

Abstract
The oncoprotein E7 from human papillomavirus (HPV) strains that confer high cancer risk mediates cell transformation by deregulating host cellular processes and activating viral gene expression through recruitment of cellular proteins such as the retinoblastoma protein (pRb) and the CREB-binding protein (CBP) and its paralog p300. Here we show that the intrinsically disordered N-terminal region of E7 from high risk HPV16 binds the TAZ2 domain of CBP with greater affinity than E7 from low risk HPV6b. HPV E7 and the tumor suppressor p53 compete for binding to TAZ2. The TAZ2 binding site in E7 overlaps the LxCxE motif that is crucial for interaction with pRb. While TAZ2 and pRb compete for binding to a monomeric E7 polypeptide, the full-length E7 dimer mediates an interaction between TAZ2 and pRb by promoting formation of a ternary complex. Cell-based assays show that expression of full-length HPV16 E7 promotes increased pRb acetylation and that this response depends both on the presence of CBP/p300 and the ability of E7 to form a dimer. These observations suggest a model for the oncogenic effect of high risk HPV16-E7. The disordered region of one E7 molecule in the homodimer interacts with the pocket domain of pRb, while the same region of the other E7 molecule binds the TAZ2 domain of CBP/p300. Through its ability to dimerize, E7 recruits CBP/p300 and pRb into a ternary complex, bringing the histone acetyltransferase domain of CBP/p300 into proximity to pRb and promoting acetylation, leading to disruption of cell cycle control.
Keywords: human papilloma virus, intrinsic protein disorder, CBP/p300, acetyltransferase, p53, NMR
Introduction
The family of double-stranded DNA human papillomaviruses (HPVs) contains more than 100 members, approximately 30 of which have been classified as either high risk or low risk for oncogenic transformation of host cells leading to human cancers [1]. High risk HPV types such as HPV16 are responsible for virtually all instances of cervical cancer, as well as a number of throat and oral cancers, while low risk forms such as HPV6b cause benign lesions that do not progress to tumors [2, 3]. High risk HPV and similar viruses, such as the adenovirus, induce tumor growth by activating appropriate host cell genes for viral replication while simultaneously suppressing apoptosis [4–7]. Viral “hijacking” of the host cell cycle leads to uncontrolled cell proliferation, ultimately resulting in tumor formation.
The oncogenic potential of different strains of HPV appears to reside in the E6 and E7 oncoproteins [5, 8], which induce host cell transformation by interacting with a wide variety of cellular proteins. While E6 and E7 function synergistically to transform the cell [5], expression of HPV16 E7 alone is sufficient to transform and immortalize epithelial cells, albeit with lower efficiency [9]. The transforming activities of E6 and E7 are dependent upon their ability to disrupt the regulatory functions of the tumor suppressors p53 and the retinoblastoma protein pRb [10, 11].
E7 is a relatively small protein consisting of three conserved regions denoted CR1, CR2 and CR3 (Figure 1a). The first two regions are intrinsically disordered [12–15], while CR3 is a zinc binding domain consisting of two β-strands and two α-helices that mediates E7 homodimer formation both in vitro and in vivo [15–20]. One of the best characterized E7 interactions is with the retinoblastoma tumor suppressor protein (pRb) [21–23]. During the normal cell cycle, pRb prevents entry into S phase by blocking activation of the E2F family of transcription factors. In HPV infected cells, E7 binds pRb, resulting in the release of E2F and premature entry into S-phase [24]. As part of this process, pRb is degraded, resulting in uncontrolled cellular proliferation [25, 26]. The efficiency of cellular transformation by the E7 oncoprotein is correlated with its pRb binding affinity [11]. Similar to other oncogenic viral proteins such as adenovirus E1A and simian virus 40 large T antigen, E7 binds the pRb pocket B domain through the LxCxE recognition motif in the CR2 region of E7 (highlighted in Figure 1a) [24]. Phosphorylation of E7 at the two conserved serine residues in CR2 (also highlighted in Figure 1a) occurs in vitro and in vivo [27–29], and has been shown to increase the affinity of E7 for pRb [30, 29]. Recent studies have revealed an additional low affinity pRb binding site in the CR3 domain that is important for E2F displacement from pRb [16, 19]. In addition to pRb and other retinoblastoma protein family members, E7 is capable of interacting with a number of other cellular targets and HPV employs this versatility to subjugate the host cell.
Figure 1.

E7 sequence alignment and analysis of TAZ1/TAZ2 domain interaction. (a) Sequence alignment for high risk HPV16 E7 and low risk HPV6b E7. The positions of the conserved regions CR1, CR2 and CR3 domains are indicated by colored bars. The LxCxE motif (the primary binding domain for pRb) and the conserved serine residues (sites for phosphorylation by CKII) are highlighted in red. (b) Domain structure of mouse CBP. (c) Interaction of full-length H6GB1-E7 immobilized on IgG agarose with the CBP TAZ1 and TAZ2 domains. (d) Interactions of H6GB1 constructs of E7(1-51), p53(13-61) and STAT1(710-750) immobilized on IgG agarose with TAZ1 and TAZ2. The pulldown assay with H6GB1 alone was used as a negative control to demonstrate that E7, STAT1 and p53 bind significantly above the background signal seen for H6GB1 alone.
The small DNA tumor viruses such as adenovirus and HPV transform cells by a common mechanism, encoding viral oncoproteins that inactivate the retinoblastoma family proteins, pRb, p107 and p130, and the tumor suppressor p53 [6]. The transforming ability of the adenovirus E1A oncoprotein depends not only upon binding to pRb, but also requires interactions with the cyclic-AMP response element binding (CREB) binding protein (CBP) and its paralog p300 to deregulate the host cell cycle and repress p53-mediated transcriptional processes [31–33]. CBP and p300 (Figure 1b) are multi-domain transcriptional co-activators that activate numerous transcriptional pathways and are key regulators of cell growth and differentiation [34, 35]. Due to their central role in regulating transcription, CBP and p300 are targeted by many viral proteins, including the E6 oncoprotein from high risk HPV [36]. HPV E7 also binds to p300 in vivo and in vitro and represses HPV E2 transcriptional activity [37]. Previous studies have suggested that E7 recruits CBP/p300 via an interaction with the TAZ1 (also known as CH1) domain [37, 38]. In the present work we undertook detailed biophysical analysis and cell-based assays to elucidate the molecular basis for interaction between HPV E7 and CBP/p300 as well as its functional outcome. We demonstrate that E7 binds preferentially and with higher affinity to the TAZ2 domain of CBP/p300, rather than to TAZ1, and show that this interaction is important for the acetylation of pRb in cells. NMR chemical shift mapping shows that E7 and the p53 transcriptional activation domain bind competitively to overlapping surfaces on TAZ2. We observe that the high-risk HPV16 E7 has a significantly higher affinity for TAZ2 than does the low-risk HPV6b E7, and show that pRb interacts with TAZ2 in the presence of the full-length E7 dimer to form a ternary complex. This raises the possibility that the oncogenic potential of the high risk HPV serotype is at least in part related to the capacity of the E7 oncoprotein to bind tightly to TAZ2 and thereby recruit pRb into a ternary complex, promoting acetylation of pRb, deregulation of the cell cycle, and pRb degradation. Understanding the structural basis for E7 control of cellular function through interaction with CBP/p300 potentially provides insight into the activity of other oncogenic viruses.
Results
E7 binds the TAZ2 domain of CBP/p300
Many oncogenic viral proteins, such as E1A from adenovirus and Tax from HTLV-1, bind to CBP/p300 via intrinsically disordered regions, and subsequently induce transformation of the cell [34, 39, 40]. Previous studies suggested that E7 binds preferentially to the TAZ1 domain of CBP/p300 [37, 38]. To confirm this result we ran pulldown assays using a protein GB1-fusion of full length high-risk HPV16 E7 (residues 1-98) incubated with the TAZ1 or TAZ2 domain of CBP in the presence of IgG resin. Surprisingly, E7 bound only weakly to TAZ1 but bound much more tightly to the TAZ2 domain (Figure 1c). In order to define the minimal TAZ2 interaction domain, a construct containing the disordered CR1 and CR2 domains, residues M1-H51, designated E7(1-51), was expressed and used to monitor TAZ2 binding. Pulldown assays using E7(1-51) fused to GB1 and incubated with either TAZ1 or TAZ2 (Figure 1d) show that E7(1-51), like the full-length E7 protein, binds preferentially to TAZ2 and only very weakly to the TAZ1 domain. Known TAZ domain binding partners STAT1 [41] and p53 [42] were used as positive controls in order to demonstrate the integrity of the TAZ domains. The band intensities show that the E7(1-51):TAZ2 interaction is of comparable affinity to that of the p53:TAZ2 and STAT1:TAZ2 complexes, suggesting that E7 could compete with these transcription factors for binding to CBP. The pulldown experiments also show that E7 is very similar to STAT1 in its ability to discriminate between the TAZ1 and TAZ2 domains (Figure 1d).
Affinity for TAZ2 depends on serotype and phosphorylation state
To compare the TAZ2 affinity for high risk and low risk E7 and determine the effect of phosphorylation on this interaction, we developed a fluorescence anisotropy assay using high risk HPV16 E7(1-51) phosphorylated by casein kinase II (CKII) at serines 31 and 32, designated ppE7(1-51). Alexa Fluor 594 was conjugated to the C-terminus of a double mutant of E7(1-51) where the native cysteine at position 24 was mutated to alanine and alanine 50 was mutated to cysteine. The fluorescent analog of this phosphorylated double mutant is designated ppE7(1-51) C24A,A50C-594. Direct titration experiments confirmed that ppE7(1-51) C24A,A50C-594 binds with high affinity (Kd = 28 ± 6 nM) to TAZ2 (Figure 2a). Binding affinities of unlabeled phosphorylated and non-phosphorylated E7 constructs from high risk HPV16 and low risk HPV6b were then determined from their ability to compete with ppE7(1-51)C24A,A50C-594 for binding to TAZ2 (Figure 2b, Supplementary Figure S1). The binding affinities calculated from these measurements are summarized in Table 1 and show that E7 from high risk HPV16 binds TAZ2 with a higher affinity compared to protein from low risk HPV6b. In addition, for both high risk and low risk HPV, phosphorylation of E7 increases the TAZ2 affinity.
Figure 2.
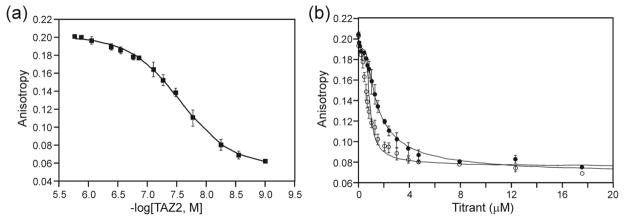
Fluorescence anisotropy analysis of the E7(1-51):TAZ2 interaction. (a) Fluorescence anisotropy results for a direct titration of fluorescently labeled ppE7(1-51)C24A,A50C-594 with TAZ2. (b) Results of fluorescence anisotropy for the competition assay of a 1:1 molar ratio sample of ppE7(1-51)C24A,A50C-594:TAZ2 with ppE7(1-51)(HPV16), open circles and ppE7(1-51)(HPV6b), closed circles.
