

Abstract
The contribution of oxidative stress to ischemic brain damage is well established. Nevertheless, for unknown reasons, several clinically tested antioxidant therapies failed to show benefits in human stroke. Based on our previous in vitro work, we hypothesized that the neuroprotective potency of antioxidants is related to their ability to limit release of the excitotoxic amino acids, glutamate and aspartate. We explored the effects of two antioxidants, tempol and edaravone, on amino acid release in the brain cortex, in a rat model of transient occlusion of the middle cerebral artery (MCAo). Amino acid levels were quantified using a microdialysis approach, with the probe positioned in the ischemic penumbra as verified by a laser Doppler technique. Two-hour MCAo triggered a dramatic increase in the levels of glutamate, aspartate, taurine and alanine. Microdialysate delivery of 10 mM tempol reduced the amino acid release by 60–80%, while matching levels of edaravone had no effect. In line with these latter data, an intracerebroventri-cular injection of tempol but not edaravone (500 nmols each, 15 minutes prior to MCAo) reduced infarction volumes by ~50% and improved neurobehavioral outcomes. In vitro assays showed that tempol was superior in removing superoxide anion, whereas edaravone was more potent in scavenging hydrogen peroxide, hydroxyl radical, and peroxynitrite. Overall, our data suggests that the neuroprotective properties of tempol are likely related to its ability to reduce tissue levels of the superoxide anion and pathological glutamate release, and, in such a way, limit progression of brain infarction within ischemic penumbra. These new findings may be instrumental in developing new antioxidant therapies for treatment of stroke.
Keywords: stroke, oxidative stress, antioxidants, superoxide anion, glutamate release, neuroprotection
Introduction
STROKE is a loss of brain functions due to interruption of cerebral blood flow. In a majority of clinical cases, strokes are initiated by thrombotic or embolic occlusion of one of the major blood vessels in the brain; however, they may also develop due to rupture of a blood vessel (hemorrhagic stroke), or complete shutdown of cerebral circulation such as in cardiac arrest [1]. Stroke represents the fourth leading cause of death in the United States, the second leading cause of mortality worldwide, and is the leading cause of adult long-term disability in the industrialized nations [2, 3]. Despite the large impact on public health, there is only one drug, tissue plasminogen activator (tPA), that is currently approved for acute stroke treatment in the U.S. Yet, due to a relatively short therapeutic window and numerous contraindications, tPA has been historically used in no more than 3–5% of stroke patients [4]. Numerous clinical trials which tested other neuroprotective treatments showed no clinical benefits in stroke patients, leaving a large unmet need for the development of new therapeutic interventions [5].
In the ischemic brain, cessation of oxygen and glucose supplies leads to a very rapid depolarization of neural cells followed by a massive release of numerous neurotransmitters via several transport mechanisms [6, 7]. The excitatory neurotransmitters glutamate and aspartate cause activation of highly abundant ionotropic receptor-channels. These receptors propagate the rapid onset of tissue depolarization and directly (the NMDA receptor subtype) or indirectly (majority of AMPA and kainate receptors) contribute to a lasting increase in cytosolic Ca2+ levels and cell damage and death termed excitotoxicity [8, 9]. The pathological rise in [Ca2+]i sets in motion multiple damaging cascades culminating in either rapid ischemic cell death via necrosis, or delayed cell death via apoptosis and the apoptosis-like process called parthanatos [6, 10, 11].
One of the critical events leading to the excitotoxic demise of neuronal cells is oxidative stress – a redox imbalance stemming from the increased production of reactive oxygen species (ROS) and reactive nitrogen species (RNS) in the ischemic tissue (reviewed in [12–14]). ROS cascades in stroke begin with formation of the superoxide anion (O2•−), which is produced in mitochondria as a result of one-electron reduction of oxygen, but also in several additional enzymatic reactions carried by NADPH oxidases, xanthine oxidase, and others. O2•− is dismutated to hydrogen peroxide (H2O2), either enzymatically or spontaneously. In the presence of transition metals, H2O2 decomposition yields the potent oxidant, hydroxyl radical (•OH). In a parallel RNS cascade, ischemia upregulates levels of the free radical signaling molecule nitric oxide (•NO) and several intermediates of its degradation. Most importantly, •NO combines in the rate-limiting reaction with O2•− generating the oxidant peroxynitrite (ONOO−), which damages cells on its own or via secondary formation of •OH and other toxic intermediates [6, 14–16]. ROS and RNS production is thought to lie downstream of the NMDA receptors because activation of these receptors and ensuing cytosolic Ca2+ overload increase mitochondrial formation of O2•− and H2O2 and stimulate several enzymes producing ROS and RNS [6, 12, 17]. Oxidative, nitrosative, and nitrative damage harms and kills neuronal cells via widespread damage to enzymes, DNA, and lipids. The major role for ROS and RNS in stroke pathology was confirmed in numerous animal studies employing antioxidants and free radical scavengers, or manipulating expression levels of enzymes, which produce or degrade ROS and RNS (see for example [18–23] and reviews [12, 13, 17]).
The extensive preclinical data on the role of free radicals and oxidative stress in ischemic brain damage have led to several clinical trials. Two of these trials, which were conducted in Japan, demonstrated modest clinical benefits of the antioxidant edaravone in stroke patients [24, 25]. In contrast, several other trials performed elsewhere showed no neuroprotective properties for other antioxidant molecules [13, 26, 27]. These latter unsuccessful studies included the most extensive to-date clinical trial, SAINT II, which tested a nitrone spin trapping agent NXY-059 or Cerovive [28]. The failure of SAINT II came as a big disappointment and solidified skepticism about clinical utility of antioxidants as neuroprotective agents [29, 30]. The reasons for clinical failure of NXY-059 continue to be debated [5, 31].
In the present study we investigated a new hypothetical mechanism that may determine the efficacy of antioxidants in stroke. As mentioned above, the traditional view is that oxidative stress represents one of the terminal steps of ischemic tissue damage, downstream of activation of glutamate receptors and elevation of intracellular [Ca2+]. Our recent in vitro and in vivo studies suggest that exogenous and endogenous oxidants, particularly H2O2, can also exert “upstream” effects and promote pathological glutamate release from glial and neuronal cells via opening of glutamate-permeable membrane channels [32–35]. Here, we used the well-established rodent model of stroke, induced by microfilament occlusion of the middle cerebral artery (MCAo), to correlate effects of antioxidants on ischemic glutamate release and their ability to reduce ischemic tissue damage. For this purpose, we selected two antioxidants, tempol and edaravone, based on previous animal studies and clinical data (see Discussion for details and references). Our new data may contribute to understanding the limited clinical efficacy of some of the previously tested antioxidant agents and provide a blueprint for selecting more effective prototypical drugs for future clinical studies.
Materials and Methods
All animal procedures in this study were in strict adherence to the Guide for the Care and Use of Laboratory Animals as adopted by the National Institutes of Health and were approved by the Institutional Animal Care and Use Committee of the Albany Medical College (animal care and use protocols #907433, 11-02003, and 11-07002).
Reagents
4-Hydroxy-2,2,6,6-tetramethylpiperidine 1-oxyl (tempol), 18α-glycyrrhetinic acid (18-αGA), L-serine-O-sulfate potassium salt (L-SOS), butylated hydroxytoluene (BHT), dimethyl sulfoxide (DMSO), hypoxanthine, xanthine oxidase (bovine milk, grade III), and dithiothreitol were obtained from Sigma-Aldrich (St Louis, MO). 4-[(2-butyl-6,7-dichloro-2-cyclopen-tyl-2,3-dihydro-1-oxo-1H-inden-5-yl)oxy]butanoic acid (DCPIB), dihydrokainate, Edaravone, and 3-morpholinylsydnoneimine chloride (SIN-1) were purchased from R&D Systems/Tocris (Ellisville, MO). Radiolabeled amino acids D-[3H]aspartate, L-[3H]glutamate, and L-[14C]cystine were acquired from PerkinElmer/New England Nuclear (Boston, MA). All cell culture reagents were from Life Systems/Invitrogen (Carlsbad, CA). All other chemicals, unless otherwise specified, were from Sigma-Aldrich and of the highest purity available.
Animal model of cerebral ischemia and implantation of microdialysis probes
Male Sprague-Dawley rats (Taconic Farms, 230–270 g) were used in all microdialysis and neuroprotection experiments. Animals were maintained on a 12/12-h light/dark cycle and allowed free access to food and water. Anesthesia was induced with 5% isoflurane in 30% O2/balance N2 and maintained through surgical procedures with 1.75–2.25% isoflurane under constant observation. To prevent excessive fluid secretion in the respiratory system and gastrointestinal tract, 0.4 mg/kg atropine sulfate was given via intramuscular injection. Animals were kept hydrated with hourly intraperitoneal (i.p.) injections of 1 mL physiological saline. Body and brain temperatures were maintained between 36.5°C to 37.5°C using a heating pad, and were measured with two probes placed rectally and in the temporalis muscle, respectively.
Middle cerebral artery occlusion (MCAo) was performed using an intraluminal suture technique as originally developed by Longa et al. [36]. The right common carotid artery and bifurcation of the external carotid and the internal carotid arteries were exposed via incision on the neck. The external carotid artery was coagulated and a standardized 4-0 microfilament with a poly-D-lysine coated tip of 0.41 or 0.43 mm diameter (Doccol Corporation, Sharon, MA) was inserted via a stump of the external carotid artery into the internal carotid artery up to 19–20 mm from the bifurcation to occlude the origin of MCA. After 120 min of ischemia, cerebral blood flow was restored by retracting the suture. The surgical wound was closed and the animal was allowed to recover in a temperature-controlled environment with the external temperature 26°C. A similar surgical procedure was performed in the sham operated group, except the suture was inserted and immediately withdrawn from the vessel without advancing it to the MCA origin.
In the microdialysis experiments, two symmetrical bilateral probes (CX-I series, 0.22×2 mm membrane, 50,000 kD molecular weight cutoff, Eicom Corporation, San Diego, CA) were lowered into the cortex through small burr holes, ~4 h before initiation of ischemia (see experimental design diagram in Fig. 1). Probe placement was done using a stereotaxic frame (David Kopf Instruments, Tujunga, CA). The position of the probes was 2 mm anteroposterior, 5 mm lateral from bregma, and 2.6 mm down from dura [37]. Artificial cerebrospinal fluid (aCSF) was perfused at a flow rate of 2 μL/min throughout the experiment. The composition of aCSF was as follows (in mM): 120 NaCl, 2.7 KCl, 1 MgSO4, 1.2 CaCl2, 25 NaHCO3, 0.05 ascorbic acid (pH=7.3). Microdialysis samples were collected every 20 minutes after a 1–1.5-hour stabilization period using a CMA 470 refrigerated fraction collector (CMA Microdialysis, Holliston, MA). Cerebral blood flow was measured using a MoorLAB laser Doppler sensor mounted next to the microdialysis probe (~1-2 mm) and acquisition module (Moor Instruments, Axminster, Devon, UK). The laser Doppler readings were used to ensure proper reduction of the blood flow rate to the levels that are expected in the ischemic penumbra. If blood flow was reduced by less than 80% (20% of the pre-ischemia values), animals were excluded from analysis on the basis of incomplete ischemia. After completion of ischemia and reperfusion in microdialysis experiments animals were euthanized with an overdose of sodium pentobarbital, brains were extracted and stained for early ischemic brain damage as described below.
FIG. 1. Effects of the antioxidants tempol and edaravone on microdialysate levels of glutamate and aspartate in rat cortex before, during and after transient occlusion of the middle cerebral artery (MCAo).

