

Abstract
AIM: To investigate the effect of Clostridium difficile (C. difficile) infection in an interleukin 10-deficient (IL-10-/-) mouse model of inflammatory bowel disease.
METHODS: Bone marrow-derived dendritic cells isolated from wild type (WT) and IL-10-/-mice were stimulated for 4 h with C. difficile toxin A (200 μg/mL), and gene expression of interferon (IFN)-γ, IL-12 and IL-23 was determined by real-time reverse transcription polymerase chain reaction. WT and IL-10-/- mice (n = 20 each) were exposed to an antibiotic cocktail for three days and then were injected with clindamycin (i.p.). Mice (n = 10 WT, 10 IL-10-/-) were then challenged with oral administration of C. difficile (1 × 105 colony forming units of strain VPI 10463). Animals were monitored daily for 7 d for signs of colitis. Colonic tissue samples were evaluated for cytokine gene expression and histopathologic analysis.
RESULTS: C. difficile toxin A treatment induced IFN-γ gene expression to a level that was significantly higher in BDMCs from IL-10-/- compared to those from WT mice (P < 0.05). However, expression of IL-12 and IL-23 was not different among the groups. Following C. difficile administration, mice developed diarrhea and lost weight within 2-3 d. Weight loss was significantly greater in IL-10-/- compared to WT mice (P < 0.05). C. difficile infection induced histopathologic features typical of colitis in both IL-10-/- and WT mice. The histopathologic severity score was significantly higher in the IL-10-/- than in WT mice (mean ± standard error; 5.50 ± 0.53 vs 2.44 ± 0.46; P < 0.05). This was accompanied by a significantly greater increase in IFN-γ gene expression in colonic tissues from IL-10-/- than from WT mice challenged with C. difficile (P < 0.05).
CONCLUSION: These results indicate that colitis is more severe after C. difficile infection in IL-10-/-mice, and that IFN-γ expression is involved in this process.
Keywords: Clostridium difficile, Inflammatory bowel disease, Interleukin 10-deficient mice, Interferon-γ, Colitis
Core tip: The results of this study indicate that Clostridium difficile (C. difficile) infection induces more severe colitis in the interleukin 10-deficient (IL-10-/-) mouse model of inflammatory bowel disease (IBD). Moreover, induction of interferon-γ gene expression was greater in the IL-10-/- mice and their bone marrow-derived dendritic cells compared to wild type mice following C. difficile infection and exposure to C. difficile toxin A, respectively. This study demonstrates the establishment of C. difficile-aggravated colitis in an IBD animal model, which provides a useful tool for studying the relationship between C. difficile and host immune response of the gut in IBD.
INTRODUCTION
Inflammatory bowel disease (IBD), which includes Crohn’s disease and ulcerative colitis, is a complex chronic inflammatory gastrointestinal disorder of unknown cause in genetically predisposed hosts[1]. There is accumulating evidence concerning the importance of intestinal microbiota in the pathogenesis of IBD, including recent culture-dependent and -independent analyses showing that patients with IBD have dysbiosis characterized by a less complex profile of commensal bacteria and a higher number of pathogenic bacteria[2].
Clostridium difficile (C. difficile) is a gram-positive, anaerobic, spore-forming bacillus. C. difficile infection (CDI) is the most common cause of nosocomial antibiotic-associated diarrhea and pseudomembranous colitis[3,4]. Disease-causing toxins released by the bacteria, toxins A and B, act directly on intestinal epithelial cells, leading to chemokine release, cell rounding and apoptosis or necrosis, resulting in inflammation and intestinal damage[5,6]. These toxins are glucosyltransferases that irreversibly inactivate Rho proteins, leading to the disruption of the cytoskeleton and tight junctions and subsequent cell death, and concurrently induce the release of interleukin (IL)-8 and intracellular adhesion molecule-1 from intestinal epithelial cells, which further leads to neutrophil adhesion, infiltration, and mucosal inflammation. Moreover, the toxins may activate immune cells and neurons once the intestinal epithelial barrier is disrupted[7].
