

Abstract
Successful human pregnancy requires extensive invasion of maternal uterine tissues by the placenta. Invasive extravillous trophoblasts derived from cytotrophoblast progenitors remodel maternal arterioles to promote blood flow to the placenta. In the pregnancy complication preeclampsia, extravillous trophoblasts invasion and vessel remodeling are frequently impaired, likely contributing to fetal underperfusion and maternal hypertension. We recently demonstrated in mouse trophoblast stem cells that hypoxia-inducible factor-2 (HIF-2)-dependent Lim domain kinase 1 (LIMK1) expression regulates invasive trophoblast differentiation by modulating the trophoblast cytoskeleton. Interestingly, in humans, LIMK1 activity promotes tumor cell invasion by modulating actin and microtubule integrity, as well as by modulating matrix metalloprotease processing. Here, we tested whether HIF-2α and LIMK1 expression patterns suggested similar roles in the human placenta. We found that LIMK1 immunoreactivity mirrored HIF-2α in the human placenta in utero and that LIMK1 activity regulated human cytotrophoblast cytoskeletal integrity, matrix metallopeptidase-9 secretion, invasion, and differentiation in vitro. Importantly, we also found that LIMK1 levels are frequently diminished in the preeclampsia setting in vivo. Our results therefore validate the use of mouse trophoblast stem cells as a discovery platform for human placentation disorders and suggest that LIMK1 activity helps promote human placental development in utero.
Hypertensive disorders of pregnancy are a significant cause of maternal and neonatal morbidity worldwide.1,2 Preeclampsia (PE), a poorly understood disorder, is characterized by maternal hypertension, proteinuria, and edema, and affects approximately 5% to 8% of all pregnancies.3 When untreated, the syndrome can trigger maternal seizures (eclampsia) and result in fetal underperfusion. The only definitive cure is delivery of the placenta, thereby contributing to the epidemic of preterm delivery.4 A leading theory regarding its etiology implicates improper early placental development5 and invasive trophoblast differentiation, followed by maternal systemic complications, via a two-stage process.6,7
Human placental development is characterized by the remarkable invasion of maternal uterine structures by trophoblasts. The histological features of this interface are diagrammed in Figure 1. Invasive extravillous trophoblasts (EVTs) derived from column cytotrophoblast (cCTB) progenitors located at the tips of anchoring villi (AV) migrate through the uterine parenchyma (interstitial invasion) in search of maternal spiral arterioles and veins (Figure 1A). This invasion peaks around 9 to 12 weeks of gestation.8 Via a process termed endovascular invasion, EVTs then breach the spiral arterioles, where they trigger the apoptotic death of resident endothelial and smooth muscle cells9 and transdifferentiate into an endothelialized trophoblast subtype that is capable of lining these vessels10 (Figure 1B). In the process, these high-resistance vessels are remodeled into low-resistance/high-capacitance conduits necessary for proper fetal perfusion and maternal hemodynamics.11 Although the interactions with veins are largely confined to the inner surface of the uterus, EVTs migrate along much of the intrauterine course of the arterioles. Endovascular invasion begins at the center of the placental bed, allowing uterine arterial blood to flow into the intervillous space by the end of the first trimester, where it bathes floating chorionic villi covered by a layer of multinucleated syncytiotrophoblasts (SynTs). SynTs perform essential transport functions of the placenta, in addition to producing multiple pregnancy hormones in humans.12 Interestingly, only about one third of the uterine spiral arteries are invaded by 18 weeks gestational age.13 In normal pregnancies at term, however, most of the spiral arteries are completely remodeled, indicating that the more lateral arteries are progressively invaded throughout the second and third trimesters.14,15
Figure 1.

A and B: Schematic representation of the human maternal–vascular interface created by the placenta as a function of trophoblast invasion. A: In the human placenta, the primary anatomical subunit is composed of the chorionic villous, which can either be floating or anchoring. Floating villi are composed of highly branched finger-like projections that are surrounded by a layer of multinucleated SynT that perform the transport functions of the placenta. B: Occasionally, the tips of villi attach to the uterus (anchoring villous), triggering the expansion of a population of cytotrophoblasts within cell columns that gives rise to invasive EVTs. Invading EVTs migrate through uterine tissues in search of uterine arterioles that are remodeled by them. During this process, EVTs breach the spiral arterioles, triggering apoptotic death of existing endothelial and smooth muscle cells, and line the spiral arterioles while transdifferentiating into an endothelialized trophoblast cell type, expressing many cell surface markers characteristic of endothelial cells. This allows for blood flow from the arterioles into the intervillous space. C: Model depicting induction of HIF activity in mouse trophoblast stem (TS) cells by ECM composition or oxygen tension ([O2]). HIF induction blocks TS cell differentiation along the SynT lineage and promotes invasive trophoblast giant cell (TGC) differentiation via LIMK1 expression and subsequent promotion of cytoskeletal integrity. BV, blood vessel.
Formation of this vascular interface relies heavily on a precisely orchestrated set of interactions between EVT-expressed adhesion molecules and the uterine extracellular matrix (ECM) and vasculature, that are directly linked to trophoblast differentiation.16–18 Interestingly, PE is associated with a dysregulated pattern of adhesion molecule expression by EVTs that is thought to be causally related to shallow placentation and incomplete vascular transformation.19 For example, in PE, EVTs fail to up-regulate expression of integrins αvβ3 and α1β1 on their cell surface.20 At a transcriptional level, EVTs express much higher levels of Id2, a negative regulator of basic helix-loop-helix family members that impairs differentiation.21,22 With regard to multinucleated SynTs that perform the transport functions of the placenta, PE is associated with disturbances in their turnover,23,24 which may be due to diminished cathepsin levels25 or reduced GCM1 expression,26 which leads to parallel reductions in SynT differentiation and fusion.27 Thus, differentiation events within both the uterus and chorionic villi may be affected, suggesting that very early differentiation events may be compromised.
Endovascular invasion by EVTs may also be compromised in PE as a result of lower levels of vascular endothelial growth (Vegf)-A and Vegfr-1, along with increased sFlt-1, a soluble VEGF receptor that acts as a VEGF antagonist.28 Interestingly, excess sFlt-1 produces a PE-like syndrome in rats.29 SynTs exhibit increased staining for endoglin, a transforming growth factor-β coreceptor, and forced expression of this molecule also produces a PE-like syndrome in rats.30 By contrast, human SynT expression of adrenomedullin (AM), a peptide vasodilator, decreases in PE,31 and mice with reduced maternal and/or fetal expression of this molecule also develop signs of this syndrome.32 AM and the related AM2 can also impact EVT invasion.33,34 Together, these findings led to the concept that PE is a complex disorder associated with an imbalance in angiogenic factors,35 suggesting inappropriate activation of placental hypoxia responses that are strong activators of angiogenic pathways.
Hypoxia-inducible factor-1 (HIF-1) is a major regulator of cellular hypoxia responses.36 A basic helix-loop-helix PAS transcription factor composed of two subunits, HIF-1α and HIF-1β/ARNT,37 this ubiquitous heterodimer is responsible for the hypoxic induction of hundreds of genes by binding to hypoxia response elements in their promoters or enhancers.38 We have shown that HIF-1β/ARNT is critical for development, particularly of the placenta, where oxygen tension regulates cell fate decisions.39–41 In mice, Arnt−/− placentas display a grossly disrupted architecture as a result of reduced progenitor proliferation and impaired vascularization. Furthermore, inactivation of the genes encoding the murine von Hippel-Lindau homolog, as well as PHD2, conditions that produce constitutively active HIF, also result in embryonic lethality that is due to impaired placental vascularization,42,43 indicating that precise regulation of HIF levels is critical for normal placentation. Interestingly, HIF deficiency also impairs trophoblast stem cell (TSC)–ECM interactions,44 suggesting direct roles in invasion.
Importantly, oxygen tension can modulate human EVT proliferation, differentiation, invasion, and ECM degradation.45–47 VEGF, VEGFR-2, and AM are induced by HIF activity,48 and Gcm-1 is regulated by oxygen.49 Additionally, oxygen tension can modulate human SynT differentiation.50 Furthermore, HIF deficiency in the mouse results in altered trophoblast differentiation and cell surface integrin expression, along with diminished VEGF expression,51 suggesting a causal link. Finally, 2-methoxyestradiol, an estrogen metabolite that can inhibit HIF activity,52 was shown to be reduced in preeclamptic women, and its deficiency causes a PE-like syndrome in mice.53 In sum, PE is associated with fundamental defects in trophoblast differentiation that negatively impact endovascular invasion, SynT formation, and placental development. Importantly, cellular hypoxia responses appear to be involved in many of these pathways.
We recently described a novel role for HIF-dependent signaling in the placenta. Specifically, we found that noncanonical HIF signaling in mouse TSCs can regulate differentiation via activation of the HIF-2–specific cytoskeletal regulatory protein, LIM domain kinase 1 (LIMK1).54 LIMK1 can modulate the actin cytoskeleton through cofilin phosphorylation,55 as well as the microtubule cytoskeleton through p25 phosphorylation.56 Cytoskeletal integrity is critical for trophoblast differentiation with invasive EVTs containing robust actin and microtubule networks, whereas in multinucleated SynTs, these are disrupted.54,57,58 Importantly, cytoskeleton disruption can redirect TSC fate along the SynT lineage. Furthermore, LIMK1 activity promotes tumor cell invasion and migration, both via cytoskeletal remodeling, as well as matrix metalloproteinase (MMP) processing,56,59 suggesting similar roles in trophoblast invasion.