A, Experimental design and schematic representation of position of microdialysis probes and laser Doppler probe in the MCAo experiments. Shaded gray (inner area) and light blue (outer area) regions approximately correspond to the ischemic core and penumbra, respectively. B, Dynamics of cerebral blood flow (CBF) in all three experimental groups. CBF was quantified using a laser Doppler probe and then normalized to the average CBF values before MCAo. C, Microdialysate levels of glutamate on ischemic and non-ischemic (contralateral) sides of the brain. Vehicle (2% DMSO), 10 mM tempol, or 10 mM edaravone were added into the aCSF as indicated. The data are the mean values ±SE in 9–10 animals per group. D, Integral values of the intra-ischemic glutamate release in the experiments presented in C. *p<0.05, DMSO vs. tempol; #p<0.05, tempol vs. edaravone. E, Microdialysate levels of aspartate on ischemic and contralateral sides of the brain. n= 9–10 per group. F, Integral values of intra-ischemic aspartate release in the experiments presented in E. *p<0.05, DMSO vs. tempol; #p<0.05, tempol vs. edaravone.
For intracerebroventricular (i.c.v.) delivery of antioxidants or vehicle, animals were placed into a stereotaxic frame. The injecting cannula was lowered through the burr hole into the right ventricle (from bregma, 0.8 mm anteroposterior; 1.5 mm lateral; 5.3 mm below the dura) and the tested agents (500 nmols each) were injected at a flow rate of 1 μL/min into the lateral ventricle over a period of 5 min. In vehicle controls, an identical volume of DMSO was delivered at the same rate. After 15 min following the drug injection, cerebral ischemia was induced as described above.
Collection and analysis of microdialysate samples
Microdialysate samples were collected every 20 min throughout experiment and refrigerated using a CMA 470 fraction collector (CMA Microdialysis) and analyzed off-line. Amino acid levels in each sample were determined by a reverse-phase HPLC using an Agilent 1200 HPLC setup and Eclipse XDB-C18 column (both from Agilent Technologies, Santa Clara CA). Precolumn derivatization was performed with a freshly prepared mix of o-phthalaldehyde and 2-mercaptoethanol in 0.4 M sodium tetraborate buffer (pH = 9.5). The amino acid derivatives were eluted with solvent containing 30 mM NaH2PO4, 1% tetrahydrofuran, 30 mM sodium acetate, 0.05% sodium azide, and increasing concentration of HPLC grade methanol (10–30%). Fluorescence signal was measured using a programmable 1200 series fluorescence detector (Agilent). Amino acid standards (L-alanine, L-aspartate, L-glutamate, and taurine), were processed in the same fashion and used to identify amino acid peaks and calculate concentrations of individual amino acids in the samples. To quantify the integral amino acid release during cerebral ischemia, we summed amino acid concentrations in all MCAo samples collected on the ischemic side of the brain and subtracted from the resulting value a sum of amino acid release values in samples collected on the contralateral (non-ischemic) side of the brain.
Quantification of infarction volumes
Brain infarction volumes were quantified at 72 h after initiation of ischemia (or at comparable time in the sham-operated animals) utilizing the 2,3,5-triphenyltetrazolium chloride (TTC) staining technique [38]. After completion of the last behavioral analysis, all animals were euthanized with an overdose of sodium pentobarbital. Brains were gently removed and sliced into 2-mm thick sections using metal matrix (Kent Scientific, Torrington, CT). Slices were incubated in 2% solution of TTC in saline phosphate for 30 min at 37°C. The red-colored region represents the viable tissue, while the infarcted area remains unstained [38]. The images of each section were captured with a digital scanner and analyzed using ImageJ software (http://imagej.nih.gov/ij/) [39]. Total infarction volumes were calculated based on values of the infarcted area in five slices per brain and their known thickness by a person who was blind to the type of treatment. To correct for brain edema, infarction volumes were additionally quantified according to the following formula: (volume of the contralateral hemisphere) minus (volume of the viable tissue in ischemic hemisphere) [40]. Brain edema values were calculated as the difference between the total volumes of contralateral and ischemic hemispheres in analyzed slices.
Behavioral evaluation of neurological deficits
Neurological deficits were evaluated in all of experimental groups 24, 48 and 72 h after initiation of ischemia using two complementary techniques described below. In order to maintain consistency and avoid bias, behavioral testing was performed by the same observer who was blind to the treatment type.
The first evaluation was done according to the method of Garcia et al. [41]. Animals were scored in the six categories: (1) spontaneous activity, (2) symmetry in the movement of four limbs, (3) forepaw stretching, (4) climbing, (5) body proprioception, and (6) response to vibrissae. The final score was the sum of six individual sub-scores. The maximum possible score was 18, corresponding to the absence of neurological deficits. Lower scores represented negative outcomes.
The second evaluation was done within a few minutes after the first battery of behavioral exams, and involved the modified adhesive removal test. This technique has been first introduced in rats by Schallert et al. [42] to measure sensorimotor asymmetries. It is now increasingly used in both rats and mice to quantify behavioral outcomes in animal stroke models [43]. Briefly, rats were removed from their cages and two 5 mm-wide tape sleeves, which were made from 3M Post-it adhesive paper, were placed around the forepaws on both the “ischemic” and “non-ischemic” side of the body. Animals then were then returned to their home cages and the time taken to remove each sleeve was recorded. For simplicity, in this manuscript we present summed times taken to remove both sleeves. The cut-off time for each measurement was set at 2 min. If animals were unable to remove adhesive on their own, they received assistance from the experimentator.
5, 5′-Dithiobis-(2-nitrobenzoic acid) (DTNB) assay of sulfhydryl groups in vivo
The total content of sulfhydryl (SH-) groups in post-ischemic tissue was measured using DTNB (Ellman’s reagent). This assay was performed as originally developed by Ando and Steiner [44] with modifications. Animals were subjected to 2-h MCAo as described above. After an additional 2-h reperfusion, animals were deeply anesthetized with sodium pentobarbital, followed by a transcardial perfusion with physiological saline. Brains were carefully removed and ~300 mg of cortical tissue was isolated from lateral cortex on both ischemic and contralateral side of the brain from the areas anatomically corresponding to core and penumbra. The tissue samples were homogenized in 10 mM sodium phosphate buffer (pH 7.4) including 2 mM EDTA, using a small sample homogenizer PRO2000 (Pro Scientific Inc., Oxford, CT). The homogenates were further diluted two-fold in 20 mM Trizma-HCl buffer (pH 8.1) containing 3 mM DTNB, 2 mM EDTA, and 2% SDS. After 30 min incubation at room temperature, the resulting absorbance of newly formed colored 2-nitro-5-thiobenzoate anion was measured at 412 nm using a BioTek ELx800 microplate reader (BioTek, Winooski, VT). The amount of free thiols in the sample was calculated using a molar absorption coefficient of 1.415×104 M−1 cm−1, and additionally verified using a standard curve generated with known concentrations of DTT. All data were normalized to the total protein content determined by a bicinchoninic acid assay (Thermo Scientific/Pierce, Rockford, IL) using bovine serum albumin as a standard according to the manufacturer’s instructions.
Preparation of primary cultures of astrocyte and microglial cells
Primary astrocyte cultures and microglial cells were prepared from brain cortical tissue of newborn Sprague-Dawley rats as previously described [34, 45]. One day-old pups were euthanized by rapid decapitation and cortical tissue was dissected from meninges, hippocampi, and basal ganglia, and minced with small scissors. Cortical cells were extracted using a 1:1 mixture of optimized MEM and recombinant protease TrypLE (both Life Technologies/Invitrogen) in the presence of bovine pancreatic DNase I (1 mg/mL, Sigma-Aldrich) at 37°C. Cells were transferred to minimal essential medium containing 10% heat-inactivated horse serum, 50 U/mL penicillin, and 50 μg/mL streptomycin. Cell were plated on poly-D-lysine coated T75 culture flasks (Techno Plastic Products, TPP, Trasadingen, Switzerland) at a density of ~200,000 cells/flask for pure astrocyte cultures or 1–1.3 million cells/flask to produce mixed glial cultures. The cultures were grown for 2–3 weeks in a humidified atmosphere of 5% CO2/95% air at 37°C. The purity of astrocyte cultures was routinely verified with staining for the astrocyte marker glial fibrillary acidic protein (GFAP) with monoclonal anti-GFAP antibody (Sigma-Aldrich, G3893) and reached ≥98%. Microglial cells were additionally purified from the mixed glial cultures by intensive shaking of T75 flasks on a titer plate shaker. The resulting cell suspensions were replated in poly-D-lysine coated 24-well plates in opti-MEM medium additionally containing B27 supplement (Life Technologies/Invitrogen) and used for glutamate transport experiments within 24 hours.
Measurements of glutamate release in rat primary astrocytes
For amino acid transport experiments astrocytes were re-plated on 18×18 mm coverslips or in 12-well multi-well culture plates. Glutamate release assays were performed as described in detail elsewhere [46]. Briefly, cultured astrocytes in 12-well plates were pre-loaded for 3 h with a non-metabolized analog of L-glutamate, D-[3H]aspartate (4 μCi/mL), which was added in the standard culture medium. D-Aspartate is taken inside cells and released via essentially the same transport mechanisms as the endogenous glutamate. Following preloading, astrocytes were washed from extracellular isotope with warm Basal medium containing (in mM): 135 NaCl, 3.8 KCl, 1.2 MgSO4, 1.3 CaCl2; 1.2 KH2PO4, 10 D-glucose, and 10 HEPES (pH adjusted to 7.4 with NaOH), and additionally preincubated for 10 min at 37°C in the same medium. To test for activation of various glutamate transport pathways, astrocytes were exposed to several types of experimental media as following. To induce glutamate release via VRAC, cells were exposed for 10 min to hypoosmotic medium, in which osmolarity was decreased 30% by reducing NaCl concentration, while all other components remained the same as in the Basal medium. To trigger opening of Cx43 hemichannels, astrocytes were incubated for 20 min in calcium/magnesium-free medium (CMF) in which CaCl2 and MgSO4 were omitted and 50 μM EDTA was added. After completion of the incubation, extracellular medium was collected into scintillation vials to measure the amount of extracellular D-[3H]aspartate. Cells were further lysed in a solution containing 2% sodium dodecylsulfate (SDS) plus 8 mM EDTA to determine the quantity of remaining intracellular isotope. Ecoscint A scintillation cocktail (National Diagnostics) was added to each sample and radioactivity levels in all fractions were measured using Tri-Carb 2900TR liquid scintillation analyzer (PerkinElmer). The rate of D-[3H]aspartate release was calculated as the ratio of extracellular [3H] label and the total isotope load (sum of the releases and intracellular [3H] counts). Antioxidants and glutamate transport inhibitors were present in the reaction media in the concentrations indicated in Results and figure legends.
In a few experiments presented in Fig. 6, kinetics of D-[3H]aspartate release were explored with high temporal resolution using a Lucite perfusion system. In these latter experiments, astrocytes grown on 18×18 mm coverslips were preloaded with D-[3H]aspartate overnight, the extracellular isotope was removed by several washes, and the coverslips were transferred into perfusion chamber. Cells were superfused with the Basal or hypoosmotic media (for composition see above). Oxidative stress was induced by adding hypoxanthine and xanthine oxidase mix as described in Results and Fig. 6. One-min superfusate fractions were collected and analyzed for [3H]-labeled amino acid content. The release rates were normalized to amino acid content inside cells and calculated as described in detail elsewhere [46].
FIG. 6. Hydrogen peroxide but not superoxide anion potently stimulates glutamate release in primary astrocyte cultures.

A, Kinetics of D-[3H]aspartate release from primary astrocytes superfused with the mix of 3 mU/mL xanthine oxidase (XO) and 300 μM hypoxanthine (HX), which produces both O2•− and H2O2. To mimic pathological swelling, cells were additionally exposed to hypoosmotic medium (HYPO, 30% reduction in medium osmolarity). n=5–6. ***p<0.001, HYPO vs. HYPO in combination with HX+XO. B, The nature of ROS contributing to stimulation of D-[3H]aspartate release was tested by adding SOD (200 U/ml), or catalase (15 μG/mL). The integral release values during 10-min periods either prior to cellular swelling (11th–20th min), or during swelling (21st–30th min) were measured as described in Materials and Methods. n= 5/group. *p<0.05, vs. HYPO alone; ###p<0.001, HYPO + catalase vs. HYPO+X/XO, or HYPO+HX/XO+SOD.