IBD is recognized as a risk factor for CDI, and a high prevalence of CDI is observed in pediatric patients with both active and inactive Crohn’s disease and ulcerative colitis[8]. C. difficile affects the course of IBD in several ways, including triggering disease flares, sustaining activity, and in some cases, acting as a ‘‘silent partner”. Moreover, CDI is associated with a longer hospital stay and a higher morbidity and mortality in patients with IBD[9,10]. Because C. difficile colitis can both mimic and precipitate an IBD flare, it is essential that clinicians are vigilant in identifying and addressing this infection[11].
C. difficile-associated disease has been studied in many animal species, including hamsters, guinea pigs, rabbits, and germ-free mice and rats. Although hamsters have been used to investigate the pathogenesis and treatment of C. difficile colitis, they have limitations in developing fulminant and lethal cecitis[12]. In recent years, mouse models of intestinal inflammation based on bacterial infections have been used to study the roles of individual bacterial species and specific bacterial components in the pathogenesis of IBD[13]. Although a recently established mouse model of C. difficile-associated disease closely resembles the human disease[14], there are no established models to study the mechanism of CDI-induced intestinal inflammation in IBD or the pathogenic role of C. difficile in mucosal immune dysregulation. In the present study, control and IL-10-deficient (IL-10-/-) mice were used to examine C. difficile toxin A-activated bone marrow-derived dendritic cells (BMDCs) and the effect of CDI on intestinal inflammation.
MATERIALS AND METHODS
Mice
Seven-week-old specific pathogen-free male C57BL/6 wild type (WT) and IL-10-/- mice used for the experiments were supplied by the Center for Animal Resource and Development (Seoul, South Korea). Mice were maintained in a controlled laboratory environment at 24 °C ± 2 °C and 50% ± 5% humidity under a 12/12 h (light/dark) cycle. Mice were given ad libitum access to irradiated mouse feed (Purina Korea, Seoul, South Korea) and 2 ppm chloride-supplemented reverse osmosis water. To prevent mice from eating their feces, a grid floor through which fecal matter could pass was installed during the rearing period. All procedures were approved by the Institutional Animal Care and Use Committee at Seoul National University (IACUC No.SNU-100318). The experiments were conducted in accordance with the August 2010 revision of The Guiding Principles for the Care and Use of Laboratory Animals (American Physiological Society).
Generation and culture of BMDCs
BMDCs were generated from the femurs of WT and IL-10-/- mice as previously described[15]. Bone marrow cells were cultured in complete media (RPMI-1640) supplemented with 10 ng/mL of murine recombinant granulocyte-macrophage colony-stimulating factor and 10 ng/mL of murine recombinant IL-4 (Peprotech, Rocky Hill, NJ, United States). The culture medium was changed after three days, and again after five days, of culture. On the seventh day of culture, non-adherent cells were collected as BMDCs. Flow cytometric analysis indicated that > 93% of the collected cells were CD11c+ and CD11b+ (BD Pharmingen, San Diego, CA, United States)[16].
Real time reverse transcription-polymerase chain reaction
After stimulating BMDCs for 4 h with 200 μg/mL of C. difficile toxin A (Sigma-Aldrich, St. Louis, MO, United States), total RNA was extracted with Trizol (Invitrogen of Thermo Fisher Scientific Inc., Waltham, MA, United States) and reverse transcribed. Amplification was performed with SYBR Green PCR Master Mix on an ABI Prism 7000 Sequence Detection System (Applied Biosystems of Thermo Fisher Scientific Inc.) using primers for interferon (IFN)-γ, IL-12p40, and IL-23p19 (Table 1). Reactions were performed in triplicate, and the data were normalized to β-actin.
Table 1.
Primer sequences
| RNA species | Oligonucleotides, 5'->3' | |
| IFN-γ | Sense | TGCATCTTGGCTTTGCAGCTCTTC |
| Antisense | GGGTTGTTGACCTCAAACTTGGCA | |
| IL-12p40 | Sense | GGAAGCACGGCAGCAGAATA |
| Antisense | AACTTGAGGGAGAAGTAGGAATGG | |
| IL-23p19 | Sense | AGCGGGACATATGAATCTACTAAGAGA |
| Antisense | GTCCTAGTAGGGAGGTGTGAAGTTG |
IFN: Interferon; IL: Interleukin.