In mouse TSCs, HIF-1α and -2α are developmentally induced in response to ECM-dependent cues downstream of MAP2K1/2-dependent MAPK3/1 (Erk2/1) activity to repress SynT formation54 (Figure 1C). In the absence of these cues, hypoxic culture conditions can act through the same kinase cascade to activate HIF-1α and, to a lesser extent, HIF-2α. It has previously been demonstrated that HIF-2α levels are also developmentally regulated in the human placenta.60 Similar to mouse TSCs, invasive CTBs and their cCTBs progenitors express high levels of this transcriptional regulator, whereas SynTs do not. We wished to determine whether in the human placenta trophoblast cytoskeletal integrity correlates with HIF-2α activity spatially and temporally, and whether LIMK1 expression also plays important roles in human placental development, as well as in PE pathogenesis. Our results suggest such a role for LIMK1 activity in cytotrophoblast invasion and differentiation, which is aberrant in PE.
Materials and Methods
Antibodies and Limk-1 Inhibitor
Rat anti-human cytokeratin monoclonal antibody, 7D3, was produced in the Fisher laboratory. Polyclonal rabbit anti–Limk-1, polyclonal rabbit anti–phospho-MAP2K1/2, and mouse monoclonal anti–p-cofilin was purchased from Cell Signaling Technology (Danvers, MA). Polyclonal rabbit anti-HIF2α (NB100-22) was purchased from Novus Biologicals (Littleton, CO). Polyclonal rabbit anti–PAPP-A was from DakoCytomation (Dako A/S, Glostrup, Denmark). Mouse monoclonal anti–α-actin was purchased from Sigma-Aldrich (Saint Louis, MO). Mouse monoclonal anti-cofilin was from BD Transduction Laboratories (San Jose, CA). Limk-1 inhibitor, BMS-5, was purchased from Synkinase (Parkville, Australia).
Tissue Sources for Immunolocalization Experiments
Placentas were obtained from normal pregnant women and patients with PE. We analyzed 10 control samples from patients with no evidence of PE, gestational hypertension, or a medical history that suggested an increased risk of developing PE. We analyzed five samples from patients with PE diagnosed according to the classic criteria originally recommended by Dr. Leon Chesley and modified by the National Institutes of Health61: no history of hypertension before pregnancy; increase in diastolic pressure of 15 mm Hg or systolic pressure of 30 mm Hg compared with blood pressure obtained before 20 weeks of gestation; proteinuria ≥0.5 g/24 hours or ≥30 mg/dL (or 1+ on urine dipstick) in a catheterized specimen; hyperuricemia >5.5 mg/dL (or 1 SD greater than the normal mean value before term); return to normal blood pressure and resolution of proteinuria by 12 weeks postpartum. Severe PE was diagnosed according to the following criteria62: systolic blood pressure of ≥160 mm Hg and/or diastolic pressure of ≥110 mm Hg; proteinuria of ≥5 g in a 24-hour period or 3+ on a urine dipstick; presence of cerebral or visual disturbances; 36 weeks gestational age or less.
Immunolocalization
Placental tissues were processed for double indirect immunolocalization as previously described.60 Tissues were fixed in 3% paraformaldehyde for 30 minutes, washed three times in PBS, infiltrated with 5% to 15% sucrose followed by optimal cutting temperature (OCT) compound medium, and frozen in liquid nitrogen. Sections (5 μm) were prepared and incubated in a mixture of anti-cytokeratin (to localize trophoblasts) and another primary antibody for 2 hours. The sections were then rinsed and incubated with the appropriate species-specific secondary antibodies conjugated to rhodamine or fluorescein. Samples were examined with a Leica epifluorescence microscope (Leica Microsystems, Buffalo Grove, IL). For immunolocalization of antigens expressed by cultured cytotrophoblasts, isolated cells were plated on coverslips coated with Matrigel (Collaborative Biomedical Products, Bedford, MA) for various periods of time, then fixed in 3% paraformaldehyde for 5 minutes and permeabilized with cold methanol for another 5 minutes. Samples were stained and analyzed as described above. Secondary antibody–alone negative control images are provided in Supplemental Figure S1.
Cytotrophoblast Isolation and Culture
Cytotrophoblasts were isolated from chorionic villi of 6- to 24-week human placentas by routine procedures established in our laboratory.60 Briefly, the placentas were obtained immediately after elective pregnancy terminations. After a series of collagenase and trypsin digestions, cytotrophoblasts were separated from contaminating cell types on Percoll gradients. Purified cells were used immediately or cultured in serum-free high-glucose medium on Matrigel-coated substrates for the times indicated.
Cell Extraction and Immunoblotting
Freshly isolated cytotrophoblasts or cytotrophoblasts cultured on Matrigel-coated wells were washed twice with PBS and extracted with 200 μL of lysis buffer [50 mmol/L Tris buffer (pH 7.6), containing 1% Nonidet P-40, 0.1% SDS, 120 mmol/L NaCl, plus EDTA-free protease inhibitor]. Cell extracts were centrifuged at 12,000 × g for 10 minutes to remove insoluble materials. Samples containing equal amounts of protein were mixed with SDS sample buffer and separated by SDS-polyacrylamide gel electrophoresis. After the proteins were transferred to nitrocellulose, the membranes were incubated first with the primary antibody, then with peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA). Immune complexes were visualized using enhanced chemiluminescence and Hyperfilm (GE Healthcare, Pittsburgh, PA).
Invasion Assay
Invasion assays were conducted as described previously.28 Briefly, isolated cytotrophoblasts (0.25 × 106) were plated on Transwell inserts (6.5 mm; Costar, Cambridge, MA) containing polycarbonate filters (pore size, 8 μm) that had been coated with Matrigel. Culture medium containing either 1 μL/mL 10 μmol/L BMS-5 or 1 μL/mL dimethyl sulfoxide was added. After 48 hours, the cultures were stained with the 7D3 antibody, which specifically reacts with human cytokeratin, to visualize the cytotrophoblasts. The filters were cut from the supports and mounted, upper surface facing down, on slides. The number of cytokeratin-positive cells and cell processes on the lower surface of the filter was counted. Each experimental condition was tested in triplicate, and the entire assay was done six times. Data were expressed as a percentage of control. The statistical significance of the data was analyzed by Student's t-test.
Results
Correlation of HIF-2α Stability and Cytoskeletal Integrity in the Human Placenta
We previously demonstrated that MAP2K1/2-dependent HIF-2 activity regulates LIMK1 expression and cytoskeletal integrity in mouse TSCs differentiated along the invasive lineage.54 The active (phosphorylated) form of the MAP2K1/2 target MAPK3/1 was identified in villous cytotrophoblasts (vCTBs) underlying SynTs before cell fusion, as well as in invasive CTBs (iCTBs) within mid-second trimester (19 weeks) human placentas (Supplemental Figure S2, A and B). Multinucleated SynTs surrounding chorionic villi, however, failed to stain for pMAPK3/1, suggesting that this pathway was differentially regulated during trophoblast differentiation. We also assessed cytoskeletal integrity in the same structures and noted that although vCTBs and invasive CTBs exhibited robust microtubule networks, SynTs did not. Specifically, α-tubulin staining was reduced in these cells, compared with all other cell types that comprise the placenta (Supplemental Figure S2, C and D). Similar results were obtained with fluorescein isothiocyanate–phalloidin staining (not shown), indicating disruption of the actin as well as the microtubule cytoskeleton on SynT formation. To more carefully delineate HIF-2α expression and localization during human placental development, and correlate its expression with cytoskeletal integrity, we analyzed first and second trimester human placentas from three different gestational ages. First, we investigated first trimester floating villi (FV) and noted that although the vCTB progenitors of multinucleated SynTs stained strongly for HIF-2α and α-tubulin, SynT formation was associated with near complete absence of HIF-2α as well as α-tubulin immunoreactivity (Figure 2). In AV, first and second trimester human placentas exhibited nuclear immunoreactivity for HIF-2α within cell columns. These same structures similarly stained strongly for α-tubulin. By the beginning of the third trimester, levels of HIF-2α were generally reduced, and we noted a more diffuse cytoplasmic as well as nuclear localization pattern, similar to what has been described in differentiated mouse TSCs.40
Figure 2.