Assay of activity of astrocytic cystine/glutamate heteroexchanger
The plasmalemmal cystine/glutamate heteroexchanger (xCT) does not accept D-aspartate as a substrate, while cytosolic L-[3H]glutamate is rapidly metabolized by astroglial cells. Therefore, we used L-[14C]cystine to measure the xCT activity in cultured astrocytes. The conditions of this assay were optimized and thoroughly validated in our previous study [46]. Briefly, astrocytes grown in 12-well plates were preincubated in the Basal medium and then transferred into LiCl medium for xCT assays. Composition of the LiCl medium was similar to that of the Basal medium, with the exception that NaCl was replaced with LiCl to suppress activity of the Na+-dependent amino acid transporters, and 0.5 mM acivicin was added to inhibit γ-glutamyl transpeptidase. pH of the medium was adjusted with CsOH. Cells were incubated for 30 min at 37°C with 0.1 μCi/mL L-[14C]cystine (~1 μM). The reaction was terminated by several washes with ice cold LiCl medium and total L-[14C]cystine accumulation was determined using scintillation counting as described above and normalized to protein content determined by the bicinchoninic acid assay.
Measurements of activity of glial GLT-1 glutamate transporter
To measure activity of the glutamate transporter GLT-1 we used primary microglial cells. Although GLT-1 is the dominant glutamate transporter in astrocytes in situ, it is functionally downregulated in astrocytic cultures, but remains expressed in primary microglia [47, 48]. To discriminate between GLT-1 and other modes of glutamate uptake we used the selective GLT-1 blocker, 1 mM dihydrokainate. The conditions of this assay were validated in our previous study [46]. Purified microglial cells were plated in 24-well plates, washed from culture media, and pre-incubated in Basal medium for 20 min at 37°. Transport measurements were initiated by replacing the Basal medium with media additionally containing 1 μCi/mL L-[3H] glutamate and 2 μM of unlabeled L-glutamate, and tested compounds as specified in the text and figure legends. The uptake reaction was terminated with 3 consecutive washes with excess of basal medium at the room temperature. Microglial cells were lysed using 2% SDS +8 mM EDTA. The glutamate uptake rates were calculated by measuring the [3H] content in cells and normalizing the uptake values to the specific activity of the isotope and protein content in wells.
Cell free assay of total antioxidant capacity with 2,2-diphenyl-1-picrylhydrazyl (DPPH)
We compared the antioxidant capacity of tempol and edaravone using the commonly utilized cell-free DPPH assay [49]. Various concentrations of antioxidants were added to the solution of 100 μM DPPH prepared in 80% methanol. The reaction mixtures were incubated at 37°C for 30 min and changes in absorbance at 490 nm were measured using a BioTek ELx800 absorbance microplate reader.
Lucigenin assay of the superoxide scavenging activity
Superoxide scavenging activity of antioxidants was determined using a lucigenin assay in a cell-free medium, which was modified from [50]. Various concentrations of antioxidants were incubated with 100 μM lucigenin, and the superoxide-producing mixture of 300 μM hypoxanthine and 10 mU/mL xanthine oxidase in 100 mM Trizma buffer (pH 7.6) for 30 min at 37°C. Cumulative chemiluminescence was measured 30 times with 60 sec intervals using a Victor3 multi-label plate reader (PerkinElmer). To ensure the specificity of the superoxide signal, control experiments were additionally performed in the absence of xanthine oxidase or in the presence of superoxide dismutase (SOD, 200 U/ml).
Amplex red assay of the H2O2 scavenging
Hydrogen peroxide scavenging capacity was measured using Amplex red reagent (Sigma-Aldrich) in a cell-free system according to manufacturer’s instructions. Various concentrations of antioxidants were incubated with 25 μM H2O2, 100 μM Amplex red reagent, and 250 mU horse radish peroxidase in PBS for 10 min at room temperature. The resulting absorbance was measured at 570 nm using a BioTek ELx800 microplate reader (BioTek, Winooski, VT, USA). H2O2 scavenging activity was calculated by comparing the resulting absorbance values to those generated with known concentrations of H2O2.
Preparation of rat brain synaptosomes
Synaptosomes were prepared from the forebrains of male Sprague Dawley rats (150–180 g) according to the method of F. Hajos [51] with modifications described elsewhere [52]. Forebrain tissue was manually homogenized in ice cold 0.32 M sucrose solution containing 5 mM HEPES (pH 7.4) in Potter-Elvehjem tissue grinder with Teflon pestle. Synaptosomes were isolated using density gradients of sucrose solutions. The final pellets obtained were synaptosomes which were resuspended in Basal medium to a protein concentration of 10–12 mg/mL and were allowed to restore their ionic gradients by incubating with continuous agitation at 37°C for 1 h.
TBARS assay of the hydroxyl radical scavenging activity
The ability of antioxidants to prevent hydroxyl radical-driven oxidation was determined using an assay of 2-thiobarbituric acid-reactive substances (TBARS), according to the method of Ohkawa et al. [53]. This assay measures formation of lipid peroxides, typically in biological membranes. Intact synaptosomes (0.5 mg protein/mL) were pre-incubated with different concentrations of antioxidants for 15 min at 37°C in the Basal medium. Lipid peroxidation was then initiated by the addition of 7.5 μM Fe2SO4 plus 1.5 mM ascorbic acid and was carried on for 30 min at 37°C. The reaction was terminated by transferring synaptosomal suspension onto ice followed by a 3-min centrifugation at 18,000 g (0–4°C). Pellets were dissolved in 1 mL of a solution containing 0.25% thiobarbituric acid, 2% SDS and 5% trichloroacetic acid, and boiled for 15 min in a water bath. After cooling, the absorbance was measured at 531 nm using a Victor3 multi-label plate reader. The concentration of TBARS was calculated using a molar extinction coefficient of 1.5×105 M−1cm−1.
Determination of antioxidant activity using peroxynitrite-dependent oxidation of dihydrorhodamine 123 (DHR123)
Peroxynitrite (ONOO−) scavenging activity was measured using dihydrorhodamine 123 (DHR123), which is readily oxidized to highly fluorescent rhodamine 123 by the intermediate products of peroxynitrite decomposition [54, 55]. In the present study, we used SIN-1 as the peroxynitrite generator in a cell-free medium. Various concentrations of antioxidants were incubated with DHR123 (10 μM) in the presence of 1 mM SIN-1 for 30 min at 37°C. Formation of rhodamine 123 was measured using a Victor3 multi-label plate reader and the excitation/emission wavelengths of 530 and 590 nm, respectively.
Statistical Analyses
All data are presented as mean values ±SE, with n referring to individual animals or independent experiments within each experimental group. Experimental groups were compared using one-way analysis of variance (ANOVA) with either Tukey or Fisher’s least significant difference (LSD) post hoc analysis for multiple comparisons. When treatment effects were analyzed over several time points (such as for blood flow measurements and in behavioral experiments) we utilized repeated measures ANOVA with effects of time, treatment and their interaction with Fisher’s LSD post hoc test. Origin 8.1 (Origin Laboratories, Northampton, MA), Prism 5.0 (GraphPad Software, San Diego, CA), or Statistica 10.0 (StatSoft, Tulsa, OK) were used for all statistical calculations. A probability of p<0.05 was considered statistically significant for all comparisons.
Results
Tempol but not edaravone decreases pathological release of glutamate and aspartate in the ischemic penumbra
In order to test the hypothetical connection between oxidative stress and release of the excitatory neurotransmitters glutamate and aspartate in ischemic brain, we sampled extracellular amino acid levels using microdialysis in conjunction with local delivery of antioxidant compounds via the microdialysis probe. To assure equilibration of tested compounds between microdialysis probe and brain tissue, they were delivered 1 h prior initiation of ischemia and throughout remaining duration of each experiment (see summary of experimental design in Fig. 1A). We placed one microdialysis probe in the cortical tissue corresponding to the ischemic penumbra, and another on the contralateral side of the brain. The contralateral probe served as a non-ischemic internal control, and allowed us to check for potential non-specific effects of all tested compounds. The proper placement of the ischemic probe within the penumbral area was confirmed in each animal by monitoring relative changes in blood flow using a laser Doppler sensor. Positioning of the microdialysis and the laser Doppler probes and the outline of the experimental design are depicted in Fig. 1A. Targeted reduction of blood flow was 80% (20% of the pre-ischemic levels) immediately after initiation of MCAo (Fig. 1B). After completion of ischemia blood circulation was completely restored in all tested groups and no difference was identified between treatments (Fig. 1B).
In the vehicle-treated animals, initiation of MCAo triggered an 8-fold increase in microdialysate levels of glutamate and an approximately 14-fold rise in the levels of aspartate on the ischemic side of the brain (Fig. 1C, E). The excitatory amino acid concentrations peaked approximately 40 min after the onset of ischemia. This was followed by a gradual decrease, and then complete return to baseline levels upon restoration of the blood flow. We observed a partial recovery of amino acid levels during ischemia in the majority, but not all animals. This paradoxical reversal of pathological amino acid release may be related to small compensatory changes in the blood flow rates seen in the ischemic penumbra (see Fig. 1B).
We next tested the effects of two antioxidants, tempol and edaravone, on amino acid release in ischemic tissue. The SOD mimetic tempol, added into the microdialysate fluid at the concentration of 10 mM, potently reduced the intra-ischemic release of glutamate and aspartate (Fig. 1C, E). Note that interstitial concentration delivered drugs is lower than in microdialysate and decreases with the distance from the probe. In contrast to tempol, edaravone (also 10 mM) had no effect on the levels of either amino acid (Fig. 1C, E). In order to quantitatively compare the release values for each amino acid, we next calculated the integral amino acid levels during ischemia. As seen in Fig. 1D, F, the effect of tempol on glutamate and aspartate levels was statistically significant, when compared to both the vehicle and edaravone groups. Importantly, we found no effects of either antioxidant on the microdialysate glutamate and aspartate values on the non-ischemic side of the brain (see contralateral traces in Fig. 1C, E). Furthermore, neither tempol nor edaravone significantly altered cerebral blood flow rates during MCAo, as measured by laser Doppler probe (Fig. 1B).
Effects of tempol and edaravone on the intra-ischemic levels of taurine and alanine
In addition to glutamate and aspartate, we measured dynamic changes in the levels of two other amino acids, taurine and alanine, in the same microdialysate samples. Unlike the excitatory amino acids, which are enriched in synaptic vesicles, taurine and alanine are abundant in the cytosol. Therefore, information on microdialysate levels of taurine and alanine can be instrumental in examining different mechanisms and sources of amino acid release. For example, pathological activation of the anion channel VRAC, which is poorly selective, has been linked to the release of all four amino acids that were measured in our assays [35, 56, 57].
As shown in Fig. 2A, MCAo led to substantial increases in the microdialysate concentrations of taurine. As in the case of the excitatory amino acids, tempol but not edaravone caused partial normalization of the intra-ischemic taurine values (Fig. 2A, B). Similar results were obtained for alanine. Alanine concentration was rapidly elevated during MCAo, and this effect was attenuated by tempol but not edaravone (Fig. 2C, D). Despite the overall similarities, the kinetics of release for taurine and alanine differed from glutamate and aspartate in at least two respects. Alanine levels continued to steadily rise during ischemia (Fig. 2C). Additionally, both taurine and alanine levels did not normalize to their baseline values even 2 h after blood flow restoration (Fig. 2A, C). These differences are likely explained by the diverse re-uptake mechanisms, such as kinetics and density of relevant amino acid transporters.
FIG. 2. Effects of tempol and edaravone on microdialysate levels of taurine and alanine before, during and after 2-h MCAo.
