

Abstract
Capillary malformation–arteriovenous malformation (CM-AVM) is an autosomal dominant blood vascular (BV) disorder characterized by CM and fast flow BV lesions. Inactivating mutations of the RASA1 gene are the cause of CM-AVM in most cases. RASA1 is a GTPase-activating protein that acts as a negative regulator of the Ras small GTP-binding protein. In addition, RASA1 performs Ras-independent functions in intracellular signal transduction. Whether CM-AVM results from loss of an ability of RASA1 to regulate Ras or loss of a Ras-independent function of RASA1 is unknown. To address this, we generated Rasa1 knockin mice with an R780Q point mutation that abrogates RASA1 catalytic activity specifically. Homozygous Rasa1R780Q/R780Q mice showed the same severe BV abnormalities as Rasa1-null mice and died midgestation. This finding indicates that BV abnormalities in CM-AVM develop as a result of loss of an ability of RASA1 to control Ras activation and not loss of a Ras-independent function of this molecule. More important, findings indicate that inhibition of Ras signaling is likely to represent an effective means of therapy for this disease.
Capillary malformation–arteriovenous malformation (CM-AVM) is an autosomal dominant blood vascular (BV) disorder that affects at least 1:100,000 individuals in Northern European populations.1–3 The pathognomonic feature of CM-AVM is the presence of one or more randomly distributed cutaneous CMs. In one third of patients, there are additional fast flow lesions that include AVM, arteriovenous fistulas, and Parkes-Weber syndrome. Complications of CM-AVM include hemorrhage, epilepsy, hydrocephalus, cardiac failure, and death. Inactivating mutations of the RASA1 gene, which encodes the p120 Ras GTPase-activating protein (p120 Ras-GAP or RASA1), are the cause of CM-AVM in most patients. Mutations are distributed throughout the length of the gene, and most are nonsense mutations, insertions, and deletions resulting in frameshifts or disruption of splice sites.1–9 Although not proved, it is likely that all of these mutations result in complete null alleles because transcripts would be rapidly degraded by nonsense-mediated RNA decay.10 Only one germline RASA1 allele is affected in CM-AVM, and it has been postulated that somatic second hit inactivating mutations in the inherited normal RASA1 allele are required for lesion development.2
As a Ras-GAP, one recognized function of RASA1 is to inactivate the Ras small GTP-binding protein.11 Ras is an intracellular membrane-tethered signaling protein that is converted from an inactive GDP-bound to an active GTP-bound state in response to growth factor stimulation.12 In its active state, Ras stimulates several different downstream enzymatic cascades that include the mitogen-activated protein kinase (MAPK) cascade and the phosphatidylinositol 3-kinase (PI3K) signaling pathway that drive cell growth, proliferation, differentiation, and survival.13–15 Ras-GAPs inactivate Ras by inserting an arginine finger, located in a GAP domain, into the Ras catalytic site, thereby increasing the ability of Ras to hydrolyze bound GTP to GDP by several orders of magnitude.16 However, RASA1 is also able to participate in growth factor receptor signal transduction independent of an ability to inactivate Ras.11
Consistent with the BV lesions in patients with CM-AVM, mice that are homozygous for a null allele of Rasa1 die by embryonic day 10.5 (E10.5) of gestation as a result of abnormal BV development.17 At E9.5, endothelial cells (ECs) in the yolk sac are seen to assemble into a honeycombed pattern but then fail to reorganize into a vascular network that supplies blood to the embryo. In the embryo proper at E9.5, a narrow and irregular dorsal aorta is observed with abnormal projecting arteries. Local rupture of blood vessels and a distended pericardial sac are also apparent. The same phenotype is observed in conditional RASA1-deficient mice in which disruption of the Rasa1 gene is restricted to ECs.18
It is unknown if dysregulated activation of the Ras pathway or loss of a Ras-independent function of RASA1 in ECs is responsible for the development of BV abnormalities in RASA1-deficient mice and humans. Therefore, to address this question, in the current studies, we generated a Rasa1R780Q knockin mouse that expresses a form of RASA1 that lacks an arginine finger. Previous studies have established that the R780Q mutation abrogates an ability of RASA1 to promote Ras hydrolysis of GTP.19 BV development in homozygous Rasa1R780Q mice was assessed.