Table 1.
Binding Analysis for TAZ2 by Fluorescence Anisotropy
| Protein | Risk | Kd (nM) |
|---|---|---|
| pp16-E7(1-51) | High | 7 ± 3 |
| 16-E7(1-51) | High | 22 ± 6 |
| pp6b-E7(1-51) | Low | 31 ± 3 |
| 6b-E7(1-51) | Low | 79 ± 10 |
NMR estimate of affinity of ppE7(1-51) for TAZ2 and TAZ1
It is clear from Table 1 and Figure 2b that the highest affinity interaction is between TAZ2 and phosphorylated E7(1-51) from high risk HPV16, but the measured affinities are close to the limits that can be reliably estimated from fluorescence competition experiments. This interaction was further analyzed and compared with TAZ1 binding by performing 1H-15N HSQC NMR titrations in which unlabeled E7 peptide from HPV16, designated ppE7(1-51), was added to uniformly 15N-labeled TAZ2 or TAZ1 (Figure a, b; full titration spectra are shown in Supplementary Figure S2). Changes in the chemical shifts of representative TAZ2 and TAZ1 residues are shown in Figure 3c and d respectively. The complete set of chemical shift titration curves for all resonances is shown in Supplementary Figure S3. The shape of the titration curves provides a graphic illustration of the differences in TAZ2 and TAZ1 binding affinity, and reveals the presence of two E7 binding sites on TAZ2. The primary binding site on TAZ2 is saturated at 1:1 molar ratio of ppE7(1-51); many of the cross peaks in the HSQC spectrum shift in fast exchange up to a 1:1 molar ratio and then their position does not change much beyond the 1:1 point (Figure 3c, blue curve). A subset of resonances is also perturbed by a weaker, secondary binding process, as exemplified by the binding curve for residue 1801 in Figure 3c. These two binding processes are indicated in Figure 3c as Kd1 and Kd2. Spectra of TAZ2 following addition of ppE7(1-51) at mole ratios of 1:1 and greater are shown in Supplementary Figure S4. By contrast, TAZ1 cross peaks shift in fast exchange over the entire titration and binding is completely saturated only at excess (~2-fold molar ratio) of ppE7(1-51) (Figure 3d). There is no evidence of a secondary binding site. The TAZ2 chemical shift titration curves were fitted to a 2-site binding model using the program nmrKd [43] yielding Kd values of 4 ± 3 nM and 33 ± 2 μM for the primary and secondary ppE7(1-51) binding processes, respectively. Although determination of nM Kd values by NMR titration can be problematic, we have shown elsewhere that it is possible to obtain accurate values when there is a secondary interaction that is coupled to binding in the high affinity site [43]. In the present work, the Kd values determined for the high affinity binding site by chemical shift titrations (4 ± 3 nM) and by fluorescence anisotropy (7 ± 3 nM) agree within experimental error. The TAZ1 titration data were fitted to a 1-site binding model, with Kd = 7.0 ± 0.3 μM at 25°C, in reasonable agreement with the affinity measured by sedimentation equilibrium (0.9 – 3.5 μM) for binding of HPV16 E7 to the p300 TAZ1 domain at 4°C [38]. Both fluorescence and NMR methods confirm that binding of HPV16 E7(1-51) to the TAZ2 domain of CBP is ~1000-fold tighter than binding to TAZ1.
Figure 3.

1H-15N HSQC analysis of the ppE7(1-51):TAZ1 and TAZ2 interaction. 1H-15N HSQC spectra of 15N TAZ1 and TAZ2 titrated with ppE7(1-51) from HPV16. Experiments were all performed at 303 K in NMR buffer (20 mM Tris, 50 mM NaCl, 1 mM DTT, pH 6.8). (a) Overlaid 750 MHz 1H-15N HSQC spectra of TAZ2 with increasing amounts of ppE7(1-51), ranging from 1:0 TAZ2:ppE7(1-51) (red) to 1:4 TAZ2:ppE7(1-51) (coral). (b) Overlaid 750 MHz 1H-15N HSQC spectra of TAZ1 with increasing amounts of ppE7(1-51), ranging from 1:0 TAZ1:ppE7(1-51) (red) to 1:3.5 TAZ1:ppE7(1-51) (coral). Chemical shift changes for representative cross peaks are denoted by arrows and labeled with the resonance assignment. (c) Changes in 15N chemical shift ΔδN of representative TAZ2 residues plotted as a function of the mole ratio of added E7(1-51). Solid lines represent fits to a 2-site binding model [43] with the primary and secondary interactions indicated by Kd1 and Kd2. (d) Changes in 15N chemical shift of a representative TAZ1 residue plotted as a function of the mole ratio of added E7(1-51). The solid line represents a fit to a single-site model.
The cross peaks in the 1H-15N HSQC spectra of the TAZ2 domain free and bound to E7(1-51) are well dispersed (Figure 3a and Supplementary Figure 2a), indicating that TAZ2 is well folded in both states. Binding of unlabeled ppE7(1-51) results in relatively large changes in the chemical shifts of specific TAZ2 residues. Figure 4a shows the chemical shift changes between free 15N-TAZ2 and a 1:1 mole ratio of added ppE7(1-51) (blue bars), representing the high-affinity interaction, and between the data at 1:1 and 1:3 mole ratios (red bars), representing the secondary interaction. Mapping of the chemical shift changes for the high-affinity interaction onto the structure of TAZ2 [44, 41] (Figure 4b) shows that the primary binding site is located in a contiguous hydrophobic pocket formed at the interface of the α1, α2 and α3 helices. The residues that exhibit chemical shift changes caused by the secondary binding process do not map to a contiguous site on the surface, but rather appear to reflect non-specific interactions at a number of sites peripheral to the primary binding site. A similar analysis of the binding site on TAZ1 (Supplementary Figure S5) reveals a much less localized binding site, with chemical shift perturbations for residues that are distributed over much of the TAZ1 surface.
Figure 4.

Changes in TAZ2 chemical shifts upon binding ppE7(1-51). (a) Histogram showing difference in the weighted average backbone HN and 15N chemical shifts <Δδ> = ½ [(ΔδHN)2 + (ΔδN/5)2]½ where ΔδHN and ΔδN correspond to the differences in amide 1H and 15N chemical shifts for a given TAZ2 residue in the free and bound states. Blue bars show shifts that occur between the free TAZ2 and a concentration ratio TAZ2:ppE7(1-51) = 1:1, corresponding to the high-affinity binding site. Red bars show shifts that occur between the 1:1 and 1:3 concentration ratios, corresponding to the low-affinity binding site (Data derived from the spectra in Supplementary Figure S4). The horizontal blue line shows a value of 1.5 x the standard deviation of the data for the high-affinity binding site. TAZ2 α-helices are indicated as gray bars. (b) Residues from TAZ2 plotted onto the TAZ2 structure (from pdb 2KA6) and colored red to indicate changes in chemical shift values upon ppE7(1-51) binding where <Δδ> is greater than 1.5 x the standard deviation for the high-affinity binding site.
The hydrophobic surface of TAZ2 that mediates the high affinity interaction with HPV16 E7 is also utilized for binding adenovirus E1A [45] and the N-terminal activation domain of the cellular transcription factor p53 [45]. NMR titrations show that p53 and E7 compete for binding to TAZ2; addition of the p53 activation domain to the 15N-HPV16 E7(1-51):TAZ2 complex displaces E7 and shifts its cross peaks back towards their positions in the spectrum of the unbound peptide (Supplementary Figure S6).
E7(1-51) remains disordered in the full-length protein
Full-length HPV16 E7 (residues 1-98) was isotopically labeled with 15N and purified for NMR analysis. The 1H-15N HSQC spectrum (Supplementary Figure S7) confirms that the 1-51 region of the full-length protein remains disordered, as in the E7(1-51) peptide, in agreement with earlier studies on full-length E7 from HPV16 and HPV45 [13, 15]. Cross peaks associated with the CR3 zinc finger dimerization domain are weak and broad (Figure S7). Concentration- and pH-dependent broadening of the CR3 resonances in full-length HPV16 E7 has recently been reported and has been attributed to flexibility and aggregation of the CR3 domain [13, 14]. Since the cross peaks for most residues in the CR1 and CR2 regions appear to coincide in the spectra of the peptide and the full length protein, and since the pulldown results were the same for E7(1-98) and E7(1-51) (Figure 1c, d), we deduced that the N-terminal region of the protein remained similarly disordered in the shorter construct and the full-length protein. Detailed NMR analysis of E7 and its interactions with TAZ2 was therefore performed using the smaller E7(1-51) peptide.
ppE7(1-51) interacts with TAZ2 through both the CR1 and CR2 Domains
Heteronuclear 3D NMR spectra of the complex of ppE7(1-51) with TAZ2 were acquired, observing either isotope-labeled TAZ2 bound to unlabeled ppE7(1-51) or labeled ppE7(1-51) bound to unlabeled TAZ2. Over 95% of the HN, N, CA, HA and CO resonances were assigned for TAZ2 in complex with ppE7(1-51). The backbone HN, N, CA, HA and CO resonances of ppE7(1-51) in complex with TAZ2 were assigned to greater than 92%. Addition of TAZ2 to 15N-labeled ppE7(1-51) causes shifts in many cross peaks in the 1H-15N HSQC spectrum, and an increase in dispersion in the 1H dimension (Figure 5a; the complete spectrum is shown in Supplementary Figure S8). Increased dispersion is a hallmark of a structural transition from a disordered state to one that is fully or partially ordered and folded on the surface of the binding partner [46]. The residues experiencing the most significant change in chemical shift bridge the CR1 and CR2 domains of E7 (Figure 5b), and include the pRb-binding LxCxE motif.
Figure 5.

NMR analysis of the interaction between 15N-ppE7(1-51) and TAZ2. (a) 600 MHz 1H-15N HSQC of 15N-ppE7(1-51) (black) and 1:1 molar ratio of 15N-ppE7(1-51) with unlabeled TAZ2 (red), with residues experiencing the greatest change in chemical shift indicated. (b) Histogram showing difference in the weighted average backbone HN and 15N chemical shifts <Δδ> = ½ [(ΔδHN)2 + (ΔδN/5)2]½ where ΔδHN and ΔδN correspond to the differences in amide 1H and 15N chemical shifts between the free ppE7(1-51) and bound ppE7(1-51) states. Conserved regions CR1, CR2 and CR3 are indicated by colored bars.