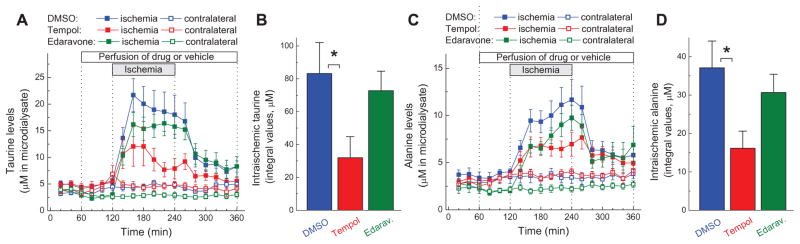
A, Microdialysate levels of taurine on ischemic and contralateral sides of the brain. n= 9–10 per group. B, Integral values of the intra-ischemic taurine release in the experiments presented in A. *p<0.05, DMSO vs. tempol. C, Microdialysate levels of alanine on ischemic and contralateral sides of the brain. n= 9–10 per group. D, Integral values of intra-ischemic alanine release in the experiments presented in C. *p<0.05, DMSO vs. tempol.
Effects of tempol and edaravone on brain infarction volumes and neurological deficits after MCAo
In order to investigate if there was a correlation between effects of antioxidants on intra-ischemic release of excitatory amino acids and neurological outcomes, we studied the effects of tempol and edaravone on MCAo-induced brain damage. For this purpose, the antioxidants were stereotactically injected into a lateral ventricle at the single bolus dose of 500 nmoles, 15 min prior to initiation of a 2-h ischemia. This mode of delivery was chosen over systemic application to assure that the intracerebral concentrations of drugs were equal and not affected by the differences in the blood-brain barrier permeability for either compound, or alterations in systemic delivery during ischemia. Following completion of ischemia, rats were allowed to survive for 3 days with daily neurobehavioral evaluation. After the last behavioral test, animals were euthanized and the extent of ischemic brain damage was quantified using a TTC staining technique. Representative TTC-stained serial coronal sections from all experimental groups are shown in Fig 3A. Compared to the vehicle-treated animals, animals treated with tempol showed a 55% reduction in the mean brain infarction volumes (151.7±39.6 vs. 333.3±24.4 mm3, p<0.01, Supplemental Fig. 1A), and 50% smaller infarction volumes when corrected for brain edema (p<0.01, Fig. 3B). Tempol reduced brain edema by 66% (39.4±14.61 vs. 116.0±25.88, p<0.05, Supplemental Fig. 1B). In contrast, edaravone was completely ineffective in reducing ischemic brain damage, and did not have significant effect on brain edema values (Fig. 3B and Supplemental Fig. 1). Because hyper- and hypothermia are known to affect final infarction values (reviewed in [58]), it is important to know that body temperature was maintained with thermosensor-regulated heating pad and did not differ between all animal groups, prior and during MCAo (data not shown). In sham operated animals infarction was not detected (Fig. 3A, B).
FIG. 3. Neuroprotective properties of tempol and edaravone assessed as drug-dependent reduction in infarction volumes at 72 h and neurological deficits at 24, 48, and 72 h after completion of 2-h MCAo.

In these experiments 5 μL of vehicle (DMSO), tempol (500 nmols), or edaravone (500 nmols) were injected into the lateral ventricle 15 min before initiation of ischemia. Sham-operated animals were subjected to the same surgical procedures without MCAo. Experimental design is depicted in diagram on the top. A, Representative images of 5 serial sections from the same brain (top to bottom) prepared 72 h after reversible MCAo and stained with TTC. B, Average infarction volumes corrected for brain edema in sham operated animals (n=6), and animals injected with vehicle (n=12), tempol (n=11), or edaravone (n=9) as indicated. N.D., infarction not detected. **p<0.01, tempol vs. either DMSO or edaravone. C, Neurobehavioral scores in the same treatment groups, which are presented in B. The score of 18 corresponds to no deficits. **p<0.01, Sham vs. all ischemia groups; #p<0.05, tempol vs. edaravone. D, Average times taken to remove adhesive tape from forepaws. ***p<0.001, Sham vs. all treatment groups; #p<0.05, tempol vs. DMSO; ##p<0.01, tempol vs. edaravone.
To further validate the neuroprotective properties of tempol, all experimental groups were subjected to two types of behavioral evaluation. We first quantified motor and sensory deficits using a testing technique developed by Garcia et al. [41] (see Materials and Methods for detailed description). In our experiments, sham-operated animals had no behavioral deficits. Compared to shams, all MCAo groups demonstrated strong and statistically significant neurological deficits (p<0.001), which persisted for the 3-day duration of the experiment (Fig. 3C). Tempol treatment produced a trend toward behavioral improvements, as compared to both vehicle- and edaravone-treated groups. Edaravone treatment produced no evidence for protection, and this was consistent with the results of TTC staining. Because of the relatively low sensitivity of this test, we additionally examined motor and sensory deficits using the modified adhesive removal test (see Materials and Methods). The latter test is more sensitive and can be successfully used to detect neurological deficits in rodents up to 6 weeks after MCAo surgeries [43]. As shown in Fig. 3D, vehicle-treated MCAo animals took much longer to remove adhesive tape from their forepaws as compared to sham operated rats (p<0.001, DMSO vs. sham for the entire 3-day testing period). Tempol treatment dramatically reduced the time required for adhesive removal (p<0.05, tempol vs. DMSO, p<0.01 tempol vs. edaravone). Animals in the edaravone-treated group showed no improvements, and were not different from vehicle controls (Fig. 3D).
Do tempol and edaravone directly affect glutamate release in glial cells?
The simplest explanation for differential efficacy of two antioxidants is the possibility that one of them (tempol) directly blocks intra-ischemic amino acid release, independent of its antioxidant activity. To test this hypothesis we examined the direct effects of both compounds on glutamate release in primary astrocyte cultures. The focus on glia is based on the prevailing idea that glial cells represent a major source of excitatory amino acid release in stroke [7, 59, 60]. In cerebral ischemia, glia, primarily astrocytes, may release glutamate via several transport pathways: (i) reversal of the glia-specific glutamate transporter GLT-1, (ii) activation of glutamate-permeable VRAC channels, (iii) opening of connexin hemichannels which in astrocytes are constituted by Cx43, and (iv) enhanced activity of the cystine/glutamate antiporter (xCT) [61–64]. Three of these transport pathways – VRAC, Cx43 hemichannels, and xCT – were of primary interest because they are activated or positively modulated by oxidative stress. The GLT-1 was also tested due to its known contribution to glutamate release in ischemic core. [65].
To activate VRAC we triggered astrocytic swelling by placing cells in hypoosmotic medium (Fig. 4A). This treatment led to a several-fold increase in the release of pre-loaded D[3H]aspartate (non-metabolized analogue of L-glutamate). Neither tempol, nor edaravone affected swelling-activated D-[3H]aspartate efflux when tested at 1 and 10 mM (i.e. up to the highest concentration used in the microdialysis experiments). In contrast, the potent VRAC blocker DCPIB completely abolished hypoosmotic D-[3H]aspartate release (Fig. 4A). The effect of DCPIB was consistent with literature findings [66] and confirmed specificity of this assay.
FIG. 4. Evaluation of direct effects of tempol and edaravone on four oxidant-sensitive transport systems contributing to release of glutamate and aspartate in primary cultures of astrocytes and microglia.

A, Effects of tempol and edaravone (1 or 10 mM) on glutamate (D[3H]aspartate) release via volume-regulated anion channel (VRAC). VRAC was activated by exposure to hypoosmotic medium. The VRAC blocker 20 μM DCPIB was used as a positive control. Data are the mean values of 6 independent experiments. ***p<0.001 vs. Basal medium; ###p<0.001 vs. release in swollen cells. B, Effects of antioxidants on D-[3H]aspartate release via connexin-43 hemichannels (Cx43). Cx hemichannels were activated by incubation in Ca2+/Mg2+-free medium (CMF). The Cx43 blocker 10 μM 18α-glycyrrhetinic acid (18-αGA) was used as a positive control. Data are the mean values of 6 experiments. ***p<0.001 vs. Basal medium; ###p<0.001 vs. release induced by CMF. C, Effect of antioxidants on function of the cystine/glutamate antiporter (xCT). The xCT activity was measured as uptake of 1 μM L[14C]cystine. The xCT blocker 1 mM L-serine-O-sulfate (L-SOS) was used as a positive control. Data are the mean values of 3 experiments. ***p<0.001 vs. Basal medium. D, Effect of antioxidants on the glutamate transporter GLT-1. The GLT-1 activity was measured as uptake of L-[3H]glutamate. The GLT-1 blocker 1 mM dihydrokainate (DHK) was used as a positive control. Data are the mean values of 5 experiments. ***p<0.001 vs. Basal uptake.
To trigger opening of connexin hemichannels, we exposed primary astrocytes to Ca2+- and Mg2+-free (CMF) medium (Fig. 4B). Our recent study validated that this treatment stimulates glutamate (D-[3H] aspartate) release that is exclusively mediated by the Cx43 protein [46]. Again, tempol was completely ineffective in inhibiting Cx43-induced amino acid release, while edaravone showed partial inhibition, but only when tested at the highest concentration, 10 mM (Fig. 4B). As a positive control we used 18α-glycyrrhetinic acid (18α-GA), a known blocker of connexin hemichannels [46]. At the concentration of 10 μM 18α-GA completely suppressed the effect of CMF on astrocytic D-[3H]aspartate release, again confirming sensitivity and specificity of this approach.
We next examined whether antioxidants have a direct effect on the cystine/glutamate antiporter, which supplies cells with cystine (oxidized cysteine) in exchange for intracellular glutamate. Oxidative stress and proinflammatory cytokines activate this system, primarily via changes in expression levels [64, 67]. To isolate the xCT activity from other amino acid transporters, we measured astrocytic uptake of L-[14C]cystine under conditions described in Materials and Methods, and validated in our preceding work [46]. As shown in Fig. 4C, tempol did not affect L-[14C]cystine transport rates. Edaravone significantly blocked L-[14C]cystine uptake, but only when used at the concentration of 10 mM. As a positive control, we utilized the xCT blocker 1 mM L-SOS [68], which suppressed transport rates by ~80%, confirming that L[14C]cystine uptake is primarily mediated by the xCT.
Finally, we measured the effects of tempol and edaravone on the activity of the GLT-1 glutamate transporter in microglia. These cells uptake glutamate via two transporters, GLT-1 and GLAST, which can be discriminated by using the well-known GLT-1 inhibitor dihydrokainate [48]. As shown in Fig. 4D, DHK blocked glutamate uptake by 40–50%, which was consistent with previous studies [46, 48]. Tempol showed no effect on microglial glutamate uptake at all concentrations tested, suggesting it does not affect the GLT-1 activity (Fig. 4D). Edaravone was not effective at the 1 mM concentration, but suppressed glutamate transport when added to the experimental media at 10 mM (Fig. 4D).
To summarize, when tested in vitro, one of the antioxidants, edaravone, showed partial “off-target” inhibition of Cx43 hemichannels, xCT, and GLT-1, when used at the highest concentration of 10 mM (Fig. 4B, C, D). This effect was not seen in vivo, likely due to dilution during the process of microdialysate delivery. In contrast, tempol produced no “off-target” effects on any of the pathologically relevant glutamate release pathways. By comparing in vitro data to the effects of the antioxidants on microdialysate amino acid levels in vivo (Figs. 1 and 2), we can conclude that the in vivo effects of tempol are likely specific and related to its antioxidant properties.