C. difficile culture
C. difficile strain VPI 10463 (ATCC 43255; American Type Tissue Culture Collection, Manassas, VA, United States) was used in the present study. Before oral administration to mice, the frozen strain was thawed and cultured in brain heart infusion-supplemented media at 37 °C for three days under anaerobic conditions.
Establishment of a C. difficile colitis model in IL-10-/- mice and experimental grouping
WT and IL-10-/- mice were randomly assigned to either a control group (n = 10 each) or a C. difficile group (n = 10 each) and fed irradiated mouse feed mixed with an antibiotic cocktail (40 mg/kg kanamycin, 3.5 mg/kg gentamicin, 4.2 mg/kg colistin, 21.5 mg/kg metronidazole, and 4.5 mg/kg vancomycin) for three days. One day after the last antibiotic cocktail, the mice were injected with clindamycin (10 mg/kg, i.p.), and WT and IL-10-/- mice in the C. difficile groups received oral administration of 1 × 105 colony forming units of C. difficile the next day (Figure 1). All mice were monitored daily for signs of colitis (e.g., diarrhea, hunched posture, wet tail, and weight loss[14]) and euthanized seven days later.
Figure 1.
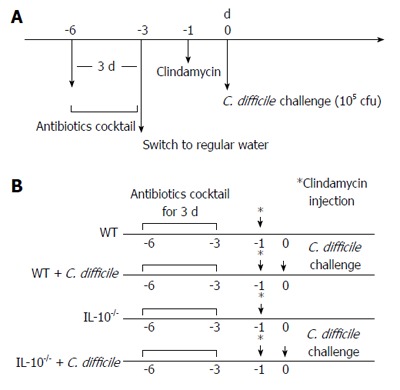
Clostridium difficile-induced colitis mouse model. A: Wild type (WT) and interleukin 10-deficient (IL-10-/-) mice were pre-treated with an antibiotic cocktail for three days followed by an i.p. injection of clindamycin two days later, before being challenged with an oral administration of Clostridium difficile (C. difficile) (cfu, colony forming units); B: The mice were divided into four groups (n = 10 each): a control and C. difficile challenge group each for WT and IL-10-/- mice. The control groups underwent all pretreatment processes in parallel with the C. difficile challenge groups, but were not administered C. difficile.
Histopathologic assessment of colonic tissue
Colonic tissues were extracted from euthanized mice and either fixed in 10% buffered formalin, embedded in paraffin and stained with hematoxylin-eosin or frozen in liquid nitrogen to quantify biochemical parameters. Histopathologic examination was performed on the proximal and distal colon of each animal by an independent pathologist who was blinded to the study methods, using previously reported classification standards[17]. Briefly, each of three factors was scored on a scale of 0 to 3 (none, mild, moderate, or severe, respectively), including neutrophil influx into tissues, colonic mucosal edema, and epithelial cell injury. The overall score was the sum of each component score. The severity of colitis was quantified by the total score.
Statistical analysis
Statistical analysis was performed using the SPSS version 19.0 statistical software (IBM, Armonk, NY, United States). Groups were compared using the Mann-Whitney U-test. All data are expressed as the mean ± standard error. P < 0.05 was considered to indicate statistical significance.
RESULTS
Cytokine gene expression in BMDCs after stimulation with C. difficile toxin A
Treatment with C. difficile toxin A significantly increased the expression of IFN-γ in BDMCs extracted from IL-10-/- mice (P < 0.05 vs WT) (Figure 2). In contrast, treatment did not affect the expression of IL-12 or IL-23 in BDMCs from WT or IL-10-/- mice.
Figure 2.
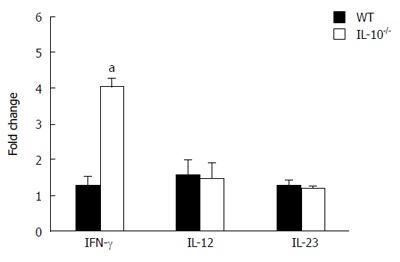
Cytokine expression in bone marrow-derived dendritic cells after Clostridium difficile toxin A stimulation. The expression levels of interferon (IFN)-γ, interleukin (IL)-12 and IL-23 mRNAs were examined in BMDCs obtained from wild type (WT) and IL-10-deficient (IL-10-/-) mice. Error bars indicate standard error of the mean; aP < 0.05 vs WT.
