: 10.1186/1471-2164-13-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Daniel R, Cai Y, Wong PM, Chung SW. Deregulation of c-abl mediated cell growth after retroviral transfer and expression of antisense sequences. Oncogene. 1995;10:1607–14. [PubMed] [Google Scholar]
- 41.Rudnicka D, Oszmiana A, Finch DK, Strickland I, Schofield DJ, Lowe DC, et al. Rituximab causes a polarisation of B cells which augments its therapeutic function in NK cell-mediated antibody-dependent cellular cytotoxicity. Blood. 2013;121:4694–702. doi: 10.1182/blood-2013-02-482570. [DOI] [PubMed] [Google Scholar]
- 42.Orange JS. Formation and function of the lytic NK-cell immunological synapse. Nat Rev Immunol. 2008;8:713–25. doi: 10.1038/nri2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Borg C, Terme M, Taïeb J, Ménard C, Flament C, Robert C, et al. Novel mode of action of c-kit tyrosine kinase inhibitors leading to NK cell-dependent antitumor effects. J Clin Invest. 2004;114:379–88. doi: 10.1172/JCI21102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ménard C, Blay J-Y, Borg C, Michiels S, Ghiringhelli F, Robert C, et al. Natural killer cell IFN-gamma levels predict long-term survival with imatinib mesylate therapy in gastrointestinal stromal tumor-bearing patients. Cancer Res. 2009;69:3563–9. doi: 10.1158/0008-5472.CAN-08-3807. [DOI] [PubMed] [Google Scholar]
- 45.Chaput N, Flament C, Locher C, Desbois M, Rey A, Rusakiewicz S, et al. Phase I clinical trial combining imatinib mesylate and IL-2: HLA-DR(+) NK cell levels correlate with disease outcome. Oncoimmunology. 2013;2:e23080. doi: 10.4161/onci.23080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pautier P, Locher C, Robert C, Deroussent A, Flament C, Le Cesne A, et al. Phase I clinical trial combining imatinib mesylate and IL-2 in refractory cancer patients: IL-2 interferes with the pharmacokinetics of imatinib mesylate. Oncoimmunology. 2013;2:e23079. doi: 10.4161/onci.23079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blake SJ, Bruce Lyons A, Fraser CK, Hayball JD, Hughes TP. Dasatinib suppresses in vitro natural killer cell cytotoxicity. Blood. 2008;111:4415–6. doi: 10.1182/blood-2008-02-138701. [DOI] [PubMed] [Google Scholar]
- 48.Salih J, Hilpert J, Placke T, Grünebach F, Steinle A, Salih HR, et al. The BCR/ABL-inhibitors imatinib, nilotinib and dasatinib differentially affect NK cell reactivity. Int J Cancer. 2010;127:2119–28. doi: 10.1002/ijc.25233. [DOI] [PubMed] [Google Scholar]
- 49.Sirvent A, Benistant C, Roche S. Cytoplasmic signalling by the c-Abl tyrosine kinase in normal and cancer cells. Biol Cell. 2008;100:617–31. doi: 10.1042/BC20080020. [DOI] [PubMed] [Google Scholar]
- 50.Wege AK, Ernst W, Eckl J, Frankenberger B, Vollmann-Zwerenz A, Männel DN, et al. Humanized tumor mice--a new model to study and manipulate the immune response in advanced cancer therapy. Int J Cancer. 2011;129:2194–206. doi: 10.1002/ijc.26159. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.