Materials and Methods
Gene Targeting
A Rasa1R780Q targeting construct was assembled in p-loxP-2FRT-PGKneo.20 A 5′ arm that spanned introns 13 through 17 of the Rasa1 gene was generated by PCR from a C57BL/6 genomic Rasa1 BAC clone and inserted into the EcoRI/KpnI sites of the vector. The primers used were as follows: 5′ arm forward, 5′-GCGCGAATTCGCGGCCGCTTAGTCTTTCAGGCATTTTATAGC-3′; and 5′ arm reverse, 5′-GCGCGGTACCGAATGCTTATTTACCAGGAGTGAC-3′. A middle segment spanning intron 17 through 18 was generated by PCR from the same Rasa1 BAC clone that contained an R780Q mutation in exon 18 that was generated by homologous recombination in Escherichia coli (CGA to CAA codon change). The middle fragment was inserted into the KpnI and 3′ BglII sites of the vector. The primers used were as follows: M forward, 5′-GCGCGGTACCAGATCTAAATATTTGAGCCTATGAGGACCATTC-3′; and M reverse, 5′-GCGCGGATCCCATATCCAACTTCACATGATGTGC-3′. A 3′ arm that spanned introns 18 through 20 was generated by PCR from the wild-type Rasa1 BAC clone and was inserted into the XhoI site of the vector. The primers used were as follows: 3′ arm forward, 5′-GCGCCTCGAGGAATTTCCCACATGGA-3′; and 3′ arm reverse, 5′-GCGCCTCGAGATATGTTGTCATGTAA-3′. The targeting construct was sequenced to verify the absence of any PCR-induced errors. The construct was electroporated into W4 embryonic stem (ES) cells that were subsequently cultured in neomycin. Genomic DNA from neomycin-resistant ES cell clones was analyzed by quantitative real-time PCR using an integration primer/probe set in which the 5′ and 3′ primers flanked the FRT-NeoR-FRT insertion site and the probe was complimentary to regions both 5′ and 3′ to the point of insertion.21 Fold differences in the amount of exon 18 target relative to that in genomic DNA prepared from wild-type W4 ES cells were calculated as described.22,23 Clones that demonstrated a twofold reduction in the intron 18 target were analyzed further by quantitative real-time PCR using a copy number primer/probe set based within exon 14 of the Rasa1 gene (Life Technologies, Carlsbad, CA). Of those clones that contained diploid amounts of the exon 14 target, a long-range PCR was performed using a Rasa1 forward primer based on the 5′ of the 5′ end of the Rasa1 targeting vector insert and a reverse primer based in intron 18. A similar long-range PCR was performed using a forward primer based in intron 18 and a reverse primer located 3′ of the 3′ end of the Rasa1 targeting vector insert. Both PCR products were sequenced to confirm the presence of the R780Q mutation within the endogenous Rasa1 locus.
Two of several correctly targeted Rasa1R780Q clones were injected into C57BL/6J X (C57BL/6J X DBA/2) blastocysts to generate chimeric mice that were then bred with C57BL/6 actin promoter-driven Flp transgenic mice to achieve germline transmission of the targeted Rasa1R780Q allele and deletion of the NeoR cassette in intron 18.24 Germline transmission and NeoR deletion were determined by PCR of tail genomic DNA prepared from agouti coat-colored progeny using a forward primer based in exon 18 and a reverse primer based 3′ to the 3′ FRT site in intron 18. Germline transmitted mice were then crossed with C57BL/6 mice and the same PCR was performed on tail DNA of progeny that had not inherited the actin-Flp transgene, also determined by PCR. This step was undertaken to obtain mice with confirmed germline deletion of the NeoR cassette within the Rasa1R780Q locus.
Embryonic Lethality
Rasa1R780Q/+ mice were crossed with Rasa1fl/fl mice with an inserted loxP sequence in intron 18.25 Rasa1R780Q/fl progeny were identified by PCR of tail genomic DNA using the same primers used above to detect deletion of the NeoR cassette. The expected PCR product sizes from the Rasa1fl and Rasa1R780Q alleles are 685 and 604 bp, respectively. Rasa1R780Q/fl mice were intercrossed, and Rasa1 genotypes of embryonic and 3-week-old pups were determined by PCR of yolk sac and tail genomic DNA, respectively, as described above. A χ2 test was used to determine the probability that the frequency of observed genotypes of pups and embryos were consistent with a mendelian pattern of inheritance. Embryo images were acquired on an Olympus SZ61 dissecting microscope equipped with a digital camera (Lumenera, Ottawa, ON). All experiments performed on mice were in compliance with the University of Michigan (Ann Arbor, MI) guidelines and were approved by the University Committee on the Use and Care of Animals.