TAZ2 and pRb form a mixture of binary complexes with ppE7(1-51) in solution
Since the NMR spectra indicate that the TAZ2 interaction site on E7 includes the LxCxE motif that interacts with the B-subunit of pRb [23], we sought to determine whether TAZ2 and pRb compete for binding to ppE7(1-51). The Kd values for binding of E7 peptides to the pocket domain of pRb (henceforth designated pRb-AB) are in the low nanomolar range [30], similar to the 6 nM Kd for binding of E7 to TAZ2 determined in this work. NMR titrations were utilized to monitor binding of unlabeled TAZ2 and pRb-AB to 15N-labeled ppE7(1-51). Many resonances of ppE7(1-51) associated with residues in or near the LxCxE motif and in the CR1 region are shifted and broadened beyond detection upon addition of pRb-AB. It was therefore necessary to work at substoichiometric molar ratios in order to maintain sufficient signal intensity to monitor spectral changes of 15N-labeled ppE7(1-51) in the presence of unlabeled pRb-AB and TAZ2. A local region of the superimposed 1H-15N HSQC spectra of ppE7(1-51) with the addition of TAZ2 and pRb is shown in Figure 6; complete spectra are shown in Supplementary Figure S9. The Glu18 and pSer31 cross peaks in the free ppE7(1-51) (black) shift upon addition of TAZ2 to the positions shown in red. Addition of pRb-AB to 15N-labeled ppE7(1-51) to a mole ratio of 1:0.5 (green spectrum) causes splitting of the pSer31 cross peak, with a new pRb-bound peak appearing at 8.85 and 117.3 ppm. The bound cross peak for Glu18 is close to its position in the free ppE7(1-51) spectrum. Addition of pRb-AB (at 1:0.5 mole ratio) to the 1:1 TAZ2:ppE7(1-51) complex causes splitting of the pSer31 cross peak (blue), with components at chemical shifts corresponding to the binary complexes of E7 with TAZ2 (8.65, 116.8 ppm; red contours) and with pRB-AB (8.85, 117.3 ppm; green contours) (Figure 6). Similar splitting is observed for the cross peaks of Glu18 (Figure 6) and the side chain of Asn29 (Figure S9), with component peaks at the positions of the binary TAZ2 and pRb-AB complexes. We thus conclude that the pRb-AB domain competes with TAZ2 for binding to the LxCxE site in ppE7(1-51), such that a mixture of the two binary complexes is formed upon addition of substoichiometric amounts of pRb-AB to the TAZ2:ppE7(1-51) complex.
Figure 6.

NMR spectra of 15N-labeled ppE7(1-51) with unlabeled TAZ2 and pRb-AB. Region of the 600 MHz 1H-15N HSQC corresponding to Glu18, pSer31 and pSer32. Free 15N-ppE7(1-51) is shown in black, a 1:1 mole ratio of 15N-ppE7(1-51):TAZ2 in red, a 1:0.5 mole ratio of 15N-ppE7(1-51):pRb-AB in green, and a 1:1:0.5 mole ratio of 15N-ppE7(1-51):TAZ2:pRb-AB in blue.
Full Length E7 mediates a ternary interaction with the TAZ2 domain of CBP/p300 and the AB subunits of pRb
E7 shares many similarities with the adenovirus oncoprotein E1A, including an LxCxE motif that mediates binding to pRb and interactions with the TAZ2 domain of CBP/p300 [20, 45, 47]. It has been demonstrated that a ternary complex is formed between pRb, E1A and the TAZ2 domain of CBP/p300 that is necessary for viral activity [45, 39, 48]. In order to determine if full length E7 behaves similarly to E1A and mediates ternary complex formation between TAZ2 and pRb, interactions between the three proteins were monitored by in vitro pulldown assays. TAZ2 immobilized on amylose resin through an N-terminal His6MBP fusion tag could pull down pRbAB when bound to full length E7 but not when bound to an E7 construct (1-51) lacking the CR3 dimerization domain (Figure 7).
Figure 7.

(a) MBP pulldown assay demonstrating the formation of a ternary complex between TAZ2 (coupled to MBP), the AB subunit of Rb and E7, either phosphorylated (by casein kinase II, CKII) or unphosphorylated. This interaction requires the presence of full length E7: the ternary complex does not form in the presence of the E7(1-51) peptide, which lacks the dimerization domain (Lanes 1 and 4). The signal for E7(1-51) is very weak in this gel, most likely because it has a slight preferential affinity for pRb and is therefore lost in the washes in the presence of pRb. When full length E7 is present, the gel demonstrates the presence of all three proteins. (b) Corresponding portions of HPLC profiles of the eluates from the MBP pulldown assay showing pRb bound to TAZ2 in the presence of both phosphorylated and unphosphorylated E7, but not in the absence of E7 or the presence of phosphorylated or unphosphorylated E7(1-51).
Formation of the ternary complex was also monitored by NMR. HSQC spectra were acquired for 15N-labeled TAZ2 upon addition of full length unlabeled E7(1-98) (unphosphorylated) and unlabeled pRb-AB at different molar ratios (Figure 8). Firstly, it is clear from Figure 8a that pRb does not bind with any measurable affinity to TAZ2 alone: an NMR experiment where unlabeled pRb-AB was added at a mole ratio of 1:1 to 15N-labeled TAZ2 showed no chemical shift change and little change in the intensity of the cross peaks. Addition of unlabeled full length E7 to 15N-labeled TAZ2 (Supplementary Figure S10a) results in broadening of many cross peaks, frequently the same peaks that showed fast-exchange chemical shift changes upon addition of ppE7(1-51) (Figure 3a). A histogram showing peak intensities for TAZ2 as E7(1-98) is added is shown in Supplementary Figure S10b. Interestingly, the peak broadening upon addition of E7(1-98) indicates that the exchange regime has changed from fast exchange for binding of E7(1-51) peptide (Figure 3a) to intermediate-slow exchange for binding of full-length E7. While this resonance broadening makes structural analysis challenging, it remains useful in terms of evaluating the binding interface.
Figure 8.
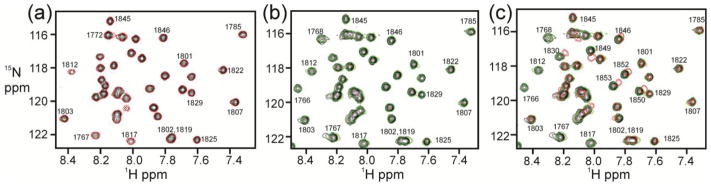
1H-15N HSQC analysis of 15N-TAZ2 with pRb in the presence and absence of E7(1-98). (a) Overlay of the 600 MHz 1H-15N HSQC spectra of 15N-TAZ2 (black) and of a 1:1 mole ratio of 15N-TAZ2:Rb-AB (red). (b) 600 MHz 1H-15N HSQC of 15N-TAZ2 (black) and 1:0.25 mole ratio of 15N-TAZ2:E7(1-98) (green). (c) 600 MHz 1H-15N HSQC of 15N-TAZ2 (black), 1:0.25 mole ratio of 15N-TAZ2:E7(1-98) (green) and 1:0.25:1 mole ratio of 15N-TAZ2:E7(1-98):Rb-AB (red).
Broadening of the TAZ2 peaks by the addition of full length E7 presents a problem for the detection of further effects on the spectrum as pRb is added: if the signals are too broad they cannot be monitored. We therefore used a mole ratio of 1:0.25 TAZ2:E7 for titration with pRb-AB, in order to maintain sufficient signal to monitor the effect of adding pRb-AB (Figure 8b). Addition of pRb-AB to 15N-TAZ2:E7(1:0.25) results in large changes in the TAZ2 HSQC spectrum (Figure 8c, Figure S11). The intensities and chemical shift changes of the cross peaks in the three spectra illustrated in Figure 8c are plotted as a function of residue number in Figure 9. While there is an overall loss in cross peak intensity upon addition of pRb-AB to TAZ2:E7(1:0.25) (red bars in Figure 9a), a subset of cross peaks is differentially broadened by pRb-AB (compare red and green bars) and some undergo significant changes in chemical shift (Figure 9b). In particular, cross peaks associated with residues near the N-terminus, in the α1 helix, and in the loop between α2 and α3 are broadened beyond detection (indicated by red bars below Figure 9a) in the presence of pRb-AB, even though these resonances remain observable even in the presence of E7(1-98) at concentrations greater than the 1:0.25 ratio used in this experiment (Supplementary Figure S10b). Residues that experience changes in chemical shift (Figure 9b) are localized in three regions of TAZ2, the C-terminal α4 helix, and parts of helices α1 and α3. Because these regions, particularly the α4 helix, are unaffected by the binding of E7 alone, they represent potential contact points between TAZ2 and pRb-AB, which occur only in the presence of the full-length E7 dimer.
Figure 9.

Location of broadened and shifted TAZ2 resonances. (a) Peak intensity ratios plotted as a function of TAZ2 residue number, comparing 1:0.25 15N-TAZ2:E7(1-98) with free 15N-TAZ2 (green) and 1:0.25:1 15N-TAZ2:E7(1-98):pRb-AB with 1:0.25 15N-TAZ2:E7(1-98) (red). Intensities were normalized to the C-terminal TAZ2 residue at position 92 before the ratio was calculated. Solid horizontal lines represent the mean value of the intensity ratios, and dotted lines represent the value of the mean minus the standard deviation of the intensity ratios. Red circles on the X-axis represent either proline residues or residues that are overlapped or have sufficiently low intensity in the free TAZ2 spectrum to be excluded from analysis. Red bars below the graph represent residues where the addition of pRb caused the resonance to be broadened beyond detection. (b) Difference in the weighted average backbone HN and 15N chemical shifts <Δδ> = ½ [(ΔδHN)2 + (ΔδN/5)2]½ where ΔδHN and ΔδN correspond to the differences in amide 1H and 15N chemical shifts in the spectrum of 1:0.25 15N-TAZ2:E7(1-98) (green cross peaks in Figure 9b) and the spectrum of 1:0.25:1 15N-TAZ2:E7(1-98):pRb-AB (red cross peaks in Figure 9b). The corresponding spectra in Supplementary Figure S11 are red and blue respectively.
The location of TAZ2 residues whose HSQC cross peaks are perturbed by the addition of E7 and pRb-AB are mapped onto the structure of the CBP TAZ2 domain in Figure 10. The TAZ2 cross peaks that undergo the most substantial broadening upon titration with full-length E7 are located in the same binding interface as observed for the ppE7(1-51) peptide (Figure 4), namely the region between the α2 and α3 helices (red area in Figure 10). The residues whose cross peaks broaden in the presence of both E7 and pRb (Figure 9a, blue areas in Figure 10) are located primarily near the N-terminus of TAZ2 and in the loops between α1-α2 and α2-α3, while the residues that undergo chemical shift changes upon addition of pRb (Figure 9b, cyan areas in Figure 10) are located in localized regions of α1 and α4. Taken together, these results suggest that the full length E7 dimer is able to mediate formation of a ternary complex with TAZ2 and pRb-AB, in which pRb interacts with TAZ2 through an interface that appears to involve primarily the loop between α2-α3 and the C-terminal helix α4 of TAZ2. We conclude that the full-length E7 dimer is absolutely required for formation of the ternary complex, firstly because there is no detectable interaction between the isolated TAZ2 and pRb domains, and secondly because the monomeric E7(1-51) peptide, which contains overlapping binding sites for TAZ2 and pRb, binds competitively to the two domains (Figure 6, S9) and is unable to support ternary complex formation.
Figure 10.
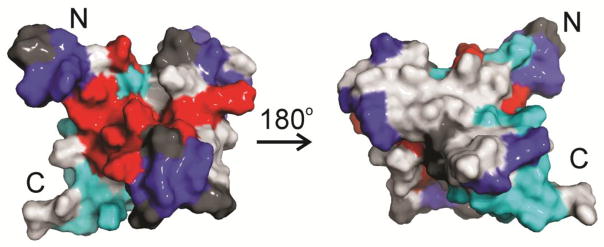
Mapping of TAZ2 residues affected by E7 and pRb-AB. Structure of TAZ2 (2KA6) showing residues affected by addition of ppE7(1-51) (red) (data from Figure 4) and by pRb-AB in the presence of E7(1-98), either broadening (blue) or peak shifts (cyan). Residues where no NMR data are available (red circles in Figure 9c) are colored dark gray.