Immunofluorescence microscopy-based analysis of HIF-2α and α-tubulin immunoreactivity in the human placenta. Placentas (6-, 15-, and 23-week-old) were stained with antibodies for HIF-2α, α-tubulin, or cytokeratin (CK). The upper row represents an assessment of expression patterns in FV at 6 weeks, whereas the lower rows represent staining patterns in AV across multiple gestational ages. As seen, vCTB progenitors of SynT exhibit HIF-2α, as well as α-tubulin, immunoreactivity, which is nearly completely lost on syncytialization. This pattern is maintained in the second trimester as well (not shown). Note the lack of HIF-2α and α-tubulin staining, with positive CK immunoreactivity, in the SynT layer immediately surrounding vCTBs (arrows). Additionally, CTBs within cell columns at 6 and 15 weeks express abundant nuclear HIF-2α immunoreactivity, as well as cytoplasmic α-tubulin immunoreactivity. Interestingly, by the end of the second trimester (23 weeks), HIF-2α immunoreactivity is more diffuse and less intense, although still greater than negative control staining (Supplemental Figure S1). CK staining demarcates all trophoblast populations.
Developmental Regulation of LIMK1 Expression in the Human Placenta
To determine whether LIMK1 was similarly expressed in the human placenta in regions also known to contain stable HIF-2α, we performed serial immunofluorescence microscopy–based analyses of human placental samples during the first two trimesters of gestation. cCTBs within AVs, previously shown to contain high levels of HIF-2α60 and verified by us (see above), also expressed LIMK1 during this time period (Figure 3). Invasive CTBs also expressed LIMK1, albeit to slightly lower levels (Figure 3). In FV (Figure 4); however, LIMK1 was expressed at generally lower levels, with reductions in LIMK1 expression noted specifically within the SynT layer (Figure 4). Importantly, this is the layer in which MAP2K1/2 activity and HIF-2α stability are also diminished. These results are consistent with the observed disruption in the cytoskeleton following cell fusion and suggest potential roles for HIF-2–dependent LIMK1 expression in promoting CTB differentiation away from SynT and toward iCTB/EVTs. Importantly, however, the regulation of HIF transcriptional activity is complex and not simply dependent on protein stability. For example, the extent of HIF-2α acetylation can regulate its activity.63 Acetyl transferase or deacetylase activity can be regulated by various environmental conditions such as oxygen or nutrient deprivation to affect transcription factor activity or stability.64 We therefore additionally tested whether inhibition of class I/II histone deacetylase (HDAC) activity with sodium butyrate or class III activity with sirtinol could modulate HIF-2 activity/stability in the placenta. HIF-2 expression in Hif-1/2α−/− TSCs promotes LIMK1 protein accumulation, which is inhibited by both classes of HDAC inhibitors (Supplemental Figure S3). Interestingly, however, whereas sodium butyrate resulted in HIF-2α protein destabilization and subsequent LIMK1 down-regulation, class III HDAC inhibition with sirtinol only resulted in impaired HIF-2 transcriptional activity without affecting its stability. These results indicate that HIF-2–dependent LIMK1 expression can be regulated by more than just HIF-2 protein stabilization and can be subjected to environmental stress-induced inputs mediated by diverse HDAC family members.
Figure 3.
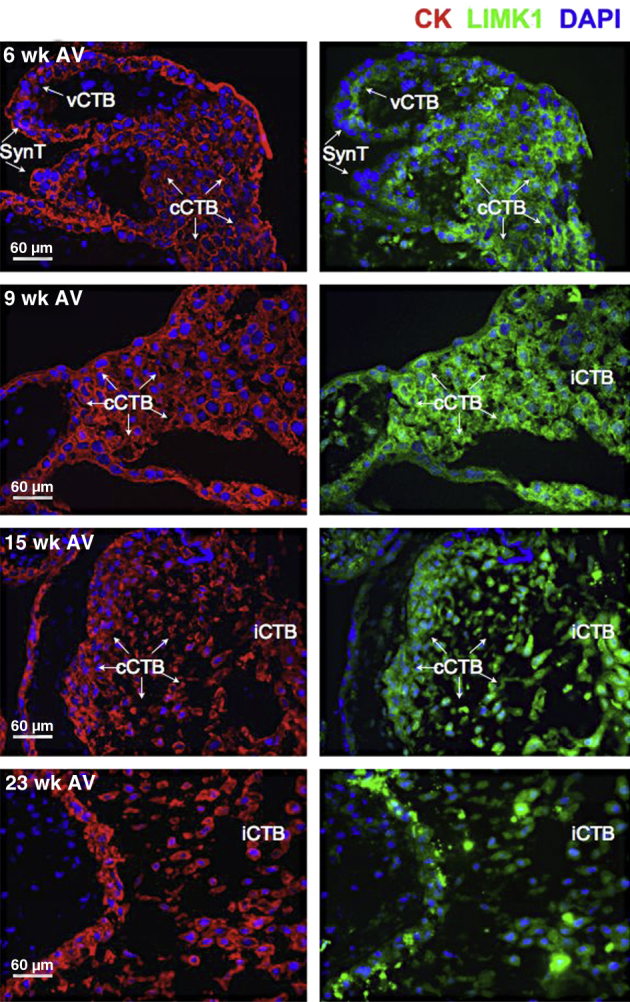
Expression pattern of LIMK1 within AV during the first two trimesters of human placental development. Immunofluorescence microscopy-based analysis of LIMK1 expression was undertaken in tissue sections of human placental segments containing AV at weeks 6, 9, 15, and 23. CK staining served as a marker of all placental cells. Nontrophoblasts do not exhibit immunoreactivity for CK. DAPI was used to stain nuclei. vCTB, SynT, and cCTBs are indicated.
Figure 4.
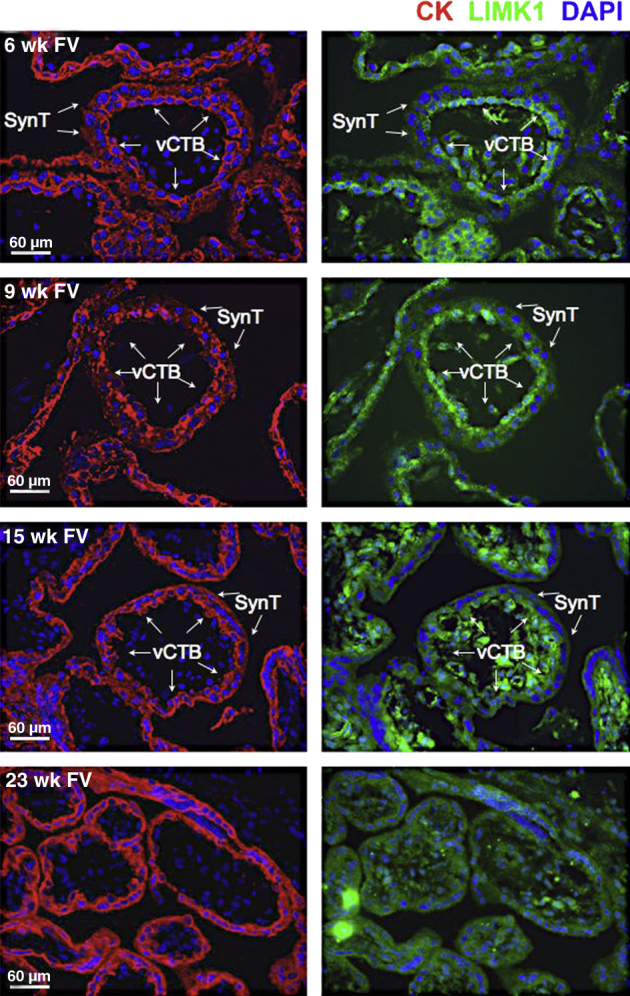
Expression pattern of LIMK1 within floating villi (FV) during the first two trimesters of human placental development. Immunofluorescence microscopy-based analysis of LIMK1 expression was undertaken in tissue sections of human segments containing FV at weeks 6, 9, 15, and 23. CK staining served as a marker of placental cells. Maternal uterine structures do not exhibit CK immunoreactivity. DAPI was used to stain nuclei. vCTBs and SynTs are indicated.
LIMK1 Activity Regulates Human CTB Cytoskeletal Integrity, Invasion, MMP Processing, and Differentiation in Vitro
To test the role of LIMK1 activity in CTBs, we cultured primary human CTBs in vitro in the presence of the specific LIMK1 inhibitor BMS-559,65 using a standard concentration (10 μmol/L) developed to detail the role of LIMK1 in cancer cell migration. LIMK1 inhibition resulted in dramatic alterations of the CTB cytoarchitecture in vitro (Figure 5A). Although control CTBs isolated from second trimester human placentas and cultured in vitro frequently exhibited robust actin stress fibers as evidenced by anti-actin staining, CTBs treated with BMS-5 did not. They continued to maintain a rounded shape with a complete absence of actin stress fibers (Figure 5A). Importantly, BMS-5–treated CTBs did not show evidence of increased apoptosis when compared with vehicle-treated controls (Supplemental Figure S4), indicating that these results were not due to drug toxicity. To confirm that BMS-5 treatment resulted in LIMK1 inhibition, we performed immunoblot-based analysis of LIMK1, as well as of its target cofilin. BMS-5 triggered noticeable decreases in cofilin phosphorylation (Figure 5B). Interestingly, total LIMK1 levels were also reduced following LIMK inhibition. LIMK1 expression is reduced in SynTs compared with iCTBs, suggesting that LIMK1 inhibition promotes differentiation along the SynT lineage. Further, suggesting an inhibition of differentiation along the iCTB lineage in vitro, LIMK inhibition also increased expression of the SynT marker PAPP-A in cultured CTBs (Figure 5, C and D). Next, we assessed whether LIMK1 inhibition could functionally impair CTB invasion through Matrigel. BMS-5 inhibited first trimester (10 weeks), as well as late second trimester (23 weeks) (Supplemental Figure S5), CTB invasion in vitro (Figure 5E). Finally, and consistent with reports that LIMK activity can regulate MMP secretion, LIMK inhibition with BMS-5 impaired MMP-9 secretion by CTBs maintained in vitro, without affecting MMP-9 expression (Figure 5F). Collectively, these results indicate that LIMK inhibition in vitro impairs CTB differentiation along the invasive lineage while disrupting cytoskeletal integrity and inhibiting MMP processing. Importantly, CTB invasion, differentiation, as well as MMP processing, are frequently impaired in the PE setting in vivo.