Western Blot Analysis
Individual E9.5 embryos were minced and digested in trypsin/EDTA (Life Technologies) at 37°C for 15 minutes. Murine embryonic fibroblasts (MEFs) were generated by culture of released cells in fibroblast media (Dulbecco’s modified Eagle’s medium, 10% fetal bovine serum, 1% penicillin/streptomycin, and 1% l-glutamine). At confluence, MEFs were harvested and 3 × 106 cells were lysed in 1% NP-40 lysis buffer. RASA1 protein abundance was determined by using Western blot analysis of cell lysates using an anti-RASA1 antibody (B4F8; Santa Cruz, Dallas, TX). Blots were stripped and reprobed with an anti–glyceraldehyde-3-phosphate dehydrogenase antibody (Santa Cruz) to ascertain equivalent protein loading.
Whole Mount Staining
E9.5 embryos were fixed in 1% paraformaldehyde in phosphate-buffered saline. Fixed embryos were stained with a rat anti-CD31 antibody (BD Biosciences, San Jose, CA), followed by a secondary goat anti-rat Ig Alexa Fluor 488 antibody (Invitrogen, Carlsbad, CA). Images of embryos were acquired on an Olympus BX60 upright fluorescence microscope equipped with a digital camera (Nikon, Tokyo, Japan).
Immunohistological Data
E9.5 embryos were fixed in formalin and embedded in paraffin. Sections (5 μm thick) were stained with a rabbit anti–phospho-extracellular signal regulated kinase (ERK) antibody (D13.14.4E; Cell Signaling, Danvers, MA), followed by biotinylated anti-rabbit Ig (Jackson Immunoresearch, West Grove, PA) and streptavidin–horseradish peroxidase with tyramide signal amplification (Perkin Elmer, Waltham, MA). Sections were then stained with a rat anti-CD31 antibody (sz31; Dianova, Hamburg, Germany), followed by secondary goat anti-rat Alexa Fluor 594 (Invitrogen). Some sections were stained with CD31 and secondary antibody alone or with hematoxylin and eosin. Images of sections were acquired as described in Whole Mount Staining.
Results
Embryonic Lethality of Homozygous Rasa1R780Q Mice
A murine Rasa1R780Q allele with an inserted NeoR cassette in intron 18 was generated by homologous recombination in ES cells (Figure 1). Of several correctly targeted euploid ES clones, two were injected into blastocysts. Chimeric mice derived from both clones were bred with transgenic mice that express the Flp recombinase under control of the actin promoter to achieve simultaneous germline transmission of the targeted Rasa1 allele and deletion of the NeoR cassette (Figure 1B). Like heterozygous Rasa1-null mice,17 heterozygous Rasa1R780Q mice are healthy and do not show any BV abnormalities.
Figure 1.

Generation of Rasa1R780Q knockin allele in mice. A: Exon-intron organization of the murine Rasa1 gene (top) and RASA1 protein (bottom). The positions of the R780Q and R780Q mutations in exon 18 and the GAP domain, respectively, are indicated. B: An R780Q targeting vector with an R780Q mutation in exon 18 and a FRT-flanked NeoR cassette in intron 18 is shown below the endogenous allele. At middle is shown the R780Q targeted allele before and after excision of the NeoR cassette achieved with an actin promoter-driven Flp transgene (Act-Flp). At bottom is depicted a Rasa1 floxed allele in which exon 18 is flanked by loxP sites. Positions of real-time PCR primer/probe pairs used in the detection of ES cell homologous recombinants and PCR primer pairs used in mouse genotyping are indicated. C2, protein kinase C2 homology; GAP, GTPase-activating protein domain; PH, pleckstrin homology; SH2, Src homology 2; SH3, Src homology 3.