Quantitative comparison of the antioxidant capacities of tempol and edaravone in vitro
Because we found a paradoxical difference in the effects of two antioxidants on excitatory amino acid release in vivo, we decided to directly compare the antioxidant capacity of tempol and edaravone in vitro, under conditions allowing for side-by-side evaluation and exposure to “individual” ROS and RNS. We started with a generic test for total antioxidant capacity, which is based on oxidation of the colored free radical molecule DPPH (see Materials and Methods). In this cell-free assay, tempol and edaravone showed very similar antioxidant potency, with edaravone being slightly more effective (Fig. 5B). This was somewhat surprising because edaravone produced no effect in microdialysis and neuroprotection experiments. Therefore, we expanded the scope of our in vitro study and evaluated relative potency of both antioxidants against several major ROS and RNS, which are thought to be critical for ischemic tissue damage. Specifically, we focused on the upstream ROS superoxide radical (O2•−), and on the products of O2•− conversions: H2O2, • OH, and ONOO− [6, 13] (see diagram in Fig. 5A).
FIG. 5. Assays of the antioxidant scavenging activity of tempol and edaravone in model oxidant-generating systems in vitro.

A, Simplified diagram depicting sources for reactive oxygen and reactive nitrogen species in the ischemic brain. B, Total antioxidant capacity of tempol and edaravone measured using a DPPH assay. Data are the mean values of 3 independent experiments. C, Effects of antioxidants on levels of O2•− that was generated by 3 mU of xanthine oxidase (XO) in the presence of 300 μM hypoxanthine (HX) and quantified using a lucigenin assay (n=4). D, As a control for the specificity of O2•− signal, lucigenin assays were additionally performed in the absence of XO or in the presence of superoxide dismutase (SOD, 200 U/ml). n=4. ***p<0.001 vs. HX+XO. E, The H2O2-scavenging capacity of antioxidants measured in solution of 25 μM H2O2 with an Amplex red assay (n=3). F, Effects of antioxidants on hydroxyl radical-induced lipid peroxidation in the presence of 7.5 μM Fe2+ and 1.5 mM ascorbate and quantified using a TBARS assay (n=4). ***p<0.001 vs. control; ##p<0.01, ###p<0.001, antioxidants vs. Fe2+ + ascorbate alone; @@p<0.01, tempol vs. edaravone. The •OH and lipid peroxide scavenger 10 μM butylated hydroxytoluene (BHT) was used as a positive control. G, Effects of antioxidants on oxidation of dihydrorhodamine123 (DHR123) in the presence of the peroxynitrite donor 1.5 mM SIN-1. n=3.
In the next set of experiments, we used hypoxanthine and xanthine oxidase to generate O2•−, and the O2•−-induced lucigenin chemiluminescence as readout. Tempol scavenged O2•− in a dose-dependent manner, and at 1 mM reduced the lucigenin signal almost completely (Fig. 5C). These findings are consistent with known properties of tempol as a superoxide dismutase mimetic [69]. In striking contrast, edaravone failed to show any effect on O2•− levels (Fig. 5C). As a control, we measured lucigenin signal when xanthine oxidase was omitted from the reaction mix, or in the presence of SOD. Under these conditions lucigenin chemiluminescence was completely inhibited (Fig. 5D), confirming specificity of the O2•− signal in our assay.
The relative H2O2-scavenging capacity was tested with exogenous H2O2 and an Amplex red H2O2 assay. At variance with the results obtained for O2•−, edaravone strongly and dose-dependently scavenged H2O2 with the EC50 of 30 μM, while tempol produced very weak scavenging activity seen only at the maximal tested concentration of 1 mM (Fig. 5E).
We further measured the antioxidant capacity of edaravone and tempol against •OH-driven lipid oxidation. •OH is a potent biological oxidant that is formed during catalytic decomposition of H2O2. To accomplish this, we quantified lipid peroxidation in rat brain synaptosomes in the presence of the •OH-generating combination of Fe2+ and ascorbate. Lipid peroxides were detected as TBARS [53]. As shown in Fig. 5F, in this detection system, 1 mM tempol and edaravone strongly reduced lipid peroxidation, to the same extent as 10 μM butylated hydroxytoluene (BHT). BHT was used as a positive control since it is well known for its ability to terminate free radical reactions in lipid phase. At a lower concentration of 100 μM, edaravone was more potent than tempol (Fig. 5F).
Finally, we performed a side-by-side comparison of the two antioxidants for their ability to prevent oxidation induced by the pathologically relevant ONOO− and its decomposition products. To this end, we monitored the oxidation of dihydrorhodamine 123 in the presence of the ONOO− donor SIN-1, which reflect the reaction of DHR123 with the products of the ONOO decomposition [54, 55]. In a cell-free system, edaravone prevented DHR123 oxidation with near complete inhibition at concentrations ≥30 μM (Fig. 5G). Tempol was only partially effective, and the incomplete (~50%) inhibition by this agent appeared to saturate at concentrations ≥100 μM.
In summary, our data on antioxidant capacity clearly show that tempol and edaravone have quite similar total antioxidant activities when compared under “ideal” conditions in a DPPH assay performed in methanol. However, when tested in vitro against a variety of pathologically relevant ROS and RNS, tempol was inferior to edaravone with one clear exception: its ability to decrease levels of the superoxide anion.
Superoxide does not directly activate glutamate release in cultured glial cells
As shown in the previous sections, tempol but not edaravone prevented pathological amino acid release in vivo. Since we found the preferential ability of tempol to reduce levels of the O2•− radical, we hypothesized that O2•− activates or potently modulates the release of glutamate via one or several transport pathways. To test this hypothesis, we exposed cultured astrocytes used as cell model to the O2•− generating mix of hypoxanthine and xanthine oxidase. To further mimic pathological conditions, we additionally exposed cells to hypoosmotic medium to trigger cell swelling because it is known that stroke causes swelling of astrocytes [70]. In this experimental paradigm, hypoxanthine/xanthine oxidase did not affect release of the glutamate analogue D-[3H]aspartate in non-swollen cells but dramatically upregulated its influx when oxidative stress was combined with hypoosmotic medium (Fig. 6A). The observed results closely resembled the previously published effects of H2O2 in cultured astrocytes and in vivo [33, 35]. Because xanthine oxidase also produces H2O2, we conducted additional control experiments. In these latter experiments, O2•− and H2O2 were scavenged by addition of SOD or catalase, respectively. As seen in Fig. 6B, catalase, but not SOD, completely reversed the effect of hypoxanthine/xanthine oxidase. These results unequivocally show that O2•− does not directly regulate glutamate transport but rather acts via formation of H2O2.
Ex vivo SH-group assay for comparing antioxidant capacity of tempol and edaravone in the ischemic tissue
The paradoxical disconnect between the results of experiments performed in vivo and in vitro made it difficult to decipher the potential mechanism (or mechanisms) of tempol neuroprotection in the MCAo model. We speculated that edaravone, although generally a more potent antioxidant, either was not equilibrated throughout the brain tissue or did not penetrate as effectively inside the intracellular compartment, as compared to tempol. To test this idea, we measured total levels of sulfhydryl (SH-) groups inside tissue samples, which were isolated from the ischemic cortex. We compared the obtained values to SH-group content in matching samples from the contralateral brain. In tissues, a large fraction of SH-groups in proteins and low molecular weight peptides, such as glutathione, is normally maintained in a reduced state. In ischemia, decline in the total SH-group content reflects intracellular oxidation by numerous ROS and RNS (see for example [71]). Here, we quantitatively analyzed SH-group levels after 2-h MCAo followed by 2 h of reperfusion. We reasoned that at this time we would see oxidative damage that occurred during both ischemia and reperfusion. We also expected that antioxidants delivered intracerebroventricularly would still be present inside the brain tissue. As shown in Fig. 7, we found a moderate but significant reduction in the SH-group content on the ischemic side of the brain.
FIG. 7. Comparison of the antioxidant capacity of tempol and edaravone in vivo based on their ability to prevent oxidation of tissue sulfhydryl (SH-) groups.

Five μL of tempol (500 nmols), edaravone (500 nmols), or vehicle (DMSO) were injected into lateral ventricle 10 min before MCAo. After 2-hr MCAo plus 2-hr reperfusion, animals were euthanized and their cortical tissue was processed for determination of the SH group content (DTNB assay). n=5 for sham, and n=6 for all ischemia groups. *p<0.05, **p<0.01, as indicated.
We found that edaravone recovered SH-group content on the ischemic side of the brain (p<0.01), while tempol did not produce statistically significant improvements (Fig. 7). These results are critical for the interpretation of the present work because they indicate that edaravone exerts higher antioxidant capacity in vivo, but paradoxically fails to attenuate glutamate release and brain damage.
Discussion
The neuroprotective properties of antioxidants in animal models of stroke have been proven beyond doubt (reviewed in [12, 13]). Yet, failure of several antioxidants and free radical scavengers in clinical trials created an uncertainty about the utility of antioxidant strategies in human stroke [29, 30]. These clinical setbacks underscore a need to improve our understanding of the molecular mechanisms, which determine the efficacy of individual antioxidants in the ischemic tissue. The traditional point of view is that ischemia initiates injurious cascades via the anoxic release of the excitatory neurotransmitter glutamate, which triggers downstream degradative processes. Oxidative, nitrosative, and nitrative stresses are thought to play a terminal role in tissue damage. The findings of this work and our previous studies [32–35] point to a more complex picture and suggest that oxidative stress also acts upstream of glutamate release, and amplifies brain damage via stimulation of one or several glutamate release pathways. Thus, we propose the existence of a pathological feed forward “loop” that involves excitatory amino acids and ROS/RNS as graphically depicted in Fig. 8 and discussed below.
FIG. 8. Working model that integrates the traditional view on the role of oxidative stress in ischemic tissue damage, and the new findings of the link between oxidative stress and pathological glutamate release.

Anoxic glutamate release is thought to initiate tissue damage via activation of ionotropic glutamate receptors (NMDA-R) and increases in cytosolic [Ca2+], which lead to terminal oxidative and nitrosative damage of neuronal cells. The relevant pathways are targeted by broad spectrum antioxidants, such as edaravone. New findings of the present study suggest that oxidative stress also acts upstream of pathological glutamate release via stimulation of glutamate permeability pathways by yet unidentified ROS intermediate (X, see text for detailed discussion). Amplification of glutamate release by the O2•− -derived ROS propagates tissue damage in the ischemic penumbra, and is preferentially targeted by the SOD mimetics tempol. Dohare et al.
Evidence that oxidative stress can modulate glutamate release in stroke
Prior studies in the field indicate that anoxic depolarization of brain tissue in stroke triggers massive release of glutamate and other neurotransmitters (see for example [61, 72–74]). Pathological release of glutamate is thought to be largely mediated by two mechanisms: activation of glutamate-permeable anion channels and the reversed operation of glial glutamate transporters, although other release routs may also contribute. The first of these pathways, anion channels are believed to be activated by cellular swelling, which represents one of the hallmarks of cerebral ischemia [60, 70]. In vitro, cell swelling causes opening of chloride/anion channel termed volume-regulated anion channel (VRAC) [62]. In vivo, non-selective VRAC blockers, including SITS, NPPB, DNDS, and tamoxifen, potently reduce glutamate release in the ischemic core and penumbra [61, 65, 74, 75]. These findings have been confirmed with the more selective VRAC inhibitor DCPIB [76]. Importantly, systemic delivery of the BBB-permeable tamoxifen, or intracerebroventricular injection of DCPIB, strongly reduces infarction volumes in rodent models of transient and permanent cerebral ischemia [76–78]. The alternative route for release, astrocytic glutamate transporter GLT-1, is thought to be prevalent in the ischemic core where severe disruption of transmembrane ionic gradients causes the transporter to operate in a reverse mode [61]. This second release mechanism is unlikely to serve as a target for therapeutic intervention. Inhibition of GLT-1 can cause undesirable effects in healthy tissue and ischemic penumbra where the “normal” mode of GLT-1 activity keeps glutamate levels low [65, 79].