C. difficile-induced weight loss
There was no difference in baseline body weights between the WT and IL-10-/- mice. C. difficile exposure resulted in weight loss within 2-3 d after administration (Figure 3). Weight loss was greater in the IL-10-/- than in the WT mice (P = 0.01), with 15% weight loss in the IL-10-/- mice by the fourth day compared to 7% loss in WT mice infected with C. difficile by the third day. The mice recovered their weight loss by the seventh day after C. difficile administration.
Figure 3.
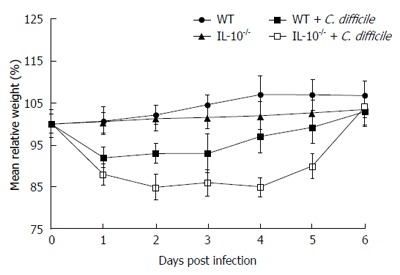
Body weight changes following Clostridium difficile challenge. Clostridium difficile (C. difficile) challenge induced a reduction in body weight that was greater in interleukin 10-deficient (IL-10-/-) compared to wild type (WT) mice. Error bars indicate standard error of the mean.
Histopathologic features of C. difficile-induced colitis
On gross examination, mice with C. difficile-induced colitis exhibited shortened colons and colonic mucosal edema. Microscopic observation confirmed the presence of desquamate or necrotic epithelial cells as well as extensive mucosal and submucosal edema, and an influx of inflammatory cells (Figure 4). Histopathologic severity scores for the controls were 0.37 ± 0.1 and 0.45 ± 0.21 for the WT and IL10-/- groups, respectively. Challenge with C. difficile significantly increased the scores in both groups (Ps < 0.05). Moreover, the severity of C. difficile-induced colitis was significantly greater in IL10-/- mice compared with WT (5.50 ± 0.53 vs 2.44 ± 0.46; P < 0.05).
Figure 4.
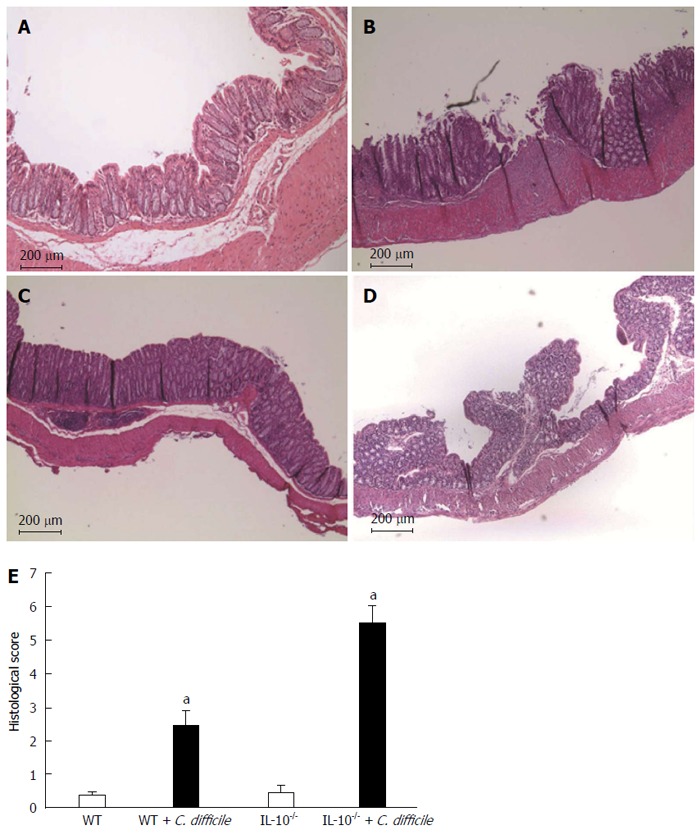
Histopathologic examination of the colonic tissue (× 100). Hematoxylin and eosin staining of colon tissues from A: Wild type (WT); B: WT challenged with Clostridium difficile (C. difficile); C: Interleukin 10-deficient (IL-10-/-); D: IL-10-/- challenged with C. difficile; E: Quantification of colitis severity. Error bars indicate standard error of the mean; aP < 0.05 vs untreated.