Heterozygous Rasa1R780Q mice were crossed to mice with a conditional Rasa1 allele in which exon 18 is flanked by loxP sites [floxed allele (fl)] (Figure 1B) to generate heterozygous Rasa1R780Q/fl mice.25 The Rasa1R780Q/fl mice were then intercrossed, and genotypes of 3-week-old progeny were determined by PCR of tail genomic DNA using forward and reverse primers based in exon 18 and intron 18, respectively, that readily distinguish between the R780Q and floxed alleles (Figures 1B and 2A). In these experiments, no homozygous Rasa1R780Q/R780Q pups were identified using parental mice derived from either R780Q targeted ES cell line (Figure 2B). This is consistent with embryonic lethality of the Rasa1R780Q mutation in homozygous form.
Figure 2.

Embryonic lethality of homozygous Rasa1R780Q mice. A:Rasa1R780Q/fl mice were intercrossed. Tail genomic DNA from progeny at 3 weeks of age was PCR amplified using genotyping primers indicated in Figure 1. Genotyping results from two Rasa1R780Q/fl (fl/rq) pups and one Rasa1fl/fl (fl/fl) pup are depicted. B: Graphs show the total number of pups of the indicated genotypes derived from intercrosses of Rasa1R780Q/fl mice determined at 3 weeks after birth and at E9.5, E11, and E13.5 of development. At E9.5, genotype frequencies are consistent with mendelian inheritance (χ2 test). At E11, E13.5, and 3 weeks of age, genotype frequencies are not consistent with mendelian inheritance (P < 0.025, P < 0.05, and P < 0.005, respectively). C and D: Light microscopic appearance (C) and hematoxylin and eosin (H&E)–stained sections (D) of Rasa1R780Q/R780Q and Rasa1R780Q/fl embryos and yolk sacs at E9.5. Features to note include reduced size and distended pericardial sac (arrow) and wrinkled appearance of the yolk sac of Rasa1R780Q/R780Q embryos. E: MEFs derived from E9.5 Rasa1R780Q/R780Q and Rasa1fl/fl embryos were analyzed for RASA1 protein abundance by using Western blot analysis. Blots were reprobed for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) to show equivalent protein loading. Shown are RASA1 amounts in MEF derived from two different embryos of each genotype. E, embryo proper; YS, yolk sac.
To determine the time of embryonic lethality, pregnancies were terminated at defined points after fertilization, embryos were harvested, and genotype was determined by PCR of yolk sacs (Figure 2B). At E9.5, homozygous Rasa1R780Q/R780Q embryos were identified in expected mendelian ratios. In contrast, at E11, a much less than expected number of Rasa1R780Q/R780Q embryos were identified (2 of 35 compared with an expected 9), and at E13.5, Rasa1R780Q/R780Q embryos were absent. These findings are consistent with midgestation lethality of Rasa1R780Q/R780Q mice, similar to that observed in Rasa1-null mice.17,18
At E9.5, Rasa1R780Q/R780Q embryos exhibited the same number of somites (20 to 25) as control Rasa1R780Q/fl and Rasa1fl/fl embryos. However, R780Q/R780Q embryos were considerably smaller than R780Q/fl and fl/fl embryos and appeared less developed (Figure 2, C and D). Other gross features of E9.5 R780Q/R780Q embryos included a distended pericardial sac and wrinkled yolk sac compared with Rasa1R780Q/fl and Rasa1fl/fl embryos (Figure 2C) (data not shown). Both of these features were noted previously in Rasa1-null embryos at E9.5.17 Furthermore, the same features were noted in the two identified Rasa1R780Q/R780Q E11 embryos (data not shown). To verify that midgestation embryonic lethality of Rasa1R780Q/R780Q embryos could be attributed specifically to the effect of the mutation on RASA1 catalytic activity and not any unforeseen influence on RASA1 protein stability, we prepared MEFs from E9.5 embryos and examined RASA1 protein abundance by using Western blot analysis. Amounts of RASA1 protein in R780Q/R780Q MEFs were unchanged compared with fl/fl MEFs (Figure 2E).