The novel finding of the present work is the prominent link between oxidative stress and glutamate release in ischemic penumbra. These findings may provide an opening for designing new neuroprotective strategies. Our prior in vitro and in vivo studies established that oxidative stress may strongly enhance glutamate release due to positive modulation of VRAC. Exogenous and endogenous ROS H2O2 strongly increases glutamate release in primary rat astrocytes and primary rat microglia, when VRAC is activated by cellular swelling [33, 34]. Similar effects have been observed with SIN-1, the chemical donor generating the RNS peroxynitrite [32]. Critically, H2O2 stimulates the endogenous glutamate release via VRAC in vivo, as measured with a microdialysis approach [35]. Since both cellular swelling and oxidative and nitrosative stress do occur in the ischemic tissue (reviewed in [13, 14, 70, 80]), the ROS/RNS-dependent amplification of the excitatory amino acid release in stroke is plausible. Our present data with the SOD mimetic tempol, which dramatically reduce glutamate release in the clinically relevant penumbral tissue, strongly support this idea. The caveat is that the broad spectrum antioxidant edaravone failed to limit glutamate release in MCAo, when delivered in compatible quantities via the same route. This latter finding is clearly at odds with the effects of H2O2 (that is scavenged by edaravone) on glutamate release in previously published model experiments in vitro and in vivo [33–35]. This discrepancy may be interpreted in two ways. Either H2O2 is not a prevalent ROS that stimulates glutamate release in stroke, or ischemic glutamate release originates from a different cellular compartment (not astrocytes) or involves a different transport mechanism (not VRAC) in penumbral tissue. Additional work will be needed to discriminate between these two possibilities, but this matter is secondary to the discovery that the extracellular glutamate levels are potently modulated by oxidative stress.
The superoxide-scavenging properties of tempol determine its superior protection against ischemic injury
We analyzed several potential differences between actions of tempol and edaravone, which may help to explain the high neuroprotective potential of tempol. The first question was whether tempol has off-target effects and directly blocks one or several glutamate release mechanisms in the ischemic tissue. As already mentioned, the ischemic glutamate release in penumbra is thought to be largely mediated by VRAC, which is selectively blocked by DCPIB [65]. The selectivity of DCPIB was, however, questioned in our recent work, which established that besides VRAC, this compound also blocks Cx43 hemichannels and the glial glutamate transporter GLT-1 [46]. Keeping these more recent findings in mind, we further tested if tempol or edaravone directly inhibit four pathologically-relevant glutamate permeability pathways. We focused our attention on VRAC, connexin hemichannels, and the cystine/glutamate antiporter, because these glutamate release mechanisms are thought to be active in hypoxia and ischemia and can be directly or indirectly activated by oxidative stress [33, 63, 64]. Additionally, we tested the effects of tempol and edaravone on GLT-1, which contributes to glutamate release in the ischemic core, and is inhibited by oxidative and nitrosative stress [81]. As summarized in Fig. 4, in the absence of oxidative stress, tempol showed no direct actions on any of the above mentioned glutamate release pathways, thus ruling out the most obvious off-target actions.
Alternative possibility was that we did not achieved therapeutic levels of edaravone in vivo, even though both tempol and edaravone were delivered in identical dosages and via the same route bypassing the blood-brain barrier. This concern was addressed in experiments in which we indirectly quantified the degree of oxidative stress by measuring tissue levels of reduced thiol (SH-) groups. As in the numerous previous studies (see for example [71, 82]), we observed the reduction of SH-group content in the ischemic tissue. The effect that we found was somewhat smaller than in the previous publications because our assays were done at a very early reperfusion time. Edaravone significantly recovered tissue SH-group content, confirming that it does diminish oxidative stress. This was qualitatively similar to findings of a recent study in which the same antioxidant was delivered systemically [82]. The inability of edaravone to protect brain tissue against ischemic damage was at odds with prior in vivo studies but can likely be explained by different dosage and route of delivery (see references and discussion below). Interestingly, i.c.v. injection of tempol did not restore SH-group levels in the ischemic tissue even if this compound was highly effective in all other assays. This is likely due to the difference in the free radical scavenging properties of two antioxidants as discussed below.
To explore if the dramatic differences between the effects of tempol and edaravone in vivo are determined by their specific antioxidant properties, we compared side-by-side the ability of these two compounds to scavenge several pathologically relevant ROS and RNS in vitro. Tempol and edaravone demonstrated similar total antioxidant capacity, when tested under the “ideal” conditions of the DPPH assay. When probed against individual ROS and RNS, edaravone was superior to tempol in scavenging H2O2, ONOO− (or products of its decomposition), and blocking formation of lipid peroxides. In contrast, tempol dose-dependently reduced reactions driven by O2•−, while edaravone failed to do so. This later effect is entirely consistent with the known superoxide dismutase mimetic activity of tempol and other structurally similar nitroxide compounds [69]. Weak scavenging of H2O2 and ONOO− may explain the inability of tempol to protect SH-groups, which are readily oxidized by the latter ROS and RNS [83]. Overall, this additional information suggests that the mechanism underlying neuroprotective properties of tempol, and the associated reduction of glutamate release, involves superoxide or the superoxide-derived intermediate that is poorly scavenged by edaravone. The “usual suspect”, H2O2, which has been previously shown to enhance glutamate release in model experiments, ought to be ruled out because of the insensitivity of glutamate release to edaravone.
Potential mechanisms for reductions in ischemic glutamate release by SOD mimetics
To further understand the role of O2•− in stimulation of glutamate release in vivo we performed model experiments in primary astrocyte cultures exposed to xanthine oxidase and hypoxanthine. Detailed analysis presented in Fig. 6 indicates that O2•− cannot directly trigger glutamate release, with or without cellular swelling, but rather stimulates such release via secondary formation of H2O2. These findings were not highly surprising because we previously established that stimulation of endogenous O2•− production in microglia does not trigger glutamate release via VRAC unless O2•− is converted to H2O2 [34]. Thus, O2•− is unlikely to be directly responsible for release of the excitatory amino acids during ischemia and this latter process is activated or modulated by other O2•−-derived intermediate or intermediates. Treatments with tempol are known to reduce production of arachidonic acid metabolites, such as prostocyclins and thromboxanes, in various preparations and in vivo, (reviewed in [84]). Since two previous studies demonstrated that the cyclooxygenase inhibitor aspirin alleviate the intraischemic glutamate release and brain damage in rat MCAo [85, 86], the link to arachidonic acid metabolism deserves further exploration. An additional mechanism that may contribute to glutamate release and involve O2•− is the formation of ONOO−, which has been found to stimulate glial VRAC in vitro [32].
Another ONOO−-related possibility that should be strongly considered is that the effects of tempol are not restricted to neuronal and/or glial cells, but also take place at the neurovascular interface. Tempol is well known for its ability to improve local blood flow via scavenging of O2•− in endothelial and smooth muscle cells. Tempol preserves the bioavailability of the vasodilator molecule •NO because in pathologies •NO is consumed when combined with O2•− to form ONOO− (for comprehensive review see [84]). Even small increases in blood flow rates in the ischemic penumbra may reduce pathological release of the excitatory amino acids. In the present experiments we observed modest spontaneous recovery of blood flow rates during MCAo (from the initial values of ~20% of the pre-ischemic controls to 25–30%), which coincided with partial restoration of the extracellular levels of glutamate and aspartate (compare Fig. 1B to 1C and 1E). The idea of protection via improved collateral blood flow is particularly attractive since it readily explains why edaravone, which unlike tempol is incapable of scavenging O2•−, is not effective. Unfortunately, we do not have direct support for the “blood flow” hypothesis. In our study, changes in blood flow rates were measured with a laser Doppler, and no statistical differences were detected between tempol-, edaravone- and vehicle-treated groups. However, the laser Doppler signal is captured from the surface pial vessels, while tempol is delivered via microdialysate probe inserted in cortical tissue. Therefore, potential effects of tempol on blood flow will have to be further explored using more advanced techniques.
Is there a therapeutic potential for SOD mimetics?
Our present findings should be placed in the context of previous work with tempol and edaravone in experimental stroke. In order to directly compare efficacies of the two antioxidants, we injected both agents into the lateral ventricle prior to ischemia, or delivered them via a microdialysis probe. Prior animal studies utilized more clinically compatible routes of administration of the same drugs. Systemic delivery of 10–100 mg/kg tempol produces significant reduction of brain infarction in both transient and permanent MCAo with the therapeutic window of up to 2 h [87, 88]. These effects have been reproduced with several structurally unrelated superoxide dismutase mimetics based on metalloporphyrins, with one caveat: the latter compounds have low BBB permeability and therefore are delivered directly inside the brain. In one study, the metalloporphyrin SOD mimetic MnTE-2-PyP decreased brain infarction volumes by >70% when administered up to 6 h after initiation of ischemia [23]. Similar outcomes have been reported for two other metalloporphyrins, with one of them AEOL 1050 reducing the infarction volumes by 43% when delivered with the delay of 7.5 h post initiation of ischemia [89]. Compared to tempol, edaravone was tested more extensively, and showed strong neuroprotective properties at the dosages of 3–10 mg/kg and with the therapeutic window of up to 6 h (see for example [90–92]). However, in clinical settings and in the majority of animal studies edaravone treatments involved multiple administrations, unlike what has been done by us.
Based on the present findings we speculate that the superoxide dismutase mimetics may have intrinsically higher neuroprotective potency in brain tissue as compared to edaravone and other clinically tested antioxidants including NXY-059. The SOD mimetics may reduce, slow down, or stop propagation of the ischemic damage via limiting the superoxide-anion dependent glutamate release in ischemic penumbra (see Fig. 8). Critically, in vivo potency of tempol and other similar in action compounds cannot be reliably predicted based on traditional in vitro screening approach. Typically, pilot in vitro experiments seek for the “ideal” antioxidant that would scavenge the most reactive ROS and RNS, such as peroxynitrite, hydroxyl radical, and lipid peroxides. As seen in the present work, protective effects of antioxidants on glutamate release, changes in blood flow, etc. cannot be captured in simple in vitro models. Further developments in our understanding of sites and mechanisms of antioxidant actions in ischemic brain may bring into clinical practice new effective stroke therapies.
Supplementary Material
Highlights.
Oxidative stress is an important contributor to brain damage in stroke;
Despite preclinical data, several tested antioxidant agents failed in human stroke trials;
We explored a new link between oxidative stress and the release of toxic neurotransmitter glutamate;
The SOD mimetic tempol reduced glutamate release and brain damage in animal stroke;
Our findings may help in developing new, more effective antioxidant therapies.
Acknowledgments
We thank Vivek Bhatty for assistance with in vivo experiments and animal care, and Dr. Sarah E. McCallum for help with establishing microdialysis technique. This study was supported in part by grant from the National Institutes of Health (R01 NS61953 to A.A.M.), the American Heart Association Student Scholarship in Cerebrovascular Disease and Stroke (A.V.), and AHA Predoctoral Fellowship (13PRE17220030 to N.H.B.).