E7 increases pRb acetylation in cells by a CBP/p300 mediated mechanism
Our in vitro measurements establish that the HPV E7 dimer mediates an interaction between the TAZ2 domain of CBP/p300 and the pocket domain of the retinoblastoma protein. In order to test whether this interaction was functional in cells, we performed pRb acetylation assays in the presence and absence of E7. pRb was immunoprecipitated (IP) from the lysates of human fibroblast cells (BJ cells) with and without full-length E7 transduction. These IP samples were then analyzed by Western blot using an anti-Rb antibody (Santa Cruz, sc-50) to measure pRb content in both the lysates and the IPs, as well as an anti-acetyl-lysine (Anti-AcK) antibody (Millipore, 06-933 or Cell Signaling, Ac-K-103) to determine the level of pRb acetylation. Cells transduced with E7 have overall decreased levels of pRb, as seen in both the lysates and the immunoprecipitated samples, due to E7-mediated pRb degradation (Figure 11a). Significantly, pRb shows increased levels of acetylation in cells transduced with E7 (Figure 11a). Comparing the measured band intensities of the Western blots for the IP assays as a ratio of acetylated pRb to the total immunoprecipitated pRb demonstrates a 4-fold increase in pRb acetylation for cells transduced with E7, compared to the control cells (Figure 11a). We observed two forms of pRb in these assays and confirmed by running the assay in the presence of alkaline phosphatase (NEB, M0371S) that the difference between the two forms is due to the phosphorylation state of pRb. Interestingly, the hypophosphorylated form of pRb (lower band) is more highly acetylated than the hyperphosphorylated form. To confirm that increased pRb acetylation results directly from E7 interaction, BJ cells were also transduced with E7Δ(21-24), which has a truncation in the LxCxE motif and does not bind pRb [49]. Transduction with E7 significantly decreases the amount of pRb present and correspondingly increases acetylation levels, while cells transduced with E7Δ(21-24) show both pRb and acetylation levels similar to the control BP cells (Figure 11a). Cell lysates were probed with an anti-E7 antibody in order to confirm that E7 and E7Δ(21-24) were expressed at similar levels (Supplementary Figure S12). Taken together, these results demonstrate that the increased levels of pRb acetylation observed in cells transduced with E7 depends on the ability of E7 to interact with pRb.
Figure 11.

Western blot analysis from immunoprecipitation assays. (a) BJ cells were transduced with control vector (BP), E7 and the pRb-binding deficient mutant E7Δ(21-24). (b) MEF cells bearing homozygous conditional knockout alleles of both CBP and p300 were transduced with E7 and then treated with Cre recombinase-expressing retrovirus to knock out CBP/p300. (c) BJ cells transduced with either the control vector (BP), E7, or the monomeric mutants E7(1-51) and E7L67R. The relative anti-AcK band intensities are the mean of two measurements from independent experiments and the error bars indicate the range of values. The intensity ratios were determined from the relative intensities of corresponding bands in the anti-AcK Western blot and in the anti-pRb Western blot, then normalized relative to the negative control (BP).
Previous studies have shown that pRb is acetylated primarily by p300 and PCAF [50, 51]. In order to determine the role of CBP/p300 in E7-induced pRb acetylation, primary mouse embryonic fibroblasts (MEF cells) bearing homozygous conditional (floxed) knockout alleles of both CBP and p300 were treated with Cre recombinase-expressing retrovirus, which effectively eliminates CBP and p300 [52, 53]. The pRb IP assays were repeated and analyzed for both pRb level and pRb acetylation (Figure 11b). Prior to Cre treatment, the cells display a similar trend to the BJ cells: E7 transduction results in significantly decreased levels of pRb and correspondingly increased levels of pRb acetylation. However, E7-induced pRb degradation and acetylation are both diminished in the Cre-treated cells lacking CBP/p300 (Figure 11b). This demonstrates that the E7-induced pRb acetylation, and its subsequent degradation, is at least partly mediated by CBP/p300.
To determine whether E7 dimerization plays a role in degradation and acetylation of pRb, BJ cells were transduced with either full-length E7, the peptide E7(1-51) (a monomeric mutant which lacks the CR3 dimerization domain but maintains the ability to bind pRb-AB), or E7L67R, which has significantly decreased propensity to dimerize but maintains a certain level of activity in cells [19]. The results presented in Figure 11c show that while pRb acetylation is enhanced in the presence of full-length E7, transduction with monomeric mutants E7(1-51) or E7L67R results in both pRb levels and pRb acetylation levels comparable to control cells. This suggests that an intact CR3 dimerization domain is necessary for E7 to promote pRb acetylation by CBP/p300 and to decrease the overall pRb levels in cells.
Discussion
Interaction of HPV16-E7 with CBP
The general transcriptional coactivators CBP and p300 play a central role in viral gene expression and host cell transformation by DNA and RNA tumor viruses [34, 54]. Adenovirus E1A, simian virus 40 (SV40) large T antigen, and the HPV E6 protein all bind to the TAZ2 domain of CBP/p300 to disrupt cellular gene regulatory programs [55, 56]. In the present work, we provide evidence of a hitherto-unreported high-affinity interaction between E7 from high risk HPV16 and the TAZ2 domain of CBP/p300. Previously reported GST-pulldown assays suggested that HPV16-E7 interacts most strongly with the N-terminal region of p300, encompassing the TAZ1 (or CH1) domain [37]. Weaker interactions were observed with a fragment that contained the TAZ2 (or CH3) domain. The TAZ1 and TAZ2 domains each bind three zinc atoms and are unfolded in the absence of metal [44, 57]. We note that the GST pulldown experiments reported in reference [37] were performed in a buffer containing 2.5 mM EDTA. At this concentration of EDTA, it is likely that the all of the zinc would be removed by the chelator and the TAZ domains of p300 would be unfolded, as shown previously for the TAZ1 domain of CBP [58]. Zinc-free TAZ1 is particularly prone to bind indiscriminately to other proteins [58], which suggests that the strong TAZ1-E7 interaction observed by Bernat et al. [37] most likely involved unfolded TAZ1. A subsequent study of the interaction of HPV E7 with p300, with no EDTA in the buffer, showed that binding to TAZ1 is relatively weak (0.9 – 3.5 μM) but did not reassess the interaction with TAZ2 [38]. Our present experiments confirm that full-length E7 also binds only weakly to the CBP TAZ1 domain, and show that an E7(1-51) peptide binds to the CBP TAZ2 domain ~1000-fold more tightly than to TAZ1. The TAZ domains of CBP and p300 have 95% (TAZ1) and 98% (TAZ2) sequence identity and CBP and p300 are expected to bind with comparable affinity to TAZ ligands.
NMR titrations mapped the site of TAZ2 interaction to the intrinsically disordered CR1 and CR2 regions of E7 (Figure 5b), which undergo full or partial folding upon binding to TAZ2. CKII phosphorylation of Ser31 and Ser32, which are adjacent to the LxCxE motif in CR2, enhances binding of HPV16-E7(1-51) to TAZ2 by 3-fold (Table 1). Phosphorylation also increases the affinity of low risk HPV6b-E7 for TAZ2, but overall the TAZ2 binding affinity of HPV6b-E7(1-51) remains significantly lower than for HPV16-E7(1-51). Phosphorylation of the CKII site in HPV E7 appears to be critical for efficient entry into S-phase and for cellular transformation [59, 60]. Changes in chemical shift upon titration of 15N-labeled TAZ2 with ppHPV16-E7(1-51) identified two binding sites, a high affinity site with Kd = 4 ± 3 nM and a secondary, low affinity site with Kd = 33 ± 2 μM. The residues that are affected by the secondary binding process are distributed in a number of sites on the surface of TAZ2, suggesting that this interaction is probably non-specific. In contrast, the high affinity ppE7(1-51) site maps to a distinct surface located in the groove between the α1, α2 and α3 helices (Figure 4b). This surface is also the binding site for adenovirus E1A and for activation domains of cellular transcription factors such as p53, STAT1 and C/EBP [45, 61, 41, 62].
Targeting of the TAZ2 domain of CBP/p300 by the oncoproteins E1A and large T antigen is required for transformation of the host cell by the adenovirus and SV40 DNA tumor viruses [63, 64]. CBP and p300 are essential coactivators of p53 mediated transcriptional processes and are required for functioning of the p53-mediated checkpoint in the G1 phase of the cell cycle [65, 66, 31, 32]. Activation requires direct interaction between the TAZ2 domain and the p53 N-terminal transactivation domain and is disrupted by E1A, which binds to TAZ2 with higher affinity (Kd ca. 5 nM [67]) than does the p53 transactivation domain (Kd 20 nM [42]). Through its ability to out-compete p53 for binding to CBP/p300, E1A inhibits both p53-mediated transcription and cell cycle arrest [31, 32]. Given the high TAZ2 binding affinity of the HPV16 E7 oncoprotein (Table 1), which is further enhanced by phosphorylation of Ser31 and Ser32 in the CKII site, it is likely that E7 fulfills a similar role in HPV infection. Indeed, HPV16 E7 expression has been shown to inhibit p53 transcriptional activity in cells [68, 69], and our data strongly suggest that competition for binding to the TAZ2 domain of CBP/p300 is a likely mechanism. Deletion of CR1 or of residues 6-10, which form part of the TAZ2 binding site (Figure 5b), abrogates cellular transformation [70]. It is notable also that E7 from the low risk HPV6b strain binds more weakly to TAZ2 (Table 1) and would therefore compete less efficiently with p53 and other cellular transcription factors.
Interactions of HPV16-E7 with TAZ2 and the Retinoblastoma Protein
Interactions between the retinoblastoma protein (pRb) and high risk forms of HPV E7 have been well documented and are necessary but not sufficient for transformation [21, 22]. Binding to pRb requires the LxCxE motif in the CR2 region of E7, and protein from high risk HPV strains bind pRb with higher affinities than that from low risk strains [22, 30]. Binding of E7 to pRb and the retinoblastoma family members p107 and p130 promotes dissociation of E2F and activates transcription of E2F-responsive genes that mediate entry of the cell into S phase, leading to uncontrolled DNA synthesis and cell proliferation [24, 71].
Adenovirus E1A functions similarly to HPV E7, binding to pRb through its CR1 and CR2 conserved regions and displacing E2F [72]. Efficient induction of cellular proliferation by E1A depends upon its ability to recruit CBP/p300 and pRb into a ternary complex, which stimulates acetylation of Lys873 and Lys874 in the C-terminal region of pRb [39, 50]. Acetylation at these sites, which can also be accomplished by P/CAF, enhances binding of pRb to Mdm2 [51]. Given the parallels between E1A and HPV16 E7 in their ability to bind both CBP/p300 and pRb, we undertook an investigation of the interactions between E7, the CBP TAZ2 domain, and the pRb pocket domain.