BV Abnormalities in Homozygous Rasa1R780Q Mice
To study potential BV abnormalities in Rasa1R780Q/R780Q mice, we performed whole mount staining of E9.5 embryos using an anti-CD31 antibody to detect ECs (Figure 3A). In yolk sacs of Rasa1fl/fl and Rasa1R780Q/fl mice, organized BV networks that were continuous with the vasculature of the embryo proper were readily identified. In contrast, no such organized vascular networks were observed in the yolk sacs of Rasa1R780Q/R780Q mice. Instead, ECs were often identified in a honeycombed pattern, as described previously in homozygous Rasa1-null mice.17 In the embryo proper of Rasa1fl/fl and Rasa1R780Q/fl mice, a normal dorsal aorta and intersegmental arteries that projected between somites were apparent. In contrast, the BV network of R780Q/R780Q embryos was abnormal (Figure 3A). In particular, the dorsal aorta showed a highly irregular shape. Furthermore, intersegmental arteries were frequently irregular in shape. To confirm these findings, serial sections of embryos were stained with CD31 antibodies to identify the dorsal aorta (Figure 3B). In control mice, the dorsal aorta was identified as a continuous structure that extended from the heart to the posterior of the embryo. In contrast, in R780Q/R780Q embryos, the dorsal aorta was severely disrupted. Similar abnormalities in BV architecture were noted previously in E9.5 homozygous Rasa1-null embryos.17
Figure 3.

Cardiovascular abnormalities in homozygous Rasa1R780Q mice. A and B: Whole mount (A) and sections (B) of E9.5 Rasa1R780Q/R780Q and Rasa1fl/fl embryos were stained with an anti-CD31 antibody to reveal blood vasculature. A: Note the organized BV network in the Rasa1fl/fl yolk sac (YS) and honeycombed appearance of blood vasculature in the Rasa1R780Q/R780Q yolk sac (asterisk). Note also irregular blood vasculature in the Rasa1R780Q/R780Q embryo proper (E). B: Note the disorganized dorsal aorta in the Rasa1R780Q/R780Q embryo compared with the Rasa1fl/fl embryo (arrows). DA, dorsal aorta; IS, intersegmental artery.
Constitutive Activation of the Ras Signaling Pathway in ECs of Homozygous Rasa1R780Q Mice
In its activated state, Ras triggers several different signaling cascades, including the MAPK pathway.12,26 To confirm that MAPK was abnormally activated in BV of homozygous Rasa1R780Q mice, sections from E9.5 embryos were stained with an antibody specific for phosphorylated activated forms of the ERK MAPK together with an anti-CD31 antibody (Figure 4). In control Rasa1fl/fl and Rasa1fl/R780Q embryos, most ECs were not stained with the anti-phosphorylated ERK (pERK) antibody. In contrast, in Rasa1R780Q/R780Q embryos, most ECs were pERK positive. Thus, MAPKs are constitutively activated in ECs of Rasa1R780Q/R780Q embryos.
Figure 4.
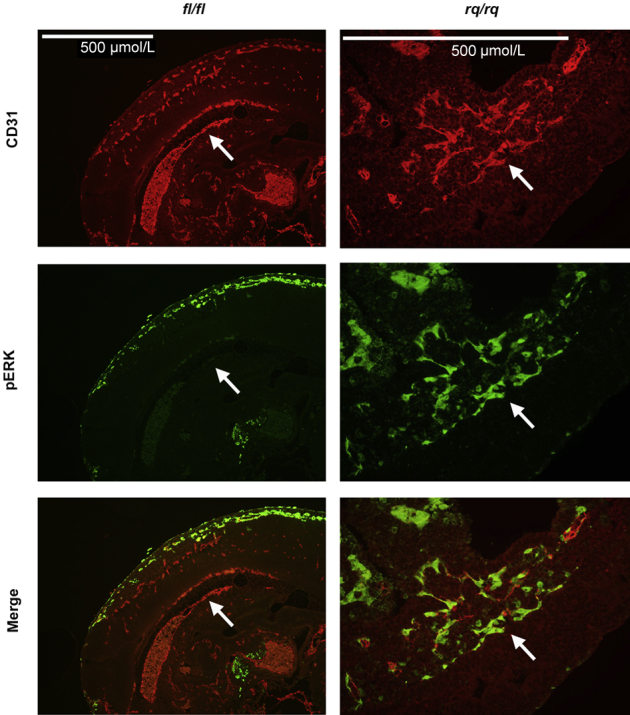
MAPK activation in homozygous Rasa1R780Q embryos. Tissue sections of E9.5 Rasa1R780Q/R780Q and Rasa1fl/fl embryos were stained with anti-CD31 (red) and anti–phospho-ERK (green) antibodies. Shown are select regions of embryos that include part of the dorsal aorta (arrows). Note the absence of pERK staining in most ECs in Rasa1fl/fl embryos and the presence of pERK staining in most ECs in Rasa1R780Q/R780Q embryos.