Figure 5.

LIMK inhibition blocks CTB invasion and differentiation in vitro. A: Primary human CTBs were stained with an antibody specific for α-actin (green) following culture without or with 10 μmol/L BMS-5 (top left panel and top right panel). CTBs frequently exhibit robust actin cytoskeletons when cultured in vitro (top left panel and top right panel), which are completely lost in the presence of LIMK-inhibitor, BMS-5 (bottom left panel and bottom right panel). B: LIMK inhibition blocks LIMK signaling. Immunoblot (IB) analysis of LIMK1 activity with (+) or without (−) 10 μmol/L LIMK inhibitor BMS-5. BMS-5 reduces cofilin phosphorylation and decreases LIMK expression, suggesting an association between LIMK activity and CTB differentiation along the iCTB lineage. C: Immunoblot analysis of PAPP-A expression in human CTBs differentiated in vitro without and with BMS-5. LIMK inhibition promotes expression of the SynT marker PAPP-A in differentiating human CTBs in vitro. D: Immunofluorescence microscopy-based analysis of LIMK expression in the second trimester (TM) floating villous. Note the robust expression in the overlying SynT layer. E: CTB invasion assay. BMS-5 impaired the invasion of CTBs derived from first trimester (10 weeks) placenta in a Matrigel-based invasion assay. F: LIMK expression impairs MMP-9 secretion by human CTBs cultured in vitro. Immunoblot assay of secreted MMP-9 in CTB cultured media (CTB-CM) versus CTB whole-cell lysate (CTB-lysate) using an MMP-9-specific antibody. As seen, LIMK-inhibition with BMS-5 does not impair MMP-9 production, but it impairs its secretion into media. Ponceau S staining of total protein content in each sample indicates equivalent loading within groups. ∗∗P < 0.01. DMSO, dimethyl sulfoxide; GAPDH, Glyceraldehyde 3-phosphate dehydrogenase; IB, immunoblot.
LIMK1 Levels Are Frequently Decreased in the Setting of Severe PE in Vivo
To test whether LIMK1 levels may be diminished in the placentas of women suffering from PE, we isolated placentas from women diagnosed with severe PE and compared their expression of LIMK1 with age-matched control placentas obtained from women without PE but who delivered prematurely due to preterm labor without overt clinical signs and symptoms of infection. iCTBs in severe PE placentas were frequently characterized by diminished LIMK1 immunoreactivity when compared with gestational age–matched controls (Figure 6A). Additionally, we performed direct comparisons of LIMK1 protein levels via immunoblotting using primary CTBs isolated from control preterm labor and severe PE placentas. CTB lysates obtained from placentas with PE frequently exhibited reduced levels of LIMK1 compared with control placentas (Figure 6B). Densitometric analysis of expression levels confirmed these findings (Figure 6C).
Figure 6.

LIMK1 expression is decreased in the preeclampsia setting in utero. Early third trimester human placental samples obtained from women diagnosed with severe preeclampsia (sPE) were compared with placental samples obtained from gestational age matched samples obtained from women who delivered due to normal preterm labor (nPTL) without overt signs of clinical infection. A: Immunofluorescence microscopy-based approaches indicate that although invasive CTBs and cCTBs in nPTL placentas exhibit robust LIMK1 immunoreactivity, the expression of LIMK1 is frequently decreased in the sPE setting in vivo. B: Immunoblot analysis of CTB lysates derived from additional nPTL and sPE placentas. C: Densitometric analysis of relative expression levels of LIMK1 in B.
Discussion
Our results suggest a potentially novel mechanism whereby HIF activity may contribute to human placental development as well as PE pathogenesis. We previously demonstrated that HIF activity is critical for placentation in the mouse.39–41 In its absence, altered trophoblast differentiation results in a grossly disrupted placental architecture and impaired formation of a maternal–fetal vascular exchange interface. Interestingly, although the deficiency of HIF-1α or -2α alone is insufficient to significantly compromise mouse placental development, combined deficiency of both subunits, or the absence of their requisite heterodimerization partner ARNT/HIF-1β, is sufficient. These results helped establish a critical role for HIF activity during placental development. More recently, we demonstrated an additional level of complexity with respect to HIF-dependent gene expression in the placenta. We showed that similar to what has been described in the developing nervous system in mice,66 and during hematopoiesis in Drosophila melanogaster,67 the HIF family of transcription factors can act via noncanonical means to activate downstream target gene expression to drive development in the mouse placenta. Specifically, we showed that the LIMK1 gene can be regulated by HIF-2α via its ability to interact with and activate MYC (alias c-MYC–)dependent transcription in mouse TSCs.54 Similar roles for HIF-2α have been shown to be operative in multiple human malignancies68 and highlight potential points of divergence between HIF-1α– and -2α–dependent activities. Here, we provide evidence that this pathway is conserved during human placental development and that LIMK1 activity promotes human CTB differentiation and invasion. Importantly, this pathway appears to be impaired in the setting of PE and may contribute to its etiology.
An additional surprising finding has been that factors other than oxygen tension can regulate HIF stability in the placenta. We previously demonstrated that trophoblast ECM composition can trigger HIF-1α and -2α subunit accumulation as a function of differentiation.54 In mouse TSCs derived on a bed of feeder cells, growth factor withdrawal induced differentiation results in robust HIF-1α and -2α stabilization and transcriptional activity independent of oxygen tension that is required during invasive trophoblast giant cell formation. Altering the ECM on which TSCs are cultured blocks this differentiation-dependent HIF induction. This has relevance for placentation because in concert with phenotypic alterations in invasive trophoblasts, the uterine ECM is remodeled to promote successful implantation and trophoblast invasion.11,69–71 This process is enhanced by trophoblast expressed MMPs—a family of enzymes that break down ECM components.72 In this capacity, multiple studies have highlighted the role of these proteases in regulating trophoblast invasion,73–88 and MMP9 deficiency in pregnant mice triggers clinical diagnostic features of PE.89 Interestingly, in addition to its well-known role regulating the cytoskeleton through cofilin56 and p2590 phosphorylation, LIMK1 also regulates MMP processing.91 Here, we show that LIMK1 plays similar roles in human CTBs and likely contributes to its ability to promote CTB invasion. Furthermore, its expression pattern in the human placenta mirrors what we described in mouse TSCs. Coupled with our observations that ECM composition can trigger differentiation-dependent HIF induction54 and that HIF activity can regulate TSC cell surface integrin localization,44 these results suggest the existence of a positive feedback loop wherein ECM composition, HIF activity, and cell surface integrin expression collectively promote the invasive phenotype in trophoblasts. We suggest that HIF-2–dependent LIMK1 expression represents one mechanism that helps propel this process by modulating CTB MMP secretion and cytoskeletal integrity.
Cytoskeletal rearrangement is central to trophoblast differentiation.57 For example, calponin 3–mediated actin cytoskeletal rearrangement promotes SynT fusion,58 whereas caspases help remodel the fodrin cytoskeleton during this process,92 and stathmin, a microtubule regulatory protein, is associated with invasive trophoblast migration.93 In TSCs, microtubule or actin cytoskeleton disruption triggers SynT formation in conditions that would otherwise result in trophoblast giant cell formation.54 Interestingly, HIF stability can also be modulated by cytoskeletal integrity,52,94 suggesting yet another feed-forward mechanism during invasive trophoblast differentiation wherein HIF activity can promote cytoskeletal stability that, in turn, helps sustain HIF activity.
Multiple studies have investigated an association between HIF function and PE pathogenesis.7,45,46,95,96 Here, we show that LIMK1 expression is frequently diminished in the severe PE setting in utero, and thus may contribute to the impaired trophoblast differentiation, invasion, and endovascular remodeling associated with this disease process, likely downstream of blunted HIF-2 activity (Figure 7). LIMK1 expression is highest within the cell column, along with HIF-2α, before iCTB migration. This may reflect the dual role of LIMK1 in the placenta: preventing SynT formation while promoting CTB invasion. Given that cytoskeletal rearrangement is central to both cell fusion as well as cell invasion, this seems reasonable. Once invasion has been initiated, perhaps other mechanisms take on a more pivotal role for maintaining invasion. Interestingly, LIMK1 activity has been shown to be important for the initiation of tumor cell invasion,59 consistent with our observations. In addition to hypoxia, alterations in placental ECM composition can modulate HIF activity and thereby direct trophoblast invasion and fate, and may actually be the prime driver of HIF activity in the placenta. It is therefore important that the presence of HIF activity not be used as the sole marker of tissue hypoxia in vivo. Additionally, post-translational modification of HIF-α subunits, including but not limited to acetylation, dramatically impact its activity and can be regulated by multiple inputs, including nutrient availability and redox state.63 Given that hypoxia, redox stress, altered ECM remodeling, and bioenergetic compromise have all been associated with PE pathogenesis, our efforts to build a more thorough picture of the role of HIF-dependent gene expression during placental development should yield novel insights into the etiology of this enigmatic pregnancy complication.