Abbreviations used
- aCSF
artificial cerebrospinal fluid
- BHT
butylated hydroxytoluene
- Cx43
connexin 43
- DCPIB
4-[(2-butyl-6,7-dichloro-2-cyclopen-tyl-2,3-dihydro-1-oxo-1H-inden-5-yl)oxy]butanoic acid
- DHK
dihydrokainate
- DHR123
dihydrorhodamine 123
- DMSO
dimethyl sulfoxide
- DPPH
2,2-diphenyl-1-picrylhydrazyl
- DTNB
5, 5′-dithiobis-(2-nitrobenzoic acid)
- DTT
dithiothreitol
- 18-αGA
18α-glycyrrhetinic acid
- i.c.v
intracerebroventricular
- MCA
middle cerebral artery
- MCAo
middle cerebral artery occlusion
- MEM
minimal essential medium
- SDS
sodium dodecyl sulfate
- SIN-1
3-morpholinosydnonimine
- SOD
superoxide dismutase
- L-SOS
L-serine O-sulfate
- TBARS
thiobarbituric acid reactive substances
- TTC
2,3,5-triphenyltetrazolium chloride
- VRAC
volume-regulated anion channel
- xCT
cystine/glutamate antiporter
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could a3ect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Brott T, Bogousslavsky J. Treatment of acute ischemic stroke. N Engl J Med. 2000;343:710–722. doi: 10.1056/NEJM200009073431007. [DOI] [PubMed] [Google Scholar]
- 2.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, Alvarado M, Anderson HR, Anderson LM, Andrews KG, Atkinson C, Baddour LM, Barker-Collo S, Bartels DH, Bell ML, Benjamin EJ, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Towfighi A, Saver JL. Stroke declines from third to fourth leading cause of death in the United States: historical perspective and challenges ahead. Stroke. 2011;42:2351–2355. doi: 10.1161/STROKEAHA.111.621904. [DOI] [PubMed] [Google Scholar]
- 4.Fang MC, Cutler DM, Rosen AB. Trends in thrombolytic use for ischemic stroke in the United States. J Hosp Med. 2010;5:406–409. doi: 10.1002/jhm.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ginsberg MD. Neuroprotection for ischemic stroke: past, present and future. Neuropharmacology. 2008;55:363–389. doi: 10.1016/j.neuropharm.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- 7.Mongin AA. Disruption of ionic and cell volume homeostasis in cerebral ischemia: he perfect storm. Pathophysiology. 2007;14:183–193. doi: 10.1016/j.pathophys.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 9.Rothman SM, Olney JW. Excitotoxicity and the NMDA receptor. Trends Neurosci. 1987;10:299–302. doi: 10.1016/0166-2236(95)93869-y. [DOI] [PubMed] [Google Scholar]
- 10.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Dawson VL, Dawson TM. Poly(ADP-ribose) signals to mitochondrial AIF: a key event in parthanatos. Exp Neurol. 2009;218:193–202. doi: 10.1016/j.expneurol.2009.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- 13.Margaill I, Plotkine M, Lerouet D. Antioxidant strategies in the treatment of stroke. Free Radic Biol Med. 2005;39:429–443. doi: 10.1016/j.freeradbiomed.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 14.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Denicola A, Freeman BA, Trujillo M, Radi R. Peroxynitrite reaction with carbon dioxide/bicarbonate: kinetics and influence on peroxynitrite-mediated oxidations. Arch Biochem Biophys. 1996;333:49–58. doi: 10.1006/abbi.1996.0363. [DOI] [PubMed] [Google Scholar]
- 16.Jourd’heuil D, Jourd’heuil FL, Kutchukian PS, Musah RA, Wink DA, Grisham MB. Reaction of superoxide, nitric oxide with peroxynitrite. Implications for peroxynitrite-mediated oxidation reactions in vivo. J Biol Chem. 2001;276:28799–28805. doi: 10.1074/jbc.M102341200. [DOI] [PubMed] [Google Scholar]
- 17.Samdani AF, Dawson TM, Dawson VL. Nitric oxide synthase in models of focal ischemia. Stroke. 1997;28:1283–1288. doi: 10.1161/01.str.28.6.1283. [DOI] [PubMed] [Google Scholar]
- 18.Cao X, Phillis JW. alpha-Phenyl-tert-butyl-nitrone reduces cortical infarct and edema in rats subjected to focal ischemia. Brain Res. 1994;644:267–272. doi: 10.1016/0006-8993(94)91689-6. [DOI] [PubMed] [Google Scholar]
- 19.Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- 20.Yang G, Chan PH, Chen J, Carlson E, Chen SF, Weinstein P, Epstein CJ, Kamii H. Human copper-zinc superoxide dismutase transgenic mice are highly resistant to reperfusion injury after focal cerebral ischemia. Stroke. 1994;25:165–170. doi: 10.1161/01.str.25.1.165. [DOI] [PubMed] [Google Scholar]
- 21.Murakami K, Kondo T, Kawase M, Li Y, Sato S, Chen SF, Chan PH. Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J Neurosci. 1998;18:205–213. doi: 10.1523/JNEUROSCI.18-01-00205.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuroda S, Tsuchidate R, Smith ML, Maples KR, Siesjo BK. Neuroprotective effects of a novel nitrone, NXY-059, after transient focal cerebral ischemia in the rat. J Cereb Blood Flow Metab. 1999;19:778–787. doi: 10.1097/00004647-199907000-00008. [DOI] [PubMed] [Google Scholar]
- 23.Mackensen GB, Patel M, Sheng H, Calvi CL, Batinic-Haberle I, Day BJ, Liang LP, Fridovich I, Crapo JD, Pearlstein RD, Warner DS. Neuroprotection from delayed postischemic administration of a metalloporphyrin catalytic antioxidant. J Neurosci. 2001;21:4582–4592. doi: 10.1523/JNEUROSCI.21-13-04582.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.The Edaravone Study Group. Effect of a novel free radical scavenger edaravone (MCI-186) on acute brain infarction. Randomized, placebo-controlled, double-blind study at multicenters. Cerebrovasc Dis. 2003;15:222–229. doi: 10.1159/000069318. [DOI] [PubMed] [Google Scholar]
- 25.Toyoda K, Fujii K, Kamouchi M, Nakane H, Arihiro S, Okada Y, Ibayashi S, Iida M. Free radical scavenger, edaravone, in stroke with internal carotid artery occlusion. J Neurol Sci. 2004;221:11–17. doi: 10.1016/j.jns.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 26.Cheng YD, Al Khoury L, Zivin JA. Neuroprotection for ischemic stroke: two decades of success and failure. NeuroRx. 2004;1:36–45. doi: 10.1602/neurorx.1.1.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ginsberg MD. Life after Cerovive. A personal perspective on ischemic neuroprotection in the post-NXY-059 era. Stroke. 2007;38:1967–1972. doi: 10.1161/STROKEAHA.106.479170. [DOI] [PubMed] [Google Scholar]
- 28.Diener HC, Lees KR, Lyden P, Grotta J, Davalos A, Davis SM, Shuaib A, Ashwood T, Wasiewski W, Alderfer V, Hardemark HG, Rodichok L. NXY-059 for the treatment of acute stroke: pooled analysis of the SAINT I and II Trials. Stroke. 2008;39:1751–1758. doi: 10.1161/STROKEAHA.107.503334. [DOI] [PubMed] [Google Scholar]
- 29.Pearson H. The bitterest pill. Nature. 2006;444:532–533. doi: 10.1038/444532a. [DOI] [PubMed] [Google Scholar]
- 30.Hill MD. Stroke: the dashed hopes of neuroprotection. Lancet Neurol. 2007;6:2–3. doi: 10.1016/S1474-4422(06)70658-5. [DOI] [PubMed] [Google Scholar]
- 31.Savitz SI. A critical appraisal of the NXY-059 neuroprotection studies for acute stroke: A need for more rigorous testing of neuroprotective agents in animal models of stroke. Exp Neurol. 2007;205:20–25. doi: 10.1016/j.expneurol.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 32.Haskew RE, Mongin AA, Kimelberg HK. Peroxynitrite enhances astrocytic volume-sensitive excitatory amino acid release via a src tyrosine kinase-dependent mechanism. J Neurochem. 2002;82:903–912. doi: 10.1046/j.1471-4159.2002.01037.x. [DOI] [PubMed] [Google Scholar]
- 33.Haskew-Layton RE, Mongin AA, Kimelberg HK. Hydrogen peroxide potentiates volume-sensitive excitatory amino acid release via a mechanism involving Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 2005;280:3548–3554. doi: 10.1074/jbc.M409803200. [DOI] [PubMed] [Google Scholar]
- 34.Harrigan TJ, Abdullaev IF, Jourd’heuil D, Mongin AA. Activation of microglia with zymosan promotes excitatory amino acid release via volume-regulated anion channels: the role of NADPH oxidases. J Neurochem. 2008;106:2449–2462. doi: 10.1111/j.1471-4159.2008.05553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haskew-Layton RE, Rudkouskaya A, Jin Y, Feustel PJ, Kimelberg HK, Mongin AA. Two distinct modes of hypoosmotic medium-induced release of excitatory amino acids and taurine in the rat brain in vivo. PLoS ONE. 2008;3:e3543. doi: 10.1371/journal.pone.0003543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 37.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. San Diego, CA: Elsevier/Academic Press; 2009. [Google Scholar]
- 38.Bederson JB, Pitts LH, Germano SM, Nishimura MC, Davis RL, Bartkowski HM. Evaluation of 2,3,5-triphenyltetrazolium chloride as a stain for detection and quantification of experimental cerebral infarction in rats. Stroke. 1986;17:1304–1308. doi: 10.1161/01.str.17.6.1304. [DOI] [PubMed] [Google Scholar]
- 39.Rasband WS. ImageJ. Bethesda, Maryland: U. S. National Institutes of Health; 1997. ( http://imagej.nih.gov/ij) [Google Scholar]
- 40.Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- 41.Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit, extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke. 1995;26:627–634. doi: 10.1161/01.str.26.4.627. [DOI] [PubMed] [Google Scholar]
- 42.Schallert T, Upchurch M, Lobaugh N, Farrar SB, Spirduso WW, Gilliam P, Vaughn D, Wilcox RE. Tactile extinction: distinguishing between sensorimotor and motor asymmetries in rats with unilateral nigrostriatal damage. Pharmacol Biochem Behav. 1982;16:455–462. doi: 10.1016/0091-3057(82)90452-x. [DOI] [PubMed] [Google Scholar]
- 43.Balkaya M, Krober JM, Rex A, Endres M. Assessing post-stroke behavior in mouse models of focal ischemia. J Cereb Blood Flow Metab. 2013;33:330–338. doi: 10.1038/jcbfm.2012.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ando Y, Steiner M. Sulfhydryl, disulfide groups of platelet membranes I. Determination of sulfhydryl groups. Biochim Biophys Acta. 1973;311:26–37. doi: 10.1016/0005-2736(73)90251-4. [DOI] [PubMed] [Google Scholar]
- 45.Hyzinski-Garcia MC, Vincent MY, Haskew-Layton RE, Dohare P, Keller RW, Jr, Mongin AA. Hypoosmotic swelling modifies glutamate-glutamine cycle in the cerebral cortex and in astrocyte cultures. J Neurochem. 2011;118:140–152. doi: 10.1111/j.1471-4159.2011.07289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bowens NH, Dohare P, Kuo YH, Mongin AA. DCPIB, the proposed selective blocker of volume-regulated anion channels, inhibits several glutamate transport pathways in glial cells. Mol Pharmacol. 2013;83:22–32. doi: 10.1124/mol.112.080457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gegelashvili G, Danbolt NC, Schousboe A. Neuronal soluble factors differentially regulate the expression of the GLT1 and GLAST glutamate transporters in cultured astroglia. J Neurochem. 1997;69:2612–2615. doi: 10.1046/j.1471-4159.1997.69062612.x. [DOI] [PubMed] [Google Scholar]
- 48.Nakajima K, Tohyama Y, Kohsaka S, Kurihara T. Ability of rat microglia to uptake extracellular glutamate. Neurosci Lett. 2001;307:171–174. doi: 10.1016/s0304-3940(01)01943-7. [DOI] [PubMed] [Google Scholar]