NMR titration experiments showed that the TAZ2 binding site and high affinity pRb site (LxCxE motif) overlap in HPV E7. As a consequence, TAZ2 and a construct containing the pRb pocket domains (pRb-AB) compete for binding to the monomeric ppE1A(1-51) peptide, producing HSQC spectra containing cross peaks that correspond to a mixture of the binary TAZ2:ppE7(1-51) and pRb-AB:ppE7(1-51) complexes (Figure 6). In contrast, the TAZ2 binding site at the C-terminal end of the CR1 region in E1A is separated from the LxCxE motif in CR2 by an approximately 40 residue spacer, allowing TAZ2 and pRb to bind simultaneously [45]. Truncation of the spacer abrogates E1A-mediated ternary complex formation and impairs E1A-induced cellular proliferation [39]. We thus conclude that appropriate spacing between the TAZ2 binding site and the pRb-binding LxCxE motif is required for simultaneous binding of these cellular proteins to a single viral polypeptide chain.
However, full-length HPV E7 is a homodimer at physiological concentrations, with two intrinsically disordered CR1-CR2 regions attached to the obligate dimeric zinc binding domain in CR3 [17, 18, 20]. We therefore asked whether full-length HPV16 E7 could mediate ternary complex formation, with TAZ2 bound to one subunit of the dimer and the pRb-AB pocket domain bound to the other. Using NMR, which is extremely sensitive to even very weak binding interactions, we showed that there is no observable interaction between pRb-AB and TAZ2 in the absence of E7 (Figure 8a). However, in the presence of the full-length HPV16 E7 dimer, formation of a ternary complex was observed both in pulldown experiments and by broadening and shifting of cross peaks in the NMR spectrum of 15N-labeled TAZ2 (Figures 7 and 8). Mapping the perturbed resonances onto the TAZ2 structure implicates a specific binding surface on TAZ2 for interactions with pRb within the ternary complex (Figure 10). The extent of ternary complex formation will clearly depend on the relative affinities for binding of TAZ2 and pRb to the E7 CR1-CR2 region; since the CR1-CR2 region of doubly phosphorylated HPV16 E7 binds to both TAZ2 and pRb with comparable affinity (Kd 7 nM (Table 1) and 2 nM [30], respectively), a substantial population of ternary complex is expected to be formed.
Interaction of E7, CBP and pRb in Cells
Our in vitro experiments show that HPV16 E7, like adenovirus E1A, is able recruit pRb and CBP/p300 into a ternary complex. By binding simultaneously to the TAZ2 domain of CBP/p300 and to the pocket domain of pRb, E1A brings pRb into close proximity with the CBP/p300 HAT domain and thereby promotes acetylation of pRb [45, 39, 50]. Given that HPV16 E7 also binds with high affinity to the TAZ2 domain, we asked whether E7 could also promote acetylation of pRb by CBP/p300 in cells. Enhanced acetylation of pRb was indeed observed when full-length HPV16 E7 was expressed in BJ or MEF fibroblast cells and acetylation levels were substantially decreased in CBP/p300−/− cells (Figure 11). No enhancement in pRb degradation or acetylation was observed in cells expressing E7 constructs that were dimerization defective. We thus conclude that acetylation and degradation of pRb in HPV16-infected cells is at least partly dependent upon CBP/p300, although it is likely that P/CAF also plays a complementary role. Our results confirm and extend a previous in vitro study showing that HPV16 E7 enhances p300-mediated acetylation of the retinoblastoma protein family member p130 [73].
In contrast to E1A, where the long spacer between the LXCXE motif in CR2 and the TAZ2 binding motif in CR1 permits the E1A monomer to bind simultaneously to pRb and TAZ2 [45], dimerization of HPV E7 through the CR3 domain is obligatory for recruitment of TAZ2 and pRb into a ternary complex. Because the binding sites for TAZ2 and pRb overlap within the CR1-CR2 region of E7, the CBP/p300 and pRb must compete to bind a monomeric E7 sequence. However, by dimerization E7 can present two binding motifs, one on each subunit, that are capable of interacting simultaneously with TAZ2 and pRb. A schematic model of such a complex, through which E7 could recruit the CBP/p300 HAT domain to acetylate the C-terminal lysine residues of pRb, is shown in Figure 12. Thus, by dimerization, E7 accomplishes what E1A achieves through widely separated binding sites in a single polypeptide chain, the ability to stimulate acetylation of pRb and thereby interfere with control of the host cell cycle.
Figure 12.

Model for pRB:CBP:E7 ternary complex formation. E7 dimerization through the CR3 domain facilitates recruitment of CBP/p300 to pRb. The LxCxE motif of one of the E7 monomer units interacts with the pRb B domain, while the other mediates a ternary interaction with the TAZ2 domain of CBP/p300 and pRb-B, bringing the HAT domain of CBP/p300 close to the pRb C-terminal region and enabling acetylation at pRb lysines 873 and 874.
High Risk vs Low Risk HPV Strains
The oncoprotein E7 from human papillomavirus induces host cell transformation by interacting with cellular proteins, including the retinoblastoma protein pRb and CBP/p300. Different strains of HPV have different risks for the formation of cancers, and at least part of this difference appears to reside in the sequence of the E7 protein [8], yet the amino acid sequences of E7 proteins from high-risk strains such as HPV16-E7 are very similar to those from low risk strains such as HPV-E6b (Figure 1a). As part of the present study, we compared the TAZ2 binding affinity of the E7 proteins from high risk HPV16 and from low risk HPV6b. Phosphorylated HPV16-E7(1-51) binds TAZ2 with higher affinity than does HPV6b-E7(1-51) (Table 1). The differences in binding affinity are amplified when differences in phosphorylation are taken into consideration. The high risk HPV16 and HPV18 E7 proteins are heavily phosphorylated by casein kinase II both in vitro and in vivo and phosphorylation is essential for S-phase entry and for cellular transformation [74, 75, 59]. CKII phosphorylation of E7 from low risk HPV6b in vitro proceeds more slowly than for high risk E7 [74, 11], and only a minor fraction of HPV6b E7 is phosphorylated in vivo [29]. Phosphorylation of the CKII motif in HPV16 E7 increases its affinity for binding to TAZ2, thereby enhancing its ability to compete with p53 and other cellular transcription factors and favoring formation of an E7:CBP/p300:pRb ternary complex. In contrast, inefficient phosphorylation of E7 from low risk HPV6b would impair its ability to inhibit p53-mediated transcriptional programs and substantially decrease the likelihood of ternary complex formation with CBP/p300 and pRb. E7 from low risk strains, such as HPV6, binds pRb with lower affinity than does HPV16-E7 and cannot target pRb for degradation [76]. These differences in binding affinity and phosphorylation state could be contributing factors to the differing oncogenic potential of the E7 oncoprotein from high and low risk HPV strains.
What role might E7-mediated pRb acetylation play in deregulation of the host cell cycle? Acetylated pRb binds more strongly to MDM2 [50]. By stimulating CBP/p300-mediated acetylation of K873 and K874, high-risk HPV16 E7 (and also adenovirus E1A) may promote interactions between pRb and MDM2 and enhance degradation of pRb by either ubiquitin-dependent or ubiquitin-independent mechanisms [77, 78], thus facilitating cell cycle progression. In addition, acetylation of pRb has been shown to inhibit phosphorylation of pRb by the cyclin-dependent kinases, [50], which may play an important role in stabilization of the E7:pRb complex. During the normal cell cycle, pRb is inactivated towards the end of G1 phase by multisite phosphorylation, which releases E2F and thereby allows progression to S phase [79]. Phosphorylation also inhibits binding of E7 and other proteins to the LxCxE site on pocket domain B [80, 81]. By recruiting the acetyltransferase activity of CBP/p300, E7 is able to promote acetylation of pRb, block normal pRb phosphorylation, and thus prevent phosphorylation-induced disruption of the E7:pRb complex. This serves as a positive feedback loop to stabilize the E7:pRb complex, leading to enhanced CBP/p300 recruitment and increased pRb acetylation. Of note, although E7-mediated stimulation of pRb acetylation may reduce phosphorylation of pRb by cyclin-dependent protein kinases, it will not inhibit cell cycle progression, as the E7-bound pRb can no longer bind to and sequester the E2F/DP transcription factors that are required for G1/S transition.
Materials and Methods
Protein expression and purification
The TAZ1 (residues 345–439) and TAZ2 (residues 1764–1855) domains of wild type mouse CBP were expressed and purified as previously described [44, 57].
The His6-GB1 fusions of E7(1-51) peptides from HPV16 and HPV6b were cloned into a co-expression vector with the CBP TAZ2 domain [82]. E7 constructs were expressed in E. coli BL21(DE3)(+DNAY) cells in M9 minimal medium. Uniformly labeled samples were prepared by supplementation with (15NH4)2SO4 and/or 13C-glucose. Cultures were grown at 37°C to OD600 of ~0.8, supplemented with 150 μM ZnSO4, induced with 0.5 mM IPTG and further incubated at 18°C for 16 hours. Pellets containing His6-GB1-E7(1- 51) fusion protein were suspended in 30 mL of 25 mM Tris-HCl (pH 8.0), 200 mM NaCl, 10 mM imidazole, 8 M urea per liter of culture and lysed by sonication. The soluble fraction was isolated by centrifugation at 20,000 × g for 30 min. Soluble His6-GB1-E7(1-51) was purified by chromatography on Ni-NTA resin equilibrated with lysis buffer and the His6-GB1 tag was removed by thrombin digestion in the column at 30°C. The cleaved E7(1-51) constructs were further purified by reversed phase HPLC, using a C4 cartridge (Waters) in standard acetonitrile/0.1% TFA mobile phase.
Full length E7 from HPV16 fused to H6GB1 for pulldown assays was expressed in E. coli BL21(DE3)(+DNAY) cells. Cells were grown at 37°C in M9 medium to OD600 of 0.4. The medium was supplemented with 150 μM ZnSO4 followed by induction with 0.5 mM IPTG, and further incubated at 16°C for 16 hours. Pellets were suspended in 30 mL of 25 mM Tris-HCl (pH 7.5), 200 mM NaCl, 10 mM imidazole and lysed by sonication. The soluble fraction was isolated by centrifugation at 15,000 × g for 30 min and purified by chromatography on Ni-NTA resin equilibrated with lysis buffer, eluted with 250 mM imidazole. The eluted protein was dialyzed into 25 mM Tris HCl (pH 7.5), 25 mM NaCl, 2 mM DTT and purified on a Q-trap HF column (GE Healthcare) with a linear gradient of 25 mM–1 M NaCl followed by size exclusion chromatography on a Superdex 75 (GE Healthcare) column equilibrated with 25 mM Tris HCl (pH 7.5), 150 mM NaCl, 1 mM DTT. Purity was assessed by SDS-PAGE gel. In order to express E7 in quantities sufficient for NMR spectroscopy, full length E7 from HPV16 was expressed in E. coli BL21(DE3)(+DNAY) cells as a fusion to maltose binding protein (MBP) using a pMAL vector with an engineered TEV cleavage site between MBP and E7. Cells were grown at 37°C in M9 media, with uniformly labeled samples supplemented with (15NH4)2SO4, to OD600 of 0.4. The medium was supplemented with 150 μM ZnSO4 followed by induction with 0.5 mM IPTG, and growth at 16°C for 16 hours. Cell pellets containing unlabeled or 15N-labeled protein were suspended in 25 mM Tris-HCl (pH 7.2), 200 mM NaCl, 10 mM DTT, 2 μg lysozyme. Soluble MBP-E7 was affinity-purified on an amylose column and MBP-bound E7 was eluted with 25 mM Tris (pH 7.2), 200 mM NaCl, 2 mM DTT, 10 mM maltose. MBP was cleaved with TEV protease and the proteins were dialyzed into 25 mM Tris (pH 7.2), 30 mM NaCl, 2 mM DTT and separated on a Q-trap HF column (GE Healthcare) with a linear gradient of 30 mM–1 M NaCl. The eluted E7 was further purified by size exclusion chromatography on a Superdex 75 (GE Healthcare) column equilibrated with 25 mM Tris (pH 7.2), 150 mM NaCl, 1 mM DTT. Purity was assessed by SDS-PAGE gel.