Figure 7.

Schematic representation of LIMK1 activity in the human placenta. During normal pregnancy, CTB progenitors of invasive extravillous trophoblasts express high levels of LIMK1 likely driven by HIF-2α activity. LIMK1 promotes cytoskeletal integrity as well as MMP processing, thereby promoting CTB invasion. CTB invasion and differentiation are linked. Therefore, LIMK1 activity also enhances CTB differentiation while inhibiting SynT formation. In the setting of preeclampsia, LIMK1 expression is frequently diminished, resulting in altered CTB cytoskeletal integrity, MMP processing, differentiation and invasive capacity. This may contribute to shallow placentation with associated impaired spiral artery remodeling and imbalances in maternal blood pressure regulation. PreE, preeclampsia.
Acknowledgments
We thank Susan Fisher and Olga Genbacev for thoughtful discussion and critical reading of the manuscript.
Footnotes
Supported by Eunice Kennedy Shriver National Institute of Child Health and Human Development/NIH through cooperative agreement U54 HD055764-07 as part of the Specialized Cooperative Centers Program in Reproduction and Infertility Research, as well as by R01 HD072455; the California Institute for Regeneration Medicine grant TB1-01194 (A.M.R.); and the 2214A-Abroad Research Fellowship for PhD students from the Scientific and Technological Research Council of Turkey (G.U.).
Disclosures: None declared.
Supplemental Data
Antibody staining controls. Adjacent sections to those shown in Figure 2 were stained with appropriate secondary antibody alone as shown and imaged in identical fashion. Note the very low-level background staining observed. A and B: The staining pattern observed in 6 weeks placenta (A) and 23 weeks placenta (B). FV, floating villi.
Immunofluorescence microscopy-based analysis of MAPK2K1/2 activity and cytoskeletal integrity in the human placenta. Placentas (19-week-old) were stained with antibodies for phospho-MAPK3/1 (A and B), or α-tubulin (α-Tub) (C and D). All slides were also stained with DAPI to identify nuclei. Expression in floating villi (FV) (A and C) is contrasted with invasive cytotrophoblast (iCTB) expression patterns (B and D). Although villous cytotrophoblasts (arrows), as well as populations of stromal cells within floating villi, show strong activation of MAPK3/1 (A) and a robust microtubule cytoskeleton (C), the overlying syncytiotrophoblasts (SynT) layer (arrowheads) does not. SynTs exhibit strong inhibition of MAPK3/1 phosphorylation, and fail to stain positively for α-tubulin. B and D: Invasive CTBs on the other hand, show strong immunoreactivity with each of these antibodies.
Regulation of HIF-2α transcriptional activity via histone deacetylase (HDAC) activity. Whole-cell lysates from Hif-1/2α−/− mouse trophoblast stem cells (TSCs) stably expressing either green fluorescent protein or HIF-2α, and either treated with or without 1.5 mmol/L inhibitors of class I/II HDAC activity [sodium butyrate (NaBu)] or 60 μmol/L class III HDAC/sirtuin inhibitor (sirtinol), were subjected to immunoblot analysis with antibodies specific for LIMK1, HIF-2α, or actin. Note the decrease of LIMK1 immunoreactivity with both inhibitor treatments. Note also, however, inhibition of HIF-2α stability only with NaBu treatment, but without a significant reduction of HIF-2α stability following sirtinol treatment. Sirtinol treatment results in a slight increase in HIF-2α molecular weight, evidenced by its slightly decreased mobility, consistent with its increased acetylation status following sirtuin inhibition.
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining of BMS-5–treated versus control dimethyl sulfoxide (DMSO)-treated cytotrophoblasts (CTBs) in vitro. Following standard TUNEL immunolabeling, numbers of TUNEL-positive (apoptotic) CTBs were counted. BMS-5 treatment (10 μmol/L) did not increase apoptotic cell death of CTBs cultured in vitro.
Cytotrophoblast (CTB) invasion assay. BMS-5 treatment (10 μmol/L) impairs second trimester CTB invasion in a Matrigel-based invasion assay. ∗∗P < 0.01.
References
- 1.Abalos E., Cuesta C., Grosso A.L., Chou D., Say L. Global and regional estimates of preeclampsia and eclampsia: a systematic review. Eur J Obstet Gynecol Reprod Biol. 2013;170:1–7. doi: 10.1016/j.ejogrb.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 2.Duley L. The global impact of pre-eclampsia and eclampsia. Semin Perinatol. 2009;33:130–137. doi: 10.1053/j.semperi.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 3.Turner J.A. Diagnosis and management of pre-eclampsia: an update. Int J Womens Health. 2010;2:327–337. doi: 10.2147/IJWH.S8550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldenberg R.L., Culhane J.F., Iams J.D., Romero R. Epidemiology and causes of preterm birth. Lancet. 2008;371:75–84. doi: 10.1016/S0140-6736(08)60074-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Norwitz E.R. Defective implantation and placentation: laying the blueprint for pregnancy complications. Reprod Biomed Online. 2007;14 Spec No 1:101–109. doi: 10.1016/S1472-6483(10)61464-2. [DOI] [PubMed] [Google Scholar]
- 6.Roberts J.M., Hubel C.A. The two stage model of preeclampsia: variations on the theme. Placenta. 2009;30 Suppl A:S32–S37. doi: 10.1016/j.placenta.2008.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Redman C.W., Sargent I.L. Latest advances in understanding preeclampsia. Science. 2005;308:1592–1594. doi: 10.1126/science.1111726. [DOI] [PubMed] [Google Scholar]
- 8.Pijnenborg R., Bland J.M., Robertson W.B., Dixon G., Brosens I. The pattern of interstitial trophoblastic invasion of the myometrium in early human pregnancy. Placenta. 1981;2:303–316. doi: 10.1016/s0143-4004(81)80027-6. [DOI] [PubMed] [Google Scholar]
- 9.Red-Horse K., Rivera J., Schanz A., Zhou Y., Winn V., Kapidzic M., Maltepe E., Okazaki K., Kochman R., Vo K.C., Giudice L., Erlebacher A., McCune J.M., Stoddart C.A., Fisher S.J. Cytotrophoblast induction of arterial apoptosis and lymphangiogenesis in an in vivo model of human placentation. J Clin Invest. 2006;116:2643–2652. doi: 10.1172/JCI27306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Damsky C.H., Fisher S.J. Trophoblast pseudo-vasculogenesis: faking it with endothelial adhesion receptors. Curr Opin Cell Biol. 1998;10:660–666. doi: 10.1016/s0955-0674(98)80043-4. [DOI] [PubMed] [Google Scholar]
- 11.Red-Horse K., Zhou Y., Genbacev O., Prakobphol A., Foulk R., McMaster M., Fisher S.J. Trophoblast differentiation during embryo implantation and formation of the maternal-fetal interface. J Clin Invest. 2004;114:744–754. doi: 10.1172/JCI22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maltepe E., Bakardjiev A.I., Fisher S.J. The placenta: transcriptional, epigenetic, and physiological integration during development. J Clin Invest. 2010;120:1016–1025. doi: 10.1172/JCI41211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pijnenborg R., Bland J.M., Robertson W.B., Brosens I. Uteroplacental arterial changes related to interstitial trophoblast migration in early human pregnancy. Placenta. 1983;4:397–413. doi: 10.1016/s0143-4004(83)80043-5. [DOI] [PubMed] [Google Scholar]
- 14.Brosens I., Pijnenborg R., Vercruysse L., Romero R. The “Great Obstetrical Syndromes” are associated with disorders of deep placentation. Am J Obstet Gynecol. 2011;204:193–201. doi: 10.1016/j.ajog.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pijnenborg R., Vercruysse L., Brosens I. Deep placentation. Best Pract Res Clin Obstet Gynaecol. 2011;25:273–285. doi: 10.1016/j.bpobgyn.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 16.Zhou Y., Fisher S.J., Janatpour M., Genbacev O., Dejana E., Wheelock M., Damsky C.H. Human cytotrophoblasts adopt a vascular phenotype as they differentiate. A strategy for successful endovascular invasion? J Clin Invest. 1997;99:2139–2151. doi: 10.1172/JCI119387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cao T.C., Thirkill T.L., Wells M., Barakat A.I., Douglas G.C. Trophoblasts and shear stress induce an asymmetric distribution of icam-1 in uterine endothelial cells. Am J Reprod Immunol. 2008;59:167–181. doi: 10.1111/j.1600-0897.2007.00542.x. [DOI] [PubMed] [Google Scholar]