Cytokine gene expression in mice with C. difficile colitis
C. difficile challenge induced an increase in IFN-γ mRNA that was ten-fold in WT mice, and significantly higher (> 100-fold) in IL-10-/- mice (P < 0.05 vs WT) (Figure 5). In addition, there was a ten-fold increase in IL-12 expression for both WT and IL-10-/- mice with C. difficile challenge, but not significant compared to non-challenge group. In contrast, there were no strain or treatment effects observed for the IL-23 mRNA expression.
Figure 5.
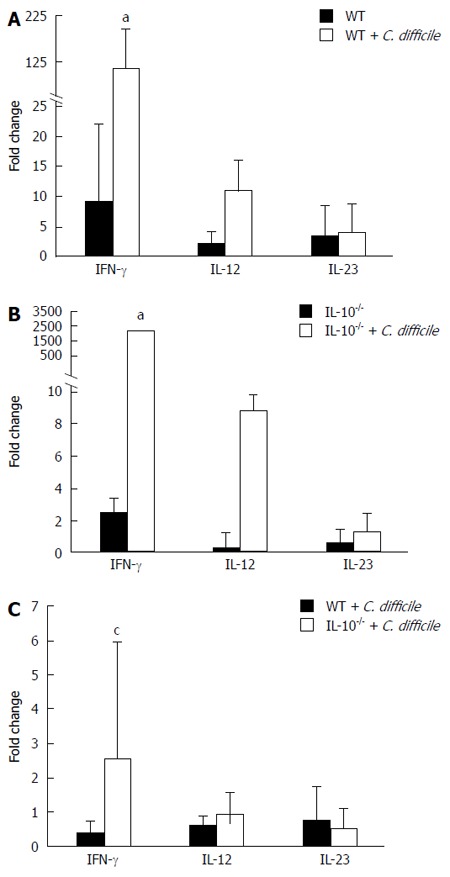
Cytokine expression in Clostridium difficile colitis. The expression of interferon (IFN)-γ, interleukin (IL)-12, and IL-23 mRNAs in colonic tissues from A: Wild type (WT) control and Clostridium difficile (C. difficile)-challenged mice; B: IL-10-deficient (IL-10-/-) control and C. difficile-challenged mice; C: Cytokine expression in the colonic tissues of WT and IL-10-/- mice after induction of C. difficile colitis. Error bars indicate standard error of the mean; aP < 0.05 vs untreated; cP < 0.05 vs WT.
DISCUSSION
Recent studies provide convincing evidence that intestinal microbiota play a critical role in the pathogenesis of IBD[2,18], and germ-free and gnotobiotic techniques have demonstrated that they are necessary for the development of intestinal inflammation[19,20]. Mouse models of gut inflammation induced by bacteria such as Citrobacter rodentium, Salmonella enterica, Bacteroides fragilis and Campylobacter sp. have been reported, which provide mechanisms for bacteria-associated gastroenteritis[13]. Although animal models of Clostridium-associated disease have been reported, there are no established models to study the mechanism of Clostridium-aggravated intestinal inflammation in IBD. The results of the present study confirm that CDI causes acute colitis within three days, and demonstrate that this was significantly more severe in a mouse model of IBD.
Various mouse models have been used to investigate IBD, the majority of which are based either on chemical induction (with dextran sodium sulfate or 2,4,6-trinitrobenzenesulfonic acid), immune cell transfer, or gene targeting. Mice deficient in IL-10 spontaneously develop IBD that progresses over several months, characterized by increased inflammatory cytokine production[21,22]. These mice rapidly develop severe, chronic IBD after treatment with piroxicam, a non-steroidal anti-inflammatory drug, as endogenous prostaglandins are important inhibitors of the development of intestinal inflammation[23]. We therefore hypothesized that CDI would have a similar effect on these mice. We observed a development of acute colitis from CDI that was amplified in IL-10-/- mice. Moreover, CDI resulted in histopathologic features typical of C. difficile colitis in humans, such as epithelial cell damage, inflammatory cells infiltration, and submucosal edema. To the best of our knowledge, this is the first animal model of established C. difficile-aggravated colitis in IBD, which provides a useful tool for studying the relationship between C. difficile and host immune responses of the gut in IBD. As CDI has emerged as a significant clinical challenge for IBD patients, the establishment of a novel relevant model will enhance our understanding of the mechanisms involved, and precipitate the development of future therapies.