The Rb central pocket (A domain, residues 372 – 577, and B domain, residues 645 – 774, connected by an engineered GSGS linker) was expressed and purified as previously described [45].
Protein concentrations were determined by absorbance at 280 nm.
In vitro GB1 pulldown assay
The in vitro GB1 pulldown assays were performed as previously described [83]. Purified H6GB1, H6GB1- E7(1-98), H6GB1-E7(1-51), H6GB1-STAT1(710-750) [41] or GB1-p53(13-61) [83] fusion proteins were added to an 8% (x/v) slurry of IgG-Sepharose 6 Fast Flow (GE Healthcare Biosciences) in assay buffer (20 mM Tris-HCl, pH 8, 150 mM NaCl, 0.1% (v/v) NP-40) and washed three times with assay buffer. Equimolar TAZ1 or TAZ2 was added to the resin-bound GB1 or GB1 fusion and incubated for 15 min at room temperature. Unbound proteins were removed by washing three times with assay buffer and bound proteins were analyzed using SDS-PAGE.
Phosphorylation of E7(1-51)
Phosphorylation of E7(1-51) was carried out as described previously [42]. Purified peptide was dissolved in 250 μL 50 mM Tris-HCl, pH 7.0 at a final concentration of approximately 2.5 mM in the presence of a 3-fold molar excess of ATP and 2 units Casein Kinase II (CKII) (New England Biosciences) per 100 μL solution. The reaction was monitored by retention time on analytical C18 HPLC. The phosphorylated peptide was then purified using a C4 semi-prep HPLC column and phosphorylation was confirmed by retention time on an analytical C18 HPLC column, MALDI-TOF mass spectrometry and HSQC analysis of the serine residues for 15N-labeled protein samples.
Fluorescence anisotropy
E7(1-51) was mutated to replace the naturally occurring Cys24 residue to alanine, followed by mutation of the alanine residue at position 50 to cysteine, E7(1-51)C24A,A50C. Alexa Fluor 594 (Molecular Probes) fluorescent dye was then attached to Cys50, which is sufficiently distant from the TAZ2 binding domain to have minimal effect on interaction. E7(1-51)C24A,A50C was then phosphorylated and labeled in 50 mM Tris, 6 M guanidine HCl, pH 7.2, using ~3-fold excess Alexa Fluor 594 dye. Labeling reactions were run for 2 h at room temperature followed by purification using an analytical C18 reverse phase HPLC column. ppE7(1-51)C24A,A50C-594 was checked for correct mass and incorporation of the Alexa dyes by both MALDI-TOF mass spectrometry and UV signal at 594 nm. Titrations in 20 mM Tris-HCl, 50 mM NaCl, 1 mM dithiothreitol (DTT), pH 7.0 at 25°C were performed by monitoring ensemble fluorescence anisotropy at constant temperature. Two titration methods were used: direct protein-ligand titration and competition binding measurements, carried out by the methods described previously [67]. For the direct titrations, the concentration of ppE7(1-51)C24A,A50C-594 was held constant at 20 nM and TAZ2 was titrated up to a final concentration of 1.7 μM. The initial sample for the indirect competition measurements consisted of a mixture of ppE7(1-51)C24A,A50C-594 at 20 nM and TAZ2 at 0.75 μM, which had been determined by the direct titration to be just beyond the saturation point. Competition with non-fluorescently labeled E7 peptides was carried out with a final titrant concentration of 20 μM.
In vitro MBP pulldown assay
H6MBP-TAZ2, H6GB1-E7(1-51), H6GB1-E7(1-98) and H6Rb-AB were purified by nickel affinity chromatography on cOmplete His-tag resin (Roche) in buffer containing 5 mM DTT and dialyzed into assay buffer (20 mM Tris pH 8, 100 mM NaCl, 1 mM DTT). E7 constructs were phosphorylated with CKII as described above and used for pulldown assays without further purification. For each pulldown assay, 60 μl amylose resin (NEB) was loaded with 4 nmol H6MBP-TAZ2 and washed with assay buffer. H6Rb-AB (3 nmol), H6GB1-E7(1-51) (4 nmol), and/or H6GB1-E7(1-98) (4 nmol) were allowed to bind for 30 minutes at room temperature in assay buffer supplemented with 5mM DTT and unbound proteins were removed by washing with assay buffer. Bound proteins were eluted with 0.1% TFA and observed by SDS PAGE or monitored by reversed phase HPLC on an analytical C4 column as previously described [41].
NMR spectroscopy
NMR experiments were conducted using either a DRX 600 MHz NMR spectrometer with cryoprobe or an Avance 750 MHz NMR spectrometer. NMR spectra were processed and analyzed using NMRPipe [84], NMRView [85], and SPARKY [86]. TAZ1 and TAZ2, the pRb pocket domain, E7 and the phosphorylated E7 peptides were dialyzed into NMR buffer (20 mM Tris, 50 mM NaCl, 1 mM DTT, pH 6.8). For Kd measurements, samples of TAZ1 or TAZ2 at 100 μM concentration were titrated with ppE7(1-51). The concentrations were confirmed for each sample using analytical HPLC and the pH was checked prior to NMR acquisition. Plots of chemical shift changes versus mole ratio of ppE7(1-51) were fitted to 1-site and 2-site binding models for TAZ1 and TAZ2, respectively [43]. Published backbone assignments for TAZ2 (residues 1764-1850) [44] were used as a basis for assignment of the longer construct employed in this work (residues 1764-1855). Backbone resonances of free ppE7(1-51) peptide, uniformly labeled with 15N and 13C, and the TAZ2:ppE7(1-51) complex were assigned using 3D HNCA, HNCACB and CBCA(CO)NH spectra [87-89]. Side chain assignments were made using 3D HCCH-COSY, HCCH-TOCSY and 15N-TOCSY-HSQC experiments [90]. For NMR experiments involving TAZ2, E7 and ppE7(1-51) with Rb, the 15N-labeled protein was maintained at a concentration of 50 μM and mixed with the other proteins in order to achieve the appropriate molar ratios.
Cell culture
Human BJ foreskin fibroblast cells or Mouse Embryonic Fibroblast (MEF) cells were maintained in Dulbecco modified Eagle medium (Gibco) supplemented with 10% calf serum. Both BJ and MEF cells stably expressing E7 proteins were generated by transduction with LXSN-E7 recombinant retroviruses as previously described [91]. Transduced cells were selected with 600 μg/ml of G418 (Sigma) until the mock-transduced cells were dead. CBP/p300 −/− cells were generated by treating the MEF cells with Cre recombinase-expressing retrovirus as previously described [52, 53], which effectively eliminates CBP and p300.
In-cell acetylation assay
For pRb immunoprecipitation and Western blotting, BJ cells with or without E7 as well as MEF cells and MEF cells treated with CRE, both with or without E7, were washed two times in cold phosphate-buffered saline (PBS), scraped and lysed by sonication in NETN buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5% NP-40) supplemented with 2 mM Na3VO4, 2 mM PMSF, 10 mM NaF, 1 mM β-glycerol phosphate, 10 mM sodium butyrate, 10 μM TSA, 1x PDx101, and one Complete Protease Inhibitor tablet (EDTA-free, Roche), followed by centrifugation at 13,000 × g for 10 min. Extracts containing 0.8 – 1.0 mg of proteins were subjected to immunoprecipitation with anti-pRb antibodies (Pharmingen 554136 for BJ cells, Santa Cruz M-153 and sc-7905 for MEF cells). Immunoconjugates were collected on protein G-agarose (Sigma), washed with lysis buffer, and resolved on 7 – 8% SDS-PAGE gels. Proteins were transferred to polyvinylidene difluoride membranes and probed with either anti-pRb antibodies (Santa Cruz sc-50 for BJ cells and sc-74562 for MEF cells), or anti-acetyl-Lysine (Ac-K) antibodies (Millipore, 06-933 for BJ cells and Cell Signaling, Ac-K-103 for MEF cells). E7 and E7Δ(21–24) were visualized by western blotting of lysis samples probed using an anti-E7 antibody (Invitrogen, 28-006). Anti-rabbit or anti-mouse immunoglobulin G1-HRP was used as the secondary reagent. Bands were detected using enhanced chemiluminescence and captured by the FluorChem HD2 Imaging System (Cell Biosciences). Band intensities were measured and corrected for background using AlphaView (AlphaInnotech).
Supplementary Material
Highlights.