- 18.Soghomonians A., Barakat A.I., Thirkill T.L., Douglas G.C. Trophoblast migration under flow is regulated by endothelial cells. Biol Reprod. 2005;73:14–19. doi: 10.1095/biolreprod.104.036509. [DOI] [PubMed] [Google Scholar]
- 19.Lim K.H., Zhou Y., Janatpour M., McMaster M., Bass K., Chun S.H., Fisher S.J. Human cytotrophoblast differentiation/invasion is abnormal in pre-eclampsia. Am J Pathol. 1997;151:1809–1818. [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou Y., Damsky C.H., Fisher S.J. Preeclampsia is associated with failure of human cytotrophoblasts to mimic a vascular adhesion phenotype. One cause of defective endovascular invasion in this syndrome? J Clin Invest. 1997;99:2152–2164. doi: 10.1172/JCI119388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Janatpour M.J., McMaster M.T., Genbacev O., Zhou Y., Dong J., Cross J.C., Israel M.A., Fisher S.J. Id-2 regulates critical aspects of human cytotrophoblast differentiation, invasion and migration. Development. 2000;127:549–558. doi: 10.1242/dev.127.3.549. [DOI] [PubMed] [Google Scholar]
- 22.Janatpour M.J., Utset M.F., Cross J.C., Rossant J., Dong J., Israel M.A., Fisher S.J. A repertoire of differentially expressed transcription factors that offers insight into mechanisms of human cytotrophoblast differentiation. Dev Genet. 1999;25:146–157. doi: 10.1002/(SICI)1520-6408(1999)25:2<146::AID-DVG9>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 23.Mayhew T.M. A stereological perspective on placental morphology in normal and complicated pregnancies. J Anat. 2009;215:77–90. doi: 10.1111/j.1469-7580.2008.00994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gauster M., Moser G., Orendi K., Huppertz B. Factors involved in regulating trophoblast fusion: potential role in the development of preeclampsia. Placenta. 2009;30 Suppl A:S49–S54. doi: 10.1016/j.placenta.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 25.Varanou A., Withington S.L., Lakasing L., Williamson C., Burton G.J., Hemberger M. The importance of cysteine cathepsin proteases for placental development. J Mol Med. 2006;84:305–317. doi: 10.1007/s00109-005-0032-2. [DOI] [PubMed] [Google Scholar]
- 26.Chen C.P., Chen C.Y., Yang Y.C., Su T.H., Chen H. Decreased placental GCM1 (glial cells missing) gene expression in pre-eclampsia. Placenta. 2004;25:413–421. doi: 10.1016/j.placenta.2003.10.014. [DOI] [PubMed] [Google Scholar]
- 27.Langbein M., Strick R., Strissel P.L., Vogt N., Parsch H., Beckmann M.W., Schild R.L. Impaired cytotrophoblast cell-cell fusion is associated with reduced Syncytin and increased apoptosis in patients with placental dysfunction. Mol Reprod Dev. 2008;75:175–183. doi: 10.1002/mrd.20729. [DOI] [PubMed] [Google Scholar]
- 28.Zhou Y., McMaster M., Woo K., Janatpour M., Perry J., Karpanen T., Alitalo K., Damsky C., Fisher S.J. Vascular endothelial growth factor ligands and receptors that regulate human cytotrophoblast survival are dysregulated in severe preeclampsia and hemolysis, elevated liver enzymes, and low platelets syndrome. Am J Pathol. 2002;160:1405–1423. doi: 10.1016/S0002-9440(10)62567-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maynard S.E., Min J.Y., Merchan J., Lim K.H., Li J., Mondal S., Libermann T.A., Morgan J.P., Sellke F.W., Stillman I.E., Epstein F.H., Sukhatme V.P., Karumanchi S.A. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111:649–658. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Venkatesha S., Toporsian M., Lam C., Hanai J., Mammoto T., Kim Y.M., Bdolah Y., Lim K.H., Yuan H.T., Libermann T.A., Stillman I.E., Roberts D., D'Amore P.A., Epstein F.H., Sellke F.W., Romero R., Sukhatme V.P., Letarte M., Karumanchi S.A. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med. 2006;12:642–649. doi: 10.1038/nm1429. [DOI] [PubMed] [Google Scholar]
- 31.Wilson C., Nikitenko L.L., Sargent I.L., Rees M.C. Adrenomedullin: multiple functions in human pregnancy. Angiogenesis. 2004;7:203–212. doi: 10.1007/s10456-004-4183-5. [DOI] [PubMed] [Google Scholar]
- 32.Li M., Yee D., Magnuson T.R., Smithies O., Caron K.M. Reduced maternal expression of adrenomedullin disrupts fertility, placentation, and fetal growth in mice. J Clin Invest. 2006;116:2653–2662. doi: 10.1172/JCI28462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chauhan M., Yallampalli U., Dong Y.L., Hankins G.D., Yallampalli C. Expression of adrenomedullin 2 (ADM2)/intermedin (IMD) in human placenta: role in trophoblast invasion and migration. Biol Reprod. 2009;81:777–783. doi: 10.1095/biolreprod.108.074419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang X., Green K.E., Yallampalli C., Dong Y.L. Adrenomedullin enhances invasion by trophoblast cell lines. Biol Reprod. 2005;73:619–626. doi: 10.1095/biolreprod.105.040436. [DOI] [PubMed] [Google Scholar]
- 35.Maynard S., Epstein F.H., Karumanchi S.A. Preeclampsia and angiogenic imbalance. Annu Rev Med. 2008;59:61–78. doi: 10.1146/annurev.med.59.110106.214058. [DOI] [PubMed] [Google Scholar]
- 36.Semenza G.L. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology (Bethesda) 2009;24:97–106. doi: 10.1152/physiol.00045.2008. [DOI] [PubMed] [Google Scholar]
- 37.Wang G.L., Jiang B.H., Rue E.A., Semenza G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaelin W.G., Jr., Ratcliffe P.J. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 39.Adelman D.M., Gertsenstein M., Nagy A., Simon M.C., Maltepe E. Placental cell fates are regulated in vivo by HIF-mediated hypoxia responses. Genes Dev. 2000;14:3191–3203. doi: 10.1101/gad.853700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maltepe E., Krampitz G.W., Okazaki K.M., Red-Horse K., Mak W., Simon M.C., Fisher S.J. Hypoxia-inducible factor-dependent histone deacetylase activity determines stem cell fate in the placenta. Development. 2005;132:3393–3403. doi: 10.1242/dev.01923. [DOI] [PubMed] [Google Scholar]
- 41.Cowden Dahl K.D., Fryer B.H., Mack F.A., Compernolle V., Maltepe E., Adelman D.M., Carmeliet P., Simon M.C. Hypoxia-inducible factors 1alpha and 2alpha regulate trophoblast differentiation. Mol Cell Biol. 2005;25:10479–10491. doi: 10.1128/MCB.25.23.10479-10491.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takeda K., Ho V.C., Takeda H., Duan L.J., Nagy A., Fong G.H. Placental but not heart defects are associated with elevated hypoxia-inducible factor alpha levels in mice lacking prolyl hydroxylase domain protein 2. Mol Cell Biol. 2006;26:8336–8346. doi: 10.1128/MCB.00425-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gnarra J.R., Ward J.M., Porter F.D., Wagner J.R., Devor D.E., Grinberg A., Emmert-Buck M.R., Westphal H., Klausner R.D., Linehan W.M. Defective placental vasculogenesis causes embryonic lethality in VHL-deficient mice. Proc Natl Acad Sci U S A. 1997;94:9102–9107. doi: 10.1073/pnas.94.17.9102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cowden Dahl K.D., Robertson S.E., Weaver V.M., Simon M.C. Hypoxia-inducible factor regulates alphavbeta3 integrin cell surface expression. Mol Biol Cell. 2005;16:1901–1912. doi: 10.1091/mbc.E04-12-1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Genbacev O., Joslin R., Damsky C.H., Polliotti B.M., Fisher S.J. Hypoxia alters early gestation human cytotrophoblast differentiation/invasion in vitro and models the placental defects that occur in preeclampsia. J Clin Invest. 1996;97:540–550. doi: 10.1172/JCI118447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Genbacev O., Zhou Y., Ludlow J.W., Fisher S.J. Regulation of human placental development by oxygen tension. Science. 1997;277:1669–1672. doi: 10.1126/science.277.5332.1669. [DOI] [PubMed] [Google Scholar]
- 47.Caniggia I., Mostachfi H., Winter J., Gassmann M., Lye S.J., Kuliszewski M., Post M. Hypoxia-inducible factor-1 mediates the biological effects of oxygen on human trophoblast differentiation through TGFbeta(3) J Clin Invest. 2000;105:577–587. doi: 10.1172/JCI8316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu C.J., Wang L.Y., Chodosh L.A., Keith B., Simon M.C. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol. 2003;23:9361–9374. doi: 10.1128/MCB.23.24.9361-9374.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCaig D., Lyall F. Hypoxia upregulates GCM1 in human placenta explants. Hypertens Pregnancy. 2009;28:457–472. doi: 10.3109/10641950802629691. [DOI] [PubMed] [Google Scholar]