Discussion
Most RASA1 mutations in CM-AVM result in premature stop codons that are predicted to cause a loss of RASA1 mRNA transcripts and RASA1 protein.2,3 Potentially, therefore, BV abnormalities in these patients could develop as a consequence of loss of an ability of RASA1 to inhibit Ras activation or loss of Ras-independent functions of RASA1. Several Ras-independent functions of RASA1 have been described.11 One Ras-independent function is directed cellular migration that has been observed in fibroblasts and is mediated by RASA1 SH2 domain recognition of the p190 RhoGAP protein that regulates actin cytoskeleton dynamics.27 It has been suggested previously that impaired directed cellular migration of ECs may underlie the pathological abnormalities in CM-AVM.28,29 Of the few RASA1 missense mutations that have been identified in CM-AVM, it is currently unknown how they would affect Ras-dependent or RAS-independent functions of RASA1 or RASA1 protein stability.
To shed light on how loss of RASA1 might lead to BV abnormalities in CM-AVM, we generated Rasa1R780Q knockin mice. Arginine 780 constitutes the arginine finger of the RASA1 GAP domain that is 100% conserved and is essential for RASA1 to promote Ras hydrolysis of bound GTP.11,16 On RASA1 interaction with Ras-GTP, the RASA1 arginine finger is inserted into the Ras active site, which permits glutamine 61 of Ras to participate in catalysis.16 As shown previously, mutation of the arginine finger to a glutamine abrogates an ability of RASA1 to promote Ras-GTP hydrolysis but does not affect overall protein structure.19 Homozygous Rasa1R780Q/R780Q mice showed abnormal BV development in both the yolk sac and embryo proper and died in midgestation at approximately E10 to E11. In these respects, the phenotype of Rasa1R780Q/R780Q mice is identical to that of Rasa1-null mice. These findings, therefore, support the contention that BV abnormalities in CM-AVM result from loss of an ability of RASA1 to regulate Ras in ECs and not from loss of a Ras-independent function of this molecule.
Consistent with the effect of the Rasa1R780Q mutation on RASA1 catalytic activity, MAPK activation was increased in ECs of Rasa1R780Q/R780Q embryos at E9.5 (Figure 4). Thus, most ECs in mutant embryos stained strongly with phospho-MAPK antibodies. In contrast, few ECs in control E9.5 embryos were phospho-MAPK positive, including in the dorsal aorta, as reported previously.30 Notably, increased pERK staining in Rasa1R780Q/R780Q embryos was largely confined to ECs, despite the broad expression of RASA1. This indicates that in non-EC types in these embryos, other Ras-GAPs compensate for the loss of RASA1 catalytic activity in the regulation of Ras-MAPK signaling. In turn, this might explain why phenotypes in mice and humans with Rasa1/RASA1 mutations are relatively restricted to the BV compartment.
Which growth factor receptors use RASA1 to inhibit Ras activation during BV development is uncertain. One recent study in zebrafish has indicated that RASA1 functions downstream of the Ephrin B4 receptor to control BV development, although this has yet to be confirmed in mammals.31 In addition, which downstream effectors of Ras drive BV abnormalities in RASA1-deficient mice and humans is currently unknown. As well as MAPK, PI3K and other downstream effectors of Ras could contribute to the development of lesions. In this regard, PI3K lies upstream of the mechanistic target of rapamycin that has been shown to be hyperactivated in CM-AVM BV.31 Potentially, therefore, inhibitors of any of MAPK, PI3K, or mechanistic target of rapamycin alone or in combination may prove effective in the treatment of BV abnormalities in this condition.
Footnotes
Supported by NIH grant RO1 HL096498 (P.D.K.) and American Heart Association grants 12PRE8850000 and 11POST7580023 (B.A.L. and P.E.L., respectively). The Transgenic Mouse Core was supported in part by the NIH through the University of Michigan's Cancer Center support grant P30 CA046592.
Disclosures: None declared.