The HPV E7 oncoprotein binds with high affinity to the TAZ2 domain of CBP/p300
E7 competes with the p53 activation domain for binding to TAZ2
The full-length E7 dimer promotes association of CBP and pRb in a ternary complex
The E7 dimer promotes pRb acetylation in cells by a CBP/p300-mediated mechanism
E7, CBP/p300, pRb interactions are potentially important for cellular transformation
Acknowledgments
This work was supported by grants CA096865 (PEW), CA131231 (PS) and CA172115 (PS) from the National Institutes of Health and by the Skaggs Institute for Chemical Biology. AJ was supported by fellowship GM095228 from the National Institutes of Health. We thank Paul K. Brindle for supplying the MEF cells bearing the homozygous conditional (floxed) knockout alleles of both CBP and p300. We thank Gerard Kroon for help with NMR experiments, Josephine Ferreon, R. Bryn Fenwick and Allan C.M. Ferreon for helpful discussions, and Euvel Manlapaz for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.de Villiers E-M, Fauquet C, Broker TR, Bernard H-U, zur Hausen H. Classification of papillomaviruses. Virology. 2004;324:17–27. doi: 10.1016/j.virol.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 2.zur Hausen H. Papillomavirus infections--a major cause of human cancers. Biochim Biophys Acta. 1996;1288:F55–78. doi: 10.1016/0304-419x(96)00020-0. [DOI] [PubMed] [Google Scholar]
- 3.zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nature Rev Cancer. 2002;2:342–50. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 4.Berk AJ. Recent lessons in gene expression, cell cycle control, and cell biology from adenovirus. Oncogene. 2005;24:7673–85. doi: 10.1038/sj.onc.1209040. [DOI] [PubMed] [Google Scholar]
- 5.Munger K, Phelps WC, Bubb V, Howley PM, Schlegel R. The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J Virol. 1989;63:4417–21. doi: 10.1128/jvi.63.10.4417-4421.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levine AJ. The common mechanisms of transformation by the small DNA tumor viruses: The inactivation of tumor suppressor gene products: p53. Virology. 2009;384:285–93. doi: 10.1016/j.virol.2008.09.034. [DOI] [PubMed] [Google Scholar]
- 7.McLaughlin-Drubin ME, Münger K. The human papillomavirus E7 oncoprotein. Virology. 2009;384:335–44. doi: 10.1016/j.virol.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barbosa MS, Vass WC, Lowy DR, Schiller JT. In vitro biological activities of the E6 and E7 genes vary among human papillomaviruses of different oncogenic potential. J Virol. 1991;65:292–8. doi: 10.1128/jvi.65.1.292-298.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halbert CL, Demers GW, Galloway DA. The E7 gene of human papillomavirus type 16 is sufficient for immortalization of human epithelial cells. J Virol. 1991;65:473–8. doi: 10.1128/jvi.65.1.473-478.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Werness BA, Levine AJ, Howley PM. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science. 1990;248:76–9. doi: 10.1126/science.2157286. [DOI] [PubMed] [Google Scholar]
- 11.Heck DV, Yee CL, Howley PM, Münger K. Efficiency of binding the retinoblastoma protein correlates with the transforming capacity of the E7 oncoproteins of the human papillomaviruses. Proc Natl Acad Sci US A. 1992;89:4442–6. doi: 10.1073/pnas.89.10.4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garcia-Alai MM, Alonso LG, dePrat-Gay G. The N-Terminal module of HPV16 E7 is an intrinsically disordered domain that confers conformational and recognition plasticity to the oncoprotein. Biochemistry. 2007;46:10405–12. doi: 10.1021/bi7007917. [DOI] [PubMed] [Google Scholar]
- 13.Calçada EO, Felli IC, Hosek T, Pierattelli R. The heterogeneous structural behavior of E7 from HPV16 revealed by NMR spectroscopy. Chembiochem. 2013;14:1876–82. doi: 10.1002/cbic.201300172. [DOI] [PubMed] [Google Scholar]
- 14.Noval MG, Gallo M, Perrone S, Salvay AG, Chemes LB, De Prat-Gay G. Conformational dissection of a viral intrinsically disordered domain involved in cellular transformation. PLoS ONE. 2013;8:e72760. doi: 10.1371/journal.pone.0072760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohlenschläger O, Seiboth T, Zengerling H, Briese L, Marchanka A, Ramachandran R, et al. Solution structure of the partially folded high-risk human papilloma virus 45 oncoprotein E7. Oncogene. 2006;25:5953–9. doi: 10.1038/sj.onc.1209584. [DOI] [PubMed] [Google Scholar]
- 16.Liu X, Clements A, Zhao K, Marmorstein R. Structure of the human papillomavirus E7 oncoprotein and its mechanism for inactivation of the retinoblastoma tumor suppressor. J Biol Chem. 2006;281:578–86. doi: 10.1074/jbc.M508455200. [DOI] [PubMed] [Google Scholar]
- 17.Clemens KE, Brent R, Gyuris J, Münger K. Dimerization of the human papillomavirus E7 oncoprotein in vivo. Virology. 1995;214:289–93. doi: 10.1006/viro.1995.9926. [DOI] [PubMed] [Google Scholar]
- 18.Zwerschke W, Joswig S, Jansen-Durr P. Identification of domains required for transcriptional activation and protein dimerization in the human papillomavirus type-16 E7 protein. Oncogene. 1996;12:213–20. [PubMed] [Google Scholar]
- 19.Todorovic B, Massimi P, Hung K, Shaw GS, Banks L, Mymryk JS. Systematic analysis of the amino acid residues of human papillomavirus type 16 E7 conserved region 3 involved in dimerization and transformation. J Virol. 2011;85:10048–57. doi: 10.1128/JVI.00643-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clements A, Johnston K, Mazzarelli JM, Ricciardi RP, Marmorstein R. Oligomerization properties of the viral oncoproteins adenovirus E1A and human papillomavirus E7 and their complexes with the retinoblastoma protein. Biochemistry. 2000;39:16033–45. doi: 10.1021/bi002111g. [DOI] [PubMed] [Google Scholar]
- 21.Dyson N, Howley PM, Münger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243:934–7. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- 22.Münger K, Werness BA, Dyson N, Phelps WC, Harlow E, Howley PM. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989;8:4099–105. doi: 10.1002/j.1460-2075.1989.tb08594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JO, Russo AA, Pavletich NP. Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature. 1998;391:859–65. doi: 10.1038/36038. [DOI] [PubMed] [Google Scholar]
- 24.Chellappan S, Kraus VB, Kroger B, Münger K, Howley PM, Phelps WC, et al. Adenovirus E1A, simian virus 40 tumor antigen, and human papillomavirus E7 protein share the capacity to disrupt the interaction between transcription factor E2F and the retinoblastoma gene product. Proc Natl Acad Sci US A. 1992;89:4549–53. doi: 10.1073/pnas.89.10.4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boyer SN, Wazer DE, Band V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996;56:4620–4. [PubMed] [Google Scholar]
- 26.Jones DL, Thompson DA, Munger K. Destabilization of the RB tumor suppressor protein and stabilization of p53 contribute to HPV type 16 E7-induced apoptosis. Virology. 1997;239:97–107. doi: 10.1006/viro.1997.8851. [DOI] [PubMed] [Google Scholar]
- 27.Smotkin D, Wettstein FO. The major human papillomavirus protein in cervical cancers is a cytoplasmic phosphoprotein. J Virol. 1987;61:1686–9. doi: 10.1128/jvi.61.5.1686-1689.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barbosa MS, Edmonds C, Fisher C, Schiller JT, Lowy DR, Vousden KH. The region of the HPV E7 oncoprotein homologous to adenovirus E1a and SV40 large T antigen contains separate domains for Rb binding and casein kinase II phosphorylation. EMBO J. 1990;9:153–60. doi: 10.1002/j.1460-2075.1990.tb08091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gage JR, Meyers C, Wettstein FO. The E7 proteins of the nononcogenic human papillomavirus type 6b (HPV-6b) and of the oncogenic HPV-16 differ in retinoblastoma protein binding and other properties. J Virol. 1990;64:723–30. doi: 10.1128/jvi.64.2.723-730.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chemes LB, Sanchez IE, Smal C, De Prat-Gay G. Targeting mechanism of the retinoblastoma tumor suppressor by a prototypical viral oncoprotein. FEBS J. 2010;277:973–88. doi: 10.1111/j.1742-4658.2009.07540.x. [DOI] [PubMed] [Google Scholar]
- 31.Lill NL, Grossman SR, Ginsberg D, DeCaprio J, Livingston DM. Binding and modulation of p53 by p300/CBP coactivators. Nature. 1997;387:823–7. doi: 10.1038/42981. [DOI] [PubMed] [Google Scholar]
- 32.Somasundaram K, El Deiry WS. Inhibition of p53-mediated transactivation and cell cycle arrest by E1A through its p300/CBP-interacting region. Oncogene. 1997;14:1047–57. doi: 10.1038/sj.onc.1201002. [DOI] [PubMed] [Google Scholar]
- 33.Frisch SM, Mymryk JS. Adenovirus-5 E1A: paradox and paradigm. Nature Rev Mol Cell Biol. 2002;3:441–52. doi: 10.1038/nrm827. [DOI] [PubMed] [Google Scholar]
- 34.Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Devel. 2000;14:1553–77. [PubMed] [Google Scholar]
- 35.Chan HM, La Thangue NB. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci. 2001;114:2363–73. doi: 10.1242/jcs.114.13.2363. [DOI] [PubMed] [Google Scholar]
- 36.Ganguly N, Parihar SP. Human papillomavirus E6 and E7 oncoproteins as risk factors for tumorigenesis. J Biosci. 2009;34:113–23. doi: 10.1007/s12038-009-0013-7. [DOI] [PubMed] [Google Scholar]
- 37.Bernat A, Avvakumov N, Mymryk JS, Banks L. Interaction between the HPV E7 oncoprotein and the transcriptional coactivator p300. Oncogene. 2003;22:7871–81. doi: 10.1038/sj.onc.1206896. [DOI] [PubMed] [Google Scholar]
- 38.Fera D, Marmorstein R. Different regions of the HPV-E7 and Ad-E1A viral oncoproteins bind competitively but through distinct mechanisms to the CH1 transactivation domain of p300. Biochemistry. 2012;51:9524–34. doi: 10.1021/bi3011863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang HG, Moran E, Yaciuk P. E1A promotes association between p300 and pRB in multimeric complexes required for normal biological activity. J Virol. 1995;69:7917–24. doi: 10.1128/jvi.69.12.7917-7924.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang J, Yamada O, Kawagishi K, Araki H, Yamaoka S, Hattori T, et al. Human T-cell leukemia virus type 1 Tax modulates interferon-α signal transduction through competitive usage of the coactivator CBP/p300. Virology. 2008;379:306–13. doi: 10.1016/j.virol.2008.06.035. [DOI] [PubMed] [Google Scholar]
- 41.Wojciak JM, Martinez-Yamout MA, Dyson HJ, Wright PE. Structural basis for recruitment of CBP/p300 coactivators by STAT1 and STAT2 transactivation domains. EMBO J. 2009;28:948–58. doi: 10.1038/emboj.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee CW, Ferreon JC, Ferreon AC, Arai M, Wright PE. Graded enhancement of p53 binding to CREB-binding protein (CBP) by multisite phosphorylation. Proc Natl Acad Sci US A. 2010;107:19290–5. doi: 10.1073/pnas.1013078107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arai M, Ferreon JC, Wright PE. Quantitative analysis of multisite protein–ligand interactions by NMR: binding of intrinsically disordered p53 transactivation subdomains with the TAZ2 domain of CBP. J Am Chem Soc. 2012;134:3792–803. doi: 10.1021/ja209936u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Guzman RN, Liu HY, Martinez-Yamout M, Dyson HJ, Wright PE. Solution structure of the TAZ2 (CH3) domain of the transcriptional adaptor protein CBP. J Mol Biol. 2000;303:243–53. doi: 10.1006/jmbi.2000.4141. [DOI] [PubMed] [Google Scholar]