- 50.Kudo Y., Boyd C.A., Sargent I.L., Redman C.W. Hypoxia alters expression and function of syncytin and its receptor during trophoblast cell fusion of human placental BeWo cells: implications for impaired trophoblast syncytialisation in pre-eclampsia. Biochim Biophys Acta. 2003;1638:63–71. doi: 10.1016/s0925-4439(03)00043-7. [DOI] [PubMed] [Google Scholar]
- 51.Maltepe E., Schmidt J.V., Baunoch D., Bradfield C.A., Simon M.C. Abnormal angiogenesis and responses to glucose and oxygen deprivation in mice lacking the protein ARNT. Nature. 1997;386:403–407. doi: 10.1038/386403a0. [DOI] [PubMed] [Google Scholar]
- 52.Mabjeesh N.J., Escuin D., LaVallee T.M., Pribluda V.S., Swartz G.M., Johnson M.S., Willard M.T., Zhong H., Simons J.W., Giannakakou P. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell. 2003;3:363–375. doi: 10.1016/s1535-6108(03)00077-1. [DOI] [PubMed] [Google Scholar]
- 53.Kanasaki K., Palmsten K., Sugimoto H., Ahmad S., Hamano Y., Xie L., Parry S., Augustin H.G., Gattone V.H., Folkman J., Strauss J.F., Kalluri R. Deficiency in catechol-O-methyltransferase and 2-methoxyoestradiol is associated with pre-eclampsia. Nature. 2008;453:1117–1121. doi: 10.1038/nature06951. [DOI] [PubMed] [Google Scholar]
- 54.Choi H.J., Sanders T.A., Tormos K.V., Ameri K., Tsai J.D., Park A.M., Gonzalez J., Rajah A.M., Liu X., Quinonez D.M., Rinaudo P.F., Maltepe E. ECM-dependent HIF induction directs trophoblast stem cell fate via LIMK1-mediated cytoskeletal rearrangement. PLoS One. 2013;8:e56949. doi: 10.1371/journal.pone.0056949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Arber S., Barbayannis F.A., Hanser H., Schneider C., Stanyon C.A., Bernard O., Caroni P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393:805–809. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- 56.Scott R.W., Olson M.F. LIM kinases: function, regulation and association with human disease. J Mol Med (Berl) 2007;85:555–568. doi: 10.1007/s00109-007-0165-6. [DOI] [PubMed] [Google Scholar]
- 57.Parast M.M., Aeder S., Sutherland A.E. Trophoblast giant-cell differentiation involves changes in cytoskeleton and cell motility. Dev Biol. 2001;230:43–60. doi: 10.1006/dbio.2000.0102. [DOI] [PubMed] [Google Scholar]
- 58.Shibukawa Y., Yamazaki N., Kumasawa K., Daimon E., Tajiri M., Okada Y., Ikawa M., Wada Y. Calponin 3 regulates actin cytoskeleton rearrangement in trophoblastic cell fusion. Mol Biol Cell. 2010;21:3973–3984. doi: 10.1091/mbc.E10-03-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scott R.W., Hooper S., Crighton D., Li A., Konig I., Munro J., Trivier E., Wickman G., Morin P., Croft D.R., Dawson J., Machesky L., Anderson K.I., Sahai E.A., Olson M.F. LIM kinases are required for invasive path generation by tumor and tumor-associated stromal cells. J Cell Biol. 2010;191:169–185. doi: 10.1083/jcb.201002041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Genbacev O., Krtolica A., Kaelin W., Fisher S.J. Human cytotrophoblast expression of the von Hippel-Lindau protein is downregulated during uterine invasion in situ and upregulated by hypoxia in vitro. Dev Biol. 2001;233:526–536. doi: 10.1006/dbio.2001.0231. [DOI] [PubMed] [Google Scholar]
- 61.ACOG practice bulletin. Diagnosis and management of preeclampsia and eclampsia. Number 33, January 2002. Obstet Gynecol. 2002;99:159–167. doi: 10.1016/s0029-7844(01)01747-1. [DOI] [PubMed] [Google Scholar]
- 62.Sibai B.M., Frangieh A.Y. Management of severe preeclampsia. Curr Opin Obstet Gynecol. 1996;8:110–113. [PubMed] [Google Scholar]
- 63.Dioum E.M., Chen R., Alexander M.S., Zhang Q., Hogg R.T., Gerard R.D., Garcia J.A. Regulation of hypoxia-inducible factor 2alpha signaling by the stress-responsive deacetylase sirtuin 1. Science. 2009;324:1289–1293. doi: 10.1126/science.1169956. [DOI] [PubMed] [Google Scholar]
- 64.Haberland M., Montgomery R.L., Olson E.N. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ross-Macdonald P., de Silva H., Guo Q., Xiao H., Hung C.Y., Penhallow B., Markwalder J., He L., Attar R.M., Lin T.A., Seitz S., Tilford C., Wardwell-Swanson J., Jackson D. Identification of a nonkinase target mediating cytotoxicity of novel kinase inhibitors. Mol Cancer Ther. 2008;7:3490–3498. doi: 10.1158/1535-7163.MCT-08-0826. [DOI] [PubMed] [Google Scholar]
- 66.Gustafsson M.V., Zheng X., Pereira T., Gradin K., Jin S., Lundkvist J., Ruas J.L., Poellinger L., Lendahl U., Bondesson M. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell. 2005;9:617–628. doi: 10.1016/j.devcel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 67.Mukherjee T., Kim W.S., Mandal L., Banerjee U. Interaction between Notch and Hif-alpha in development and survival of Drosophila blood cells. Science. 2011;332:1210–1213. doi: 10.1126/science.1199643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huang L.E. Carrot and stick: HIF-alpha engages c-Myc in hypoxic adaptation. Cell Death Differ. 2008;15:672–677. doi: 10.1038/sj.cdd.4402302. [DOI] [PubMed] [Google Scholar]
- 69.Singh H., Aplin J.D. Adhesion molecules in endometrial epithelium: tissue integrity and embryo implantation. J Anat. 2009;215:3–13. doi: 10.1111/j.1469-7580.2008.01034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fitzgerald J.S., Germeyer A., Huppertz B., Jeschke U., Knofler M., Moser G., Scholz C., Sonderegger S., Toth B., Markert U.R. Governing the invasive trophoblast: current aspects on intra- and extracellular regulation. Am J Reprod Immunol. 2010;63:492–505. doi: 10.1111/j.1600-0897.2010.00824.x. [DOI] [PubMed] [Google Scholar]
- 71.Lim H.J., Wang H. Uterine disorders and pregnancy complications: insights from mouse models. J Clin Invest. 2010;120:1004–1015. doi: 10.1172/JCI41210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cockle J.V., Gopichandran N., Walker J.J., Levene M.I., Orsi N.M. Matrix metalloproteinases and their tissue inhibitors in preterm perinatal complications. Reprod Sci. 2007;14:629–645. doi: 10.1177/1933719107304563. [DOI] [PubMed] [Google Scholar]
- 73.Morgan M., Kniss D., McDonnell S. Expression of metalloproteinases and their inhibitors in human trophoblast continuous cell lines. Exp Cell Res. 1998;242:18–26. doi: 10.1006/excr.1997.3929. [DOI] [PubMed] [Google Scholar]
- 74.Seval Y., Akkoyunlu G., Demir R., Asar M. Distribution patterns of matrix metalloproteinase (MMP)-2 and -9 and their inhibitors (TIMP-1 and TIMP-2) in the human decidua during early pregnancy. Acta Histochem. 2004;106:353–362. doi: 10.1016/j.acthis.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 75.Xu P., Wang Y.L., Zhu S.J., Luo S.Y., Piao Y.S., Zhuang L.Z. Expression of matrix metalloproteinase-2, -9, and -14, tissue inhibitors of metalloproteinase-1, and matrix proteins in human placenta during the first trimester. Biol Reprod. 2000;62:988–994. doi: 10.1095/biolreprod62.4.988. [DOI] [PubMed] [Google Scholar]
- 76.Wang H., Li Q., Shao L., Zhu C. Expression of matrix metalloproteinase-2, -9, -14, and tissue inhibitors of metalloproteinase-1, -2, -3 in the endometrium and placenta of rhesus monkey (Macaca mulatta) during early pregnancy. Biol Reprod. 2001;65:31–40. doi: 10.1095/biolreprod65.1.31. [DOI] [PubMed] [Google Scholar]
- 77.Wang H., Wen Y., Mooney S., Li H., Behr B., Polan M.L. Matrix metalloproteinase and tissue inhibitor of matrix metalloproteinase expression in human preimplantation embryos. Fertil Steril. 2003;80 Suppl 2:736–742. doi: 10.1016/s0015-0282(03)00782-9. [DOI] [PubMed] [Google Scholar]
- 78.Isaka K., Usuda S., Ito H., Sagawa Y., Nakamura H., Nishi H., Suzuki Y., Li Y.F., Takayama M. Expression and activity of matrix metalloproteinase 2 and 9 in human trophoblasts. Placenta. 2003;24:53–64. doi: 10.1053/plac.2002.0867. [DOI] [PubMed] [Google Scholar]