- 49.Blois MS. Antioxidant determinations by the use of a stable free radical. Nature. 1958;181:1199–1200. [Google Scholar]
- 50.Storch J, Ferber E. Detergent-amplified chemiluminescence of lucigenin for determination of superoxide anion production by NADPH oxidase and xanthine oxidase. Anal Biochem. 1988;169:262–267. doi: 10.1016/0003-2697(88)90283-7. [DOI] [PubMed] [Google Scholar]
- 51.Hajos F. An improved method for the preparation of synaptosomal fractions in high purity. Brain Res. 1975;93:485–489. doi: 10.1016/0006-8993(75)90186-9. [DOI] [PubMed] [Google Scholar]
- 52.Rudkouskaya A, Sim V, Shah AA, Feustel PJ, Jourd’heuil D, Mongin AA. Long-lasting inhibition of presynaptic metabolism and neurotransmitter release by protein S-nitrosylation. Free Radic Biol Med. 2010;49:757–769. doi: 10.1016/j.freeradbiomed.2010.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 1979;95:351–358. doi: 10.1016/0003-2697(79)90738-3. [DOI] [PubMed] [Google Scholar]
- 54.Crow JP. Dichlorodihydrofluorescein and dihydrorhodamine 123 are sensitive indicators of peroxynitrite in vitro: implications for intracellular measurement of reactive nitrogen and oxygen species. Nitric Oxide. 1997;1:145–157. doi: 10.1006/niox.1996.0113. [DOI] [PubMed] [Google Scholar]
- 55.Kalyanaraman B, Darley-Usmar V, Davies KJ, Dennery PA, Forman HJ, Grisham MB, Mann GE, Moore K, Roberts LJ, Ischiropoulos H. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic Biol Med. 2012;52:1–6. doi: 10.1016/j.freeradbiomed.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Banderali U, Roy G. Anion channels for amino-acids in Mdck cells. Am J Physiol. 1992;263:C1200–C1207. doi: 10.1152/ajpcell.1992.263.6.C1200. [DOI] [PubMed] [Google Scholar]
- 57.Kirk K, Strange K. Functional properties and physiological roles of organic solute channels. Annu Rev Physiol. 1998;60:719–739. doi: 10.1146/annurev.physiol.60.1.719. [DOI] [PubMed] [Google Scholar]
- 58.van der Worp HB, Sena ES, Donnan GA, Howells DW, Macleod MR. Hypothermia in animal models of acute ischaemic stroke: a systematic review and meta-analysis. Brain. 2007;130:3063–3074. doi: 10.1093/brain/awm083. [DOI] [PubMed] [Google Scholar]
- 59.Kimelberg HK, Mongin AA. Swelling-activated release of excitatory amino acids in the brain: Relevance for pathophysiology. Contrib Nephrol. 1998;123:240–257. doi: 10.1159/000059916. [DOI] [PubMed] [Google Scholar]
- 60.Kimelberg HK. Astrocytic swelling in cerebral ischemia as a possible cause of injury and target for therapy. Glia. 2005;50:389–397. doi: 10.1002/glia.20174. [DOI] [PubMed] [Google Scholar]
- 61.Seki Y, Feustel PJ, Keller RW, Jr, Tranmer BI, Kimelberg HK. Inhibition of ischemia-induced glutamate release in rat striatum by dihydrokinate and an anion channel blocker. Stroke. 1999;30:433–440. doi: 10.1161/01.str.30.2.433. [DOI] [PubMed] [Google Scholar]
- 62.Kimelberg HK, Goderie SK, Higman S, Pang S, Waniewski RA. Swelling-induced release of glutamate, aspartate, and taurine from astrocyte cultures. J Neurosci. 1990;10:1583–1591. doi: 10.1523/JNEUROSCI.10-05-01583.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ye ZC, Wyeth MS, Baltan-Tekkok S, Ransom BR. Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J Neurosci. 2003;23:3588–3596. doi: 10.1523/JNEUROSCI.23-09-03588.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fogal B, Li J, Lobner D, McCullough LD, Hewett SJ. System x(c)- activity and astrocytes are necessary for interleukin-1 beta-mediated hypoxic neuronal injury. J Neurosci. 2007;27:10094–10105. doi: 10.1523/JNEUROSCI.2459-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Feustel PJ, Jin Y, Kimelberg HK. Volume-regulated anion channels are the predominant contributors to release of excitatory amino acids in the ischemic cortical penumbra. Stroke. 2004;35:1164–1168. doi: 10.1161/01.STR.0000124127.57946.a1. [DOI] [PubMed] [Google Scholar]
- 66.Abdullaev IF, Rudkouskaya A, Schools GP, Kimelberg HK, Mongin AA. Pharmacological comparison of swelling-activated excitatory amino acid release and Cl- currents in rat cultured astrocytes. J Physiol. 2006;572:677–689. doi: 10.1113/jphysiol.2005.103820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bannai S, Sato H, Ishii T, Sugita Y. Induction of cystine transport activity in human fibroblasts by oxygen. J Biol Chem. 1989;264:18480–18484. [PubMed] [Google Scholar]
- 68.Patel SA, Warren BA, Rhoderick JF, Bridges RJ. Differentiation of substrate and non-substrate inhibitors of transport system xc(−): an obligate exchanger of L-glutamate and L-cystine. Neuropharmacology. 2004;46:273–284. doi: 10.1016/j.neuropharm.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 69.Krishna MC, Russo A, Mitchell JB, Goldstein S, Dafni H, Samuni A. Do nitroxide antioxidants act as scavengers of O2-or as SOD mimics? J Biol Chem. 1996;271:26026–26031. doi: 10.1074/jbc.271.42.26026. [DOI] [PubMed] [Google Scholar]
- 70.Mongin AA, Kimelberg HK. Astrocytic swelling in neuropathology. In: Kettenmann H, Ransom BR, editors. Neuroglia. Oxford/New York: Oxford University Press; 2005. pp. 550–562. [Google Scholar]
- 71.Shivakumar BR, Kolluri SV, Ravindranath V. Glutathione and protein thiol homeostasis in brain during reperfusion after cerebral ischemia. J Pharmacol Exp Ther. 1995;274:1167–1173. [PubMed] [Google Scholar]
- 72.Benveniste H, Drejer J, Schousboe A, Diemer NH. Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J Neurochem. 1984;43:1369–1374. doi: 10.1111/j.1471-4159.1984.tb05396.x. [DOI] [PubMed] [Google Scholar]
- 73.Hagberg H, Lehmann A, Sandberg M, Nystrom B, Jacobson I, Hamberger A. Ischemia-induced shift of inhibitory and excitatory amino acids from intra- to extracellular compartments. J Cereb Blood Flow Metab. 1985;5:413–419. doi: 10.1038/jcbfm.1985.56. [DOI] [PubMed] [Google Scholar]
- 74.Phillis JW, Song D, O’Regan MH. Inhibition by anion channel blockers of ischemia-evoked release of excitotoxic and other amino acids from rat cerebral cortex. Brain Res. 1997;758:9–16. doi: 10.1016/s0006-8993(97)00155-8. [DOI] [PubMed] [Google Scholar]
- 75.Phillis JW, Song D, O’Regan MH. Tamoxifen, a chloride channel blocker, reduces glutamate and aspartate release from the ischemic cerebral cortex. Brain Res. 1998;780:352–355. doi: 10.1016/s0006-8993(97)01352-8. [DOI] [PubMed] [Google Scholar]
- 76.Zhang Y, Zhang H, Feustel PJ, Kimelberg HK. DCPIB, a specific inhibitor of volume regulated anion channels (VRACs), reduces infarct size in MCAo and the release of glutamate in the ischemic cortical penumbra. Exp Neurol. 2008;210:514–520. doi: 10.1016/j.expneurol.2007.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kimelberg HK, Feustel PJ, Jin Y, Paquette J, Boulos A, Keller RW, Jr, Tranmer BI. Acute treatment with tamoxifen reduces ischemic damage following middle cerebral artery occlusion. Neuroreport. 2000;11:2675–2679. doi: 10.1097/00001756-200008210-00014. [DOI] [PubMed] [Google Scholar]
- 78.Kimelberg HK, Jin Y, Charniga C, Feustel PJ. Neuroprotective activity of tamoxifen in permanent focal ischemia. J Neurosurg. 2003;99:138–142. doi: 10.3171/jns.2003.99.1.0138. [DOI] [PubMed] [Google Scholar]
- 79.Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 80.Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- 81.Trotti D, Rossi D, Gjesdal O, Levy LM, Racagni G, Danbolt NC, Volterra A. Peroxynitrite inhibits glutamate transporter subtypes. J Biol Chem. 1996;271:5976–5979. doi: 10.1074/jbc.271.11.5976. [DOI] [PubMed] [Google Scholar]
- 82.Ahmad A, Khan MM, Javed H, Raza SS, Ishrat T, Khan MB, Safhi MM, Islam F. Edaravone ameliorates oxidative stress associated cholinergic dysfunction and limits apoptotic response following focal cerebral ischemia in rat. Mol Cell Biochem. 2012;367:215–225. doi: 10.1007/s11010-012-1335-6. [DOI] [PubMed] [Google Scholar]
- 83.Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite-induced membrane lipid peroxidation: the cytotoxic potential of superoxide and nitric oxide. Arch Biochem Biophys. 1991;288:481–487. doi: 10.1016/0003-9861(91)90224-7. [DOI] [PubMed] [Google Scholar]
- 84.Wilcox CS, Pearlman A. Chemistry and antihypertensive effects of tempol and other nitroxides. Pharmacol Rev. 2008;60:418–469. doi: 10.1124/pr.108.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.De Cristobal J, Moro MA, Davalos A, Castillo J, Leza JC, Camarero J, Colado MI, Lorenzo P, Lizasoain I. Neuroprotective effect of aspirin by inhibition of glutamate release after permanent focal cerebral ischaemia in rats. J Neurochem. 2001;79:456–459. doi: 10.1046/j.1471-4159.2001.00600.x. [DOI] [PubMed] [Google Scholar]
- 86.Berger C, Stauder A, Xia F, Sommer C, Schwab S. Neuroprotection and glutamate attenuation by acetylsalicylic acid in temporary but not in permanent cerebral ischemia. Exp Neurol. 2008;210:543–548. doi: 10.1016/j.expneurol.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 87.Rak R, Chao DL, Pluta RM, Mitchell JB, Oldfield EH, Watson JC. Neuroprotection by the stable nitroxide Tempol during reperfusion in a rat model of transient focal ischemia. J Neurosurg. 2000;92:646–651. doi: 10.3171/jns.2000.92.4.0646. [DOI] [PubMed] [Google Scholar]
- 88.Leker RR, Teichner A, Lavie G, Shohami E, Lamensdorf I, Ovadia H. The nitroxide antioxidant tempol is cerebroprotective against focal cerebral ischemia in spontaneously hypertensive rats. Exp Neurol. 2002;176:355–363. doi: 10.1006/exnr.2002.7910. [DOI] [PubMed] [Google Scholar]
- 89.Sheng H, Enghild JJ, Bowler R, Patel M, Batinic-Haberle I, Calvi CL, Day BJ, Pearlstein RD, Crapo JD, Warner DS. Effects of metalloporphyrin catalytic antioxidants in experimental brain ischemia. Free Radic Biol Med. 2002;33:947–961. doi: 10.1016/s0891-5849(02)00979-6. [DOI] [PubMed] [Google Scholar]
- 90.Shichinohe H, Kuroda S, Yasuda H, Ishikawa T, Iwai M, Horiuchi M, Iwasaki Y. Neuroprotective effects of the free radical scavenger Edaravone (MCI-186) in mice permanent focal brain ischemia. Brain Res. 2004;1029:200–206. doi: 10.1016/j.brainres.2004.09.055. [DOI] [PubMed] [Google Scholar]
- 91.Zhang N, Komine-Kobayashi M, Tanaka R, Liu M, Mizuno Y, Urabe T. Edaravone reduces early accumulation of oxidative products and sequential inflammatory responses after transient focal ischemia in mice brain. Stroke. 2005;36:2220–2225. doi: 10.1161/01.STR.0000182241.07096.06. [DOI] [PubMed] [Google Scholar]
- 92.Xiao B, Bi FF, Hu YQ, Tian FF, Wu ZG, Mujlli HM, Ding L, Zhou XF. Edaravone neuroprotection effected by suppressing the gene expression of the Fas signal pathway following transient focal ischemia in rats. Neurotox Res. 2007;12:155–162. doi: 10.1007/BF03033912. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.