Cytokines play a central role in the development and control of intestinal inflammation, particularly IL-12 and IL-23, which share the p40 subunit[24]. A pathogenic role for IL-23 signaling in CDI has been recently reported in both human and murine models[25]. Both IL-23 and IL-12 have been demonstrated as more important than IFN-γ for inducing chronic inflammation in IL-10-/- mice[26,27]. However, in the present study, we observed a more dramatic increase in IFN-γ expression in the colonic tissues of IL-10-/- mice infected with C. difficile. Nonetheless, these results are consistent with a previous report in which IFN-γ played a decisive role in C. difficile toxin A-mediated enteritis through acute inflammatory reaction with neutrophil infiltration[28]. Indeed, results from the in vitro analysis in the present study confirmed that C. difficile toxin A induces IFN-γ expression in BMDCs. IFN-γ triggers the secretion of pro-inflammatory mediators by activated macrophages, and regulates the expression of adhesion molecules[29,30]. IFN-γ also increases the permeability of intestinal epithelial cells[31-33], which can occur via widening of the tight junctions, without triggering necrosis[34]. IFN-γ activation may be associated with the aggravation of colitis in IL-10-/- mice infected with C. difficile, as a greater increase in IFN-γ expression was observed in these mice. It is plausible that an acute inflammatory reaction such as heavy neutrophil infiltration was induced by CDI infection in the IL-10-/- mice that is distinct from the chronic inflammatory reaction in colitis.
Dendritic cells play a critical role in the pathogenesis of IBD due to the link between innate and adaptive immunity[35,36]. A recent paper demonstrated that surface layer proteins from C. difficile induce inflammatory and regulatory cytokines (IL-1β and IL-6) in human monocytes and dendritic cells[37]. In addition, it has been reported that C. difficile toxin A promotes dendritic cell maturation through p38 mitogen-activated protein kinase, an inhibitor of IκB, and the NF-κB signaling pathway[38]. In the present study, we show that toxin A increased the expression of IFN-γ in BMDCs. However, further studies are needed to explore the specific mechanisms of IFN-γ activation in dendritic cells by C. difficile toxin A.
The present study presents a model of CDI-aggravated colitis, which provides an additional platform for evaluation of other substances that can exacerbate colitis. For example, some pre- and probiotics, advocated for their potential benefits in IBD, pouchitis and diarrhea, do not alleviate all symptoms. Intestinal inflammation was not reduced with Lactobacillus fermentum BR11 and fructo-oligosaccharide in an experimental model of colitis[39]. However, administration of a cocktail consisting of six bacterial strains, including Anaerostipes sp., has been shown to relieve chronic CDI in mice[40]. In another study, Lactobacillus acidophilus was shown to modulate the virulence of C. difficile[41].
In conclusion, the results of this study indicate that CDI induces more severe acute colitis in the IL-10-/- model of chronic IBD. Moreover, this exacerbation likely involves IFN-γ, as expression of this cytokine was dramatically enhanced. Use of this novel mouse model will allow further investigation into mechanisms associated with CDI-induced aggravation of intestinal inflammation in IBD and the pathogenic role of C. difficile in mucosal immune dysregulation.
COMMENTS
Background
Inflammatory bowel disease (IBD), including Crohn’s disease and ulcerative colitis, is a complex chronic inflammatory gastrointestinal disorder of unknown cause in a genetically predisposed host. There is accumulating evidence concerning the importance of microbiota in the pathogenesis of IBD, and infection with Clostridium difficile (C. difficile) is associated with a higher morbidity and mortality in IBD patients. However, the pathogenic role of C. difficile is still unknown.