References
- 1.Eerola I., Boon L.M., Mulliken J.B., Burrows P.E., Dompmartin A., Watanabe S., Vanwijck R., Vikkula M. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73:1240–1249. doi: 10.1086/379793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Revencu N., Boon L.M., Mulliken J.B., Enjolras O., Cordisco M.R., Burrows P.E., Clapuyt P., Hammer F., Dubois J., Baselga E., Brancati F., Carder R., Quintal J.M., Dallapiccola B., Fischer G., Frieden I.J., Garzon M., Harper J., Johnson-Patel J., Labreze C., Martorell L., Paltiel H.J., Pohl A., Prendiville J., Quere I., Siegel D.H., Valente E.M., Van Hagen A., Van Hest L., Vaux K.K., Vicente A., Weibel L., Chitayat D., Vikkula M. Parkes Weber syndrome, vein of Galen aneurysmal malformation, and other fast-flow vascular anomalies are caused by RASA1 mutations. Hum Mutat. 2008;29:959–965. doi: 10.1002/humu.20746. [DOI] [PubMed] [Google Scholar]
- 3.Revencu N., Boon L.M., Mendola A., Cordisco M.R., Dubois J., Clapuyt P. RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum Mutat. 2013;34:1632–1641. doi: 10.1002/humu.22431. [DOI] [PubMed] [Google Scholar]
- 4.Bayrak-Toydemir P, Stevenson D: RASA1-Related Disorders. Edited by RA Pagon, MP Adam, HH Ardinger, et al. In GeneReviews [Internet]. Copyright University of Washington, Seattle. 2011. Available at http://www.ncbi.nlm.nih.gov/books/NBK52764, last revised December 19, 2013
- 5.de Wijn R.S., Oduber C.E., Breugem C.C., Alders M., Hennekam R.C., van der Horst C.M. Phenotypic variability in a family with capillary malformations caused by a mutation in the RASA1 gene. Eur J Med Genet. 2012;55:191–195. doi: 10.1016/j.ejmg.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 6.Hershkovitz D., Bercovich D., Sprecher E., Lapidot M. RASA1 mutations may cause hereditary capillary malformations without arteriovenous malformations. Br J Dermatol. 2008;158:1035–1040. doi: 10.1111/j.1365-2133.2008.08493.x. [DOI] [PubMed] [Google Scholar]
- 7.Hershkovitz D., Bergman R., Sprecher E. A novel mutation in RASA1 causes capillary malformation and limb enlargement. Arch Dermatol Res. 2008;300:385–388. doi: 10.1007/s00403-008-0842-5. [DOI] [PubMed] [Google Scholar]
- 8.Thiex R., Mulliken J.B., Revencu N., Boon L.M., Burrows P.E., Cordisco M., Dwight Y., Smith E.R., Vikkula M., Orbach D.B. A novel association between RASA1 mutations and spinal arteriovenous anomalies. AJNR Am J Neuroradiol. 2010;31:775–779. doi: 10.3174/ajnr.A1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wooderchak-Donahue W., Stevenson D.A., McDonald J., Grimmer J.F., Gedge F., Bayrak-Toydemir P. RASA1 analysis: clinical and molecular findings in a series of consecutive cases. Eur J Med Genet. 2011;55:91–95. doi: 10.1016/j.ejmg.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 10.Conti E., Izaurralde E. Nonsense-mediated mRNA decay: molecular insights and mechanistic variations across species. Curr Opin Cell Biol. 2005;17:316–325. doi: 10.1016/j.ceb.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 11.King P.D., Lubeck B.A., Lapinski P.E. Nonredundant functions for Ras GTPase-activating proteins in tissue homeostasis. Sci Signal. 2013;6:re1. doi: 10.1126/scisignal.2003669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buday L., Downward J. Many faces of Ras activation. Biochim Biophys Acta. 2008;1786:178–187. doi: 10.1016/j.bbcan.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 13.Castellano E., Downward J. Role of RAS in the regulation of PI 3-kinase. Curr Top Microbiol Immunol. 2010;346:143–169. doi: 10.1007/82_2010_56. [DOI] [PubMed] [Google Scholar]
- 14.Castellano E., Downward J. RAS interaction with PI3K: more than just another effector pathway. Genes Cancer. 2011;2:261–274. doi: 10.1177/1947601911408079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rubinfeld H., Seger R. The ERK cascade: a prototype of MAPK signaling. Mol Biotechnol. 2005;31:151–174. doi: 10.1385/MB:31:2:151. [DOI] [PubMed] [Google Scholar]