- 45.Ferreon JC, Martinez-Yamout MA, Dyson HJ, Wright PE. Structural basis for subversion of cellular control mechanisms by the adenoviral E1A oncoprotein. Proc Natl Acad Sci US A. 2009;106:13260–5. doi: 10.1073/pnas.0906770106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yao J, Dyson HJ, Wright PE. Chemical shift dispersion and secondary structure prediction in unfolded and partly folded proteins. FEBS Lett. 1997;419:285–9. doi: 10.1016/s0014-5793(97)01474-9. [DOI] [PubMed] [Google Scholar]
- 47.Singh M, Krajewski M, Mikolajka A, Holak TA. Molecular determinants for the complex formation between the retinoblastoma protein and LXCXE sequences. J Biol Chem. 2005;280:37868–76. doi: 10.1074/jbc.M504877200. [DOI] [PubMed] [Google Scholar]
- 48.Deng Q, Li Y, Tedesco D, Liao R, Fuhrmann G, Sun P. The ability of E1A to rescue ras-induced premature senescence and confer transformation relies on inactivation of both p300/CBP and Rb family proteins. Cancer Res. 2005;65:8298–307. doi: 10.1158/0008-5472.CAN-05-0054. [DOI] [PubMed] [Google Scholar]
- 49.Helt AM, Galloway DA. Destabilization of the retinoblastoma tumor suppressor by human papillomavirus type 16 E7 is not sufficient to overcome cell cycle arrest in human keratinocytes. J Virol. 2001;75:6737–47. doi: 10.1128/JVI.75.15.6737-6747.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chan HM, Krstic-Demonacos M, Smith L, Demonacos C, La Thangue NB. Acetylation control of the retinoblastoma tumour-suppressor protein. Nature Cell Biol. 2001;3:667–74. doi: 10.1038/35083062. [DOI] [PubMed] [Google Scholar]
- 51.Nguyen DX, Baglia LA, Huang SM, Baker CM, McCance DJ. Acetylation regulates the differentiation-specific functions of the retinoblastoma protein. EMBO J. 2004;23:1609–18. doi: 10.1038/sj.emboj.7600176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kasper LH, Lerach S, Wang J, Wu S, Jeevan T, Brindle PK. CBP/p300 double null cells reveal effect of coactivator level and diversity on CREB transactivation. EMBO J. 2010;29:3660–72. doi: 10.1038/emboj.2010.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kasper LH, Thomas MC, Zambetti GP, Brindle PK. Double null cells reveal that CBP and p300 are dispensable for p53 targets p21 and Mdm2 but variably required for target genes of other signaling pathways. Cell Cycle. 2011;10:212–21. doi: 10.4161/cc.10.2.14542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hottiger MO, Nabel GJ. Viral replication and the coactivators p300 and CBP. Trends Microbiol. 2000;8:560–5. doi: 10.1016/s0966-842x(00)01874-6. [DOI] [PubMed] [Google Scholar]
- 55.Arany Z, Newsome D, Oldread E, Livingston DM, Eckner R. A family of transcriptional adaptor proteins targeted by the E1A oncoprotein. Nature. 1995;374:81–4. doi: 10.1038/374081a0. [DOI] [PubMed] [Google Scholar]
- 56.Eckner R, Ewen ME, Newsome D, Gerdes M, DeCaprio JA, Lawrence JB, et al. Molecular cloning and functional analysis of the adenovirus E1A- associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Devel. 1994;8:869–84. doi: 10.1101/gad.8.8.869. [DOI] [PubMed] [Google Scholar]
- 57.De Guzman RN, Wojciak JM, Martinez-Yamout MA, Dyson HJ, Wright PE. CBP/p300 TAZ1 domain forms a structured scaffold for ligand binding. Biochemistry. 2005;44:490–7. doi: 10.1021/bi048161t. [DOI] [PubMed] [Google Scholar]
- 58.Matt T, Martinez-Yamout MA, Dyson HJ, Wright PE. The CBP/p300 TAZ1 domain in its native state is not a binding partner of MDM2. Biochem J. 2004;381:685–91. doi: 10.1042/BJ20040564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chien W-M, Parker JN, Schmidt-Grimminger D-C, Broker TR, Chow LT. Casein kinase II phosphorylation of the human papillomavirus-18 E7 protein is critical for promoting S-phase entry. Cell Growth Differ. 2000;11:425–35. [PubMed] [Google Scholar]
- 60.Firzlaff JM, Luscher B, Eisenman RN. Negative charge at the casein kinase II phosphorylation site is important for transformation but not for Rb protein binding by the E7 protein of human papillomavirus type 16. Proc Natl Acad Sci U S A. 1991;88:5187–91. doi: 10.1073/pnas.88.12.5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Feng H, Jenkins LMM, Durell SR, Hayashi R, Mazur SJ, Cherry S, et al. Structural basis for p300 Taz2-p53 TAD1 binding and modulation by phosphorylation. Structure. 2009;17:202–10. doi: 10.1016/j.str.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bhaumik P, Davis J, Tropea JE, Cherry S, Johnson PF, Miller M. Structural insights into interactions of C/EBP transcriptional activators with the Taz2 domain of p300. Acta Cryst D. 2014;70:1914–21. doi: 10.1107/S1399004714009262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Howe JA, Mymryk JS, Egan C, Branton PE, Bayley ST. Retinoblastoma growth suppressor and a 300-kDa protein appear to regulate cellular DNA synthesis. Proc Natl Acad Sci US A. 1990;87:5883–7. doi: 10.1073/pnas.87.15.5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eckner R, Ludlow JW, Lill NL, Oldread E, Arany Z, Modjtahedi N, et al. Association of p300 and CBP with simian virus 40 large T antigen. Mol Cell Biol. 1996;16:3454–64. doi: 10.1128/mcb.16.7.3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Avantaggiati ML, Ogryzko V, Gardner K, Giordano A, Levine A, Kelly K. Recruitment of p300/CBP in p53-dependent signal pathways. Cell. 1997;89:1175–84. doi: 10.1016/s0092-8674(00)80304-9. [DOI] [PubMed] [Google Scholar]
- 66.Gu W, Shi XL, Roeder RG. Synergistic activation of transcription by CBP and p53. Nature. 1997;387:819–23. doi: 10.1038/42972. [DOI] [PubMed] [Google Scholar]
- 67.Ferreon AC, Ferreon JC, Wright PE, Deniz AA. Modulation of allostery by protein intrinsic disorder. Nature. 2013;498:390–4. doi: 10.1038/nature12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Massimi P, Banks L. Repression of p53 Transcriptional Activity by the HPV E7 Proteins. Virology. 1997;227:255–9. doi: 10.1006/viro.1996.8315. [DOI] [PubMed] [Google Scholar]
- 69.Eichten A, Westfall M, Pietenpol JA, Münger K. Stabilization and functional impairment of the tumor suppressor p53 by the human papillomavirus type 16 E7 oncoprotein. Virology. 2002;295:74–85. doi: 10.1006/viro.2002.1375. [DOI] [PubMed] [Google Scholar]
- 70.Phelps WC, Münger K, Yee CL, Barnes JA, Howley PM. Structure-function analysis of the human papillomavirus type 16 E7 oncoprotein. J Virol. 1992;66:2418–27. doi: 10.1128/jvi.66.4.2418-2427.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wu EW, Clemens KE, Heck DV, Münger K. The human papillomavirus E7 oncoprotein and the cellular transcription factor E2F bind to separate sites on the retinoblastoma tumor suppressor protein. J Virol. 1993;67:2402–7. doi: 10.1128/jvi.67.4.2402-2407.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fattaey AR, Harlow E, Helin K. Independent regions of adenovirus E1A are required for binding to and dissociation of E2F-protein complexes. Mol Cell Biol. 1993;13:7267–77. doi: 10.1128/mcb.13.12.7267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schwarze F, Meraner J, Lechner M, Loidl A, Stasyk T, Laich A, et al. Cell cycle-dependent acetylation of Rb2/p130 in NIH3T3 cells. Oncogene. 2010;29:5755–60. doi: 10.1038/onc.2010.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Barbosa MS, Edmonds C, Fisher C, Schiller JT, Lowy DR, Vousden KH. The region of the HPV E7 oncoprotein homologous to adenovirus E1a and SV40 large T antigen contains separate domains for Rb binding and casein kinase II phosphorylation. The EMBO journal. 1990;9:153–60. doi: 10.1002/j.1460-2075.1990.tb08091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Massimi P, Banks L. Differential phosphorylation of the HPV-16 E7 oncoprotein during the cell cycle. Virology. 2000;276:388–94. doi: 10.1006/viro.2000.0514. [DOI] [PubMed] [Google Scholar]
- 76.Zhang B, Chen W, Roman A. The E7 proteins of low- and high-risk human papillomaviruses share the ability to target the pRB family member p130 for degradation. Proc Natl Acad Sci US A. 2006;103:437–42. doi: 10.1073/pnas.0510012103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sdek P, Ying H, Chang DLF, Qiu W, Zheng H, Touitou R, et al. MDM2 promotes proteasome-dependent ubiquitin-independent degradation of retinoblastoma protein. Mol Cell. 2005;20:699–708. doi: 10.1016/j.molcel.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 78.Uchida C, Miwa S, Kitagawa K, Hattori T, Isobe T, Otani S, et al. Enhanced Mdm2 activity inhibits pRB function via ubiquitin-dependent degradation. EMBO J. 2005;24:160–9. doi: 10.1038/sj.emboj.7600486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rubin SM. Deciphering the retinoblastoma protein phosphorylation code. Trends Biochem Sci. 2013;38:12–9. doi: 10.1016/j.tibs.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Burke JR, Hura GL, Rubin SM. Structures of inactive retinoblastoma protein reveal multiple mechanisms for cell cycle control. Genes Devel. 2012;26:1156–66. doi: 10.1101/gad.189837.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rubin SM, Gall AL, Zheng N, Pavletich NP. Structure of the Rb C-terminal domain bound to E2F1-DP1: a mechanism for phosphorylation-induced E2F release. Cell. 2005;123:1093–106. doi: 10.1016/j.cell.2005.09.044. [DOI] [PubMed] [Google Scholar]
- 82.Sugase K, Landes MA, Wright PE, Martinez-Yamout M. Overexpression of post-translationally modified peptides in Escherichia coli by co-expression with modifying enzymes. Prot. Express. Purif. 2008;57:108–15. doi: 10.1016/j.pep.2007.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Legge GB, Martinez-Yamout MA, Hambly DM, Trinh T, Lee BM, Dyson HJ, et al. ZZ domain of CBP: an unusual zinc finger fold in a protein interaction module. J Mol Biol. 2004;343:1081–93. doi: 10.1016/j.jmb.2004.08.087. [DOI] [PubMed] [Google Scholar]
- 84.Delaglio F, Grzesiek S, Vuister GW, Guang Z, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–93. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 85.Johnson BA, Blevins RA. NMRView: A computer program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4:603–14. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 86.Goddard TD, Kneller DG. SPARKY. Vol. 3. University of California; San Francisco: 2006. [Google Scholar]
- 87.Grzesiek S, Bax A. Correlating backbone amide and side chain resonances in larger proteins by multiple relayed triple resonance NMR. J Am Chem Soc. 1992;114:6291–3. [Google Scholar]
- 88.Grzesiek S, Bax A. Improved 3D triple-resonance NMR techniques applied to a 31 kDa protein. J Magn Reson. 1992;96:432–40. [Google Scholar]
- 89.Wittekind M, Mueller L. HNCACB, a high-sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonances with the alpha- and beta-carbon resonances in proteins. J Magn Reson. 1993;101:201–5. [Google Scholar]
- 90.Bax A, Clore GM, Gronenborn AM. 1H-1H correlation via isotropic mixing of 13C magnetization, a new three-dimensional approach for assigning 1H and 13C spectra of 13C-enriched proteins. J. Magn. Reson. 1990;88:425–31. [Google Scholar]
- 91.Sun P, Dong P, Dai K, Hannon GJ, Beach D. p53-independent role of MDM2 in TGF-beta1 resistance. Science. 1998;282:2270–2. doi: 10.1126/science.282.5397.2270. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.