- 79.Cetin I., Huppertz B., Burton G., Cuckle H., Gonen R., Lapaire O., Mandia L., Nicolaides K., Redman C., Soothill P., Spencer K., Thilaganathan B., Williams D., Meiri H. Pregenesys pre-eclampsia markers consensus meeting: what do we require from markers, risk assessment and model systems to tailor preventive strategies? Placenta. 2011;32 Suppl:S4–S16. doi: 10.1016/j.placenta.2010.11.022. [DOI] [PubMed] [Google Scholar]
- 80.Nawrocki B., Polette M., Marchand V., Maquoi E., Beorchia A., Tournier J.M., Foidart J.M., Birembaut P. Membrane-type matrix metalloproteinase-1 expression at the site of human placentation. Placenta. 1996;17:565–572. doi: 10.1016/s0143-4004(96)80073-7. [DOI] [PubMed] [Google Scholar]
- 81.Fisher S.J., Leitch M.S., Kantor M.S., Basbaum C.B., Kramer R.H. Degradation of extracellular matrix by the trophoblastic cells of first-trimester human placentas. J Cell Biochem. 1985;27:31–41. doi: 10.1002/jcb.240270105. [DOI] [PubMed] [Google Scholar]
- 82.Librach C.L., Werb Z., Fitzgerald M.L., Chiu K., Corwin N.M., Esteves R.A., Grobelny D., Galardy R., Damsky C.H., Fisher S.J. 92-kD type IV collagenase mediates invasion of human cytotrophoblasts. J Cell Biol. 1991;113:437–449. doi: 10.1083/jcb.113.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Estelles A., Gilabert J., Aznar J., Loskutoff D.J., Schleef R.R. Changes in the plasma levels of type 1 and type 2 plasminogen activator inhibitors in normal pregnancy and in patients with severe preeclampsia. Blood. 1989;74:1332–1338. [PubMed] [Google Scholar]
- 84.Estelles A., Gilabert J., Keeton M., Eguchi Y., Aznar J., Grancha S., Espna F., Loskutoff D.J., Schleef R.R. Altered expression of plasminogen activator inhibitor type 1 in placentas from pregnant women with preeclampsia and/or intrauterine fetal growth retardation. Blood. 1994;84:143–150. [PubMed] [Google Scholar]
- 85.Graham C.H., McCrae K.R. Altered expression of gelatinase and surface-associated plasminogen activator activity by trophoblast cells isolated from placentas of preeclamptic patients. Am J Obstet Gynecol. 1996;175:555–562. doi: 10.1053/ob.1996.v175.a74404. [DOI] [PubMed] [Google Scholar]
- 86.Autio-Harmainen H., Hurskainen T., Niskasaari K., Hoyhtya M., Tryggvason K. Simultaneous expression of 70 kilodalton type IV collagenase and type IV collagen alpha 1 (IV) chain genes by cells of early human placenta and gestational endometrium. Lab Invest. 1992;67:191–200. [PubMed] [Google Scholar]
- 87.Hurskainen T., Hoyhtya M., Tuuttila A., Oikarinen A., Autio-Harmainen H. mRNA expressions of TIMP-1, -2, and -3 and 92-KD type IV collagenase in early human placenta and decidual membrane as studied by in situ hybridization. J Histochem Cytochem. 1996;44:1379–1388. doi: 10.1177/44.12.8985130. [DOI] [PubMed] [Google Scholar]
- 88.Hiden U., Ghaffari-Tabrizi N., Gauster M., Tam-Amersdorfer C., Cetin I., Dieber-Rotheneder M., Lang U., Desoye G. Membrane-type matrix metalloproteinase 1 regulates trophoblast functions and is reduced in fetal growth restriction. Am J Pathol. 2013;182:1563–1571. doi: 10.1016/j.ajpath.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 89.Plaks V., Rinkenberger J., Dai J., Flannery M., Sund M., Kanasaki K., Ni W., Kalluri R., Werb Z. Matrix metalloproteinase-9 deficiency phenocopies features of preeclampsia and intrauterine growth restriction. Proc Natl Acad Sci U S A. 2013;110:11109–11114. doi: 10.1073/pnas.1309561110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Acevedo K., Li R., Soo P., Suryadinata R., Sarcevic B., Valova V.A., Graham M.E., Robinson P.J., Bernard O. The phosphorylation of p25/TPPP by LIM kinase 1 inhibits its ability to assemble microtubules. Exp Cell Res. 2007;313:4091–4106. doi: 10.1016/j.yexcr.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 91.Tapia T., Ottman R., Chakrabarti R. LIM kinase1 modulates function of membrane type matrix metalloproteinase 1: implication in invasion of prostate cancer cells. Mol Cancer. 2011;10:6. doi: 10.1186/1476-4598-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gauster M., Siwetz M., Orendi K., Moser G., Desoye G., Huppertz B. Caspases rather than calpains mediate remodelling of the fodrin skeleton during human placental trophoblast fusion. Cell Death Differ. 2010;17:336–345. doi: 10.1038/cdd.2009.133. [DOI] [PubMed] [Google Scholar]
- 93.Yoshie M., Kashima H., Bessho T., Takeichi M., Isaka K., Tamura K. Expression of stathmin, a microtubule regulatory protein, is associated with the migration and differentiation of cultured early trophoblasts. Hum Reprod. 2008;23:2766–2774. doi: 10.1093/humrep/den317. [DOI] [PubMed] [Google Scholar]
- 94.Escuin D., Kline E.R., Giannakakou P. Both microtubule-stabilizing and microtubule-destabilizing drugs inhibit hypoxia-inducible factor-1alpha accumulation and activity by disrupting microtubule function. Cancer Res. 2005;65:9021–9028. doi: 10.1158/0008-5472.CAN-04-4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rolfo A., Many A., Racano A., Tal R., Tagliaferro A., Ietta F., Wang J., Post M., Caniggia I. Abnormalities in oxygen sensing define early and late onset preeclampsia as distinct pathologies. PLoS One. 2010;5:e13288. doi: 10.1371/journal.pone.0013288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Caniggia I., Winter J., Lye S.J., Post M. Oxygen and placental development during the first trimester: implications for the pathophysiology of pre-eclampsia. Placenta. 2000;21 Suppl A:S25–S30. doi: 10.1053/plac.1999.0522. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Antibody staining controls. Adjacent sections to those shown in Figure 2 were stained with appropriate secondary antibody alone as shown and imaged in identical fashion. Note the very low-level background staining observed. A and B: The staining pattern observed in 6 weeks placenta (A) and 23 weeks placenta (B). FV, floating villi.
Immunofluorescence microscopy-based analysis of MAPK2K1/2 activity and cytoskeletal integrity in the human placenta. Placentas (19-week-old) were stained with antibodies for phospho-MAPK3/1 (A and B), or α-tubulin (α-Tub) (C and D). All slides were also stained with DAPI to identify nuclei. Expression in floating villi (FV) (A and C) is contrasted with invasive cytotrophoblast (iCTB) expression patterns (B and D). Although villous cytotrophoblasts (arrows), as well as populations of stromal cells within floating villi, show strong activation of MAPK3/1 (A) and a robust microtubule cytoskeleton (C), the overlying syncytiotrophoblasts (SynT) layer (arrowheads) does not. SynTs exhibit strong inhibition of MAPK3/1 phosphorylation, and fail to stain positively for α-tubulin. B and D: Invasive CTBs on the other hand, show strong immunoreactivity with each of these antibodies.
Regulation of HIF-2α transcriptional activity via histone deacetylase (HDAC) activity. Whole-cell lysates from Hif-1/2α−/− mouse trophoblast stem cells (TSCs) stably expressing either green fluorescent protein or HIF-2α, and either treated with or without 1.5 mmol/L inhibitors of class I/II HDAC activity [sodium butyrate (NaBu)] or 60 μmol/L class III HDAC/sirtuin inhibitor (sirtinol), were subjected to immunoblot analysis with antibodies specific for LIMK1, HIF-2α, or actin. Note the decrease of LIMK1 immunoreactivity with both inhibitor treatments. Note also, however, inhibition of HIF-2α stability only with NaBu treatment, but without a significant reduction of HIF-2α stability following sirtinol treatment. Sirtinol treatment results in a slight increase in HIF-2α molecular weight, evidenced by its slightly decreased mobility, consistent with its increased acetylation status following sirtuin inhibition.
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining of BMS-5–treated versus control dimethyl sulfoxide (DMSO)-treated cytotrophoblasts (CTBs) in vitro. Following standard TUNEL immunolabeling, numbers of TUNEL-positive (apoptotic) CTBs were counted. BMS-5 treatment (10 μmol/L) did not increase apoptotic cell death of CTBs cultured in vitro.
Cytotrophoblast (CTB) invasion assay. BMS-5 treatment (10 μmol/L) impairs second trimester CTB invasion in a Matrigel-based invasion assay. ∗∗P < 0.01.