- 16.Scheffzek K., Ahmadian M.R., Kabsch W., Wiesmuller L., Lautwein A., Schmitz F., Wittinghofer A. The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science. 1997;277:333–338. doi: 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
- 17.Henkemeyer M., Rossi D.J., Holmyard D.P., Puri M.C., Mbamalu G., Harpal K., Shih T.S., Jacks T., Pawson T. Vascular system defects and neuronal apoptosis in mice lacking ras GTPase-activating protein. Nature. 1995;377:695–701. doi: 10.1038/377695a0. [DOI] [PubMed] [Google Scholar]
- 18.Lapinski P.E., Kwon S., Lubeck B.A., Wilkinson J.E., Srinivasan R.S., Sevick-Muraca E., King P.D. RASA1 maintains the lymphatic vasculature in a quiescent functional state in mice. J Clin Invest. 2012;122:733–747. doi: 10.1172/JCI46116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miao W., Eichelberger L., Baker L., Marshall M.S. p120 Ras GTPase-activating protein interacts with Ras-GTP through specific conserved residues. J Biol Chem. 1996;271:15322–15329. doi: 10.1074/jbc.271.26.15322. [DOI] [PubMed] [Google Scholar]
- 20.Charles M.A., Saunders T.L., Wood W.M., Owens K., Parlow A.F., Camper S.A., Ridgway E.C., Gordon D.F. Pituitary-specific Gata2 knockout: effects on gonadotrope and thyrotrope function. Mol Endocrinol. 2006;20:1366–1377. doi: 10.1210/me.2005-0378. [DOI] [PubMed] [Google Scholar]
- 21.Soliman G.A., Ishida-Takahashi R., Gong Y., Jones J.C., Leshan R.L., Saunders T.L., Fingar D.C., Myers M.G., Jr. A simple qPCR-based method to detect correct insertion of homologous targeting vectors in murine ES cells. Transgenic Res. 2007;16:665–670. doi: 10.1007/s11248-007-9110-2. [DOI] [PubMed] [Google Scholar]
- 22.Oliver J.A., Stolberg V.R., Chensue S.W., King P.D. IL-4 acts as a potent stimulator of IFN-gamma expression in CD8+ T cells through STAT6-dependent and independent induction of Eomesodermin and T-bet. Cytokine. 2012;57:191–199. doi: 10.1016/j.cyto.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmittgen T.D., Livak K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 24.Rodriguez C.I., Buchholz F., Galloway J., Sequerra R., Kasper J., Ayala R., Stewart A.F., Dymecki S.M. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat Genet. 2000;25:139–140. doi: 10.1038/75973. [DOI] [PubMed] [Google Scholar]
- 25.Lapinski P.E., Bauler T.J., Brown E.J., Hughes E.D., Saunders T.L., King P.D. Generation of mice with a conditional allele of the p120 Ras GTPase-activating protein. Genesis. 2007;45:762–767. doi: 10.1002/dvg.20354. [DOI] [PubMed] [Google Scholar]
- 26.Chang L., Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 27.Kulkarni S.V., Gish G., van der Geer P., Henkemeyer M., Pawson T. Role of p120 Ras-GAP in directed cell movement. J Cell Biol. 2000;149:457–470. doi: 10.1083/jcb.149.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dejana E., Tournier-Lasserve E., Weinstein B.M. The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. Dev Cell. 2009;16:209–221. doi: 10.1016/j.devcel.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 29.Uebelhoer M., Boon L.M., Vikkula M. Vascular anomalies: from genetics toward models for therapeutic trials. Cold Spring Harb Perspect Med. 2012;2 doi: 10.1101/cshperspect.a009688. pii:a009688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lan Y., He W., Li Z., Wang Y., Wang J., Gao J., Wang W., Cheng T., Liu B., Yang X. Endothelial Smad4 restrains the transition to hematopoietic progenitors via suppression of ERK activation. Blood. 2014;123:2161–2171. doi: 10.1182/blood-2013-09-526053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kawasaki J., Aegerter S., Fevurly R.D., Mammoto A., Mammoto T., Sahin M., Mably J.D., Fishman S.J., Chan J. RASA1 functions in EPHB4 signaling pathway to suppress endothelial mTORC1 activity. J Clin Invest. 2014;124:2774–2784. doi: 10.1172/JCI67084. [DOI] [PMC free article] [PubMed] [Google Scholar]