Research frontiers
Although animal models of C. difficile-associated disease have been reported, there are no established models to study the mechanism of C. difficile-aggravated intestinal inflammation in IBD. This study demonstrates C. difficile infection in a model of spontaneous, chronic IBD, for the purpose of exploring mechanisms of intestinal inflammation.
Innovations and breakthroughs
Although previous mouse models of C. difficile-associated disease closely resemble the human disease, there are no animal models of C. difficile-aggravated IBD-like colitis. The present study utilizes interleukin 10-deficient (IL-10-/-) mice, which express a spontaneous, chronic form of IBD, and investigates the effect and mechanism of C. difficile-induced colitis. In vitro and in vivo experiments implicate the involvement of interferon (IFN)-γ in this process. Furthermore, the results establish a model of C. difficile-aggravated intestinal inflammation in IBD.
Applications
This study establishes an animal model of C. difficile-aggravated colitis in IBD, which provides a useful tool for studying the relationship between C. difficile infection and host immune response within IBD-affected intestinal mucosa.
Terminology
IL-10 is a cytokine with potent anti-inflammatory activity. IL-10-/- mice develop a spontaneous, chronic IBD-like colitis that can take months to develop. IFN-γ is a cytokine important for innate and adaptive immune response to infection. IL-12 and IL-23 are inflammatory cytokines important for the differentiation of T-cells.
Peer review
The authors examined the effect of C. difficile toxin A on cytokine expression in bone marrow-derived dendritic cells (BMDCs) and C. difficile infection on intestinal inflammation in IL-10-/- mice. Toxin A and C. difficile infection significantly induced IFN-γ gene expression in BMDCs and colonic tissues of the IL-10-/- mice. In addition, the IL-10-/- mice developed more severe colitis following C. difficile infection, compared to wild type mice. These results establish a model of C. difficile-aggravated IBD-like colitis, and suggest a role for IFN-γ in this process.
Footnotes
P- Reviewer: Howarth GS, Matsuda A, Sadik R S- Editor: Qi Y L- Editor: A E- Editor: Wang CH
References
- 1.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 2.Chassaing B, Darfeuille-Michaud A. The commensal microbiota and enteropathogens in the pathogenesis of inflammatory bowel diseases. Gastroenterology. 2011;140:1720–1728. doi: 10.1053/j.gastro.2011.01.054. [DOI] [PubMed] [Google Scholar]
- 3.Bartlett JG, Chang TW, Gurwith M, Gorbach SL, Onderdonk AB. Antibiotic-associated pseudomembranous colitis due to toxin-producing clostridia. N Engl J Med. 1978;298:531–534. doi: 10.1056/NEJM197803092981003. [DOI] [PubMed] [Google Scholar]
- 4.Kyne L, Hamel MB, Polavaram R, Kelly CP. Health care costs and mortality associated with nosocomial diarrhea due to Clostridium difficile. Clin Infect Dis. 2002;34:346–353. doi: 10.1086/338260. [DOI] [PubMed] [Google Scholar]
- 5.Kelly CP, LaMont JT. Clostridium difficile infection. Annu Rev Med. 1998;49:375–390. doi: 10.1146/annurev.med.49.1.375. [DOI] [PubMed] [Google Scholar]
- 6.Kelly CP, Pothoulakis C, LaMont JT. Clostridium difficile colitis. N Engl J Med. 1994;330:257–262. doi: 10.1056/NEJM199401273300406. [DOI] [PubMed] [Google Scholar]
- 7.Sun X, Savidge T, Feng H. The enterotoxicity of Clostridium difficile toxins. Toxins (Basel) 2010;2:1848–1880. doi: 10.3390/toxins2071848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mann EA, Saeed SA. Gastrointestinal infection as a trigger for inflammatory bowel disease. Curr Opin Gastroenterol. 2012;28:24–29. doi: 10.1097/MOG.0b013e32834c453e. [DOI] [PubMed] [Google Scholar]
- 9.Ananthakrishnan AN, McGinley EL, Binion DG. Excess hospitalisation burden associated with Clostridium difficile in patients with inflammatory bowel disease. Gut. 2008;57:205–210. doi: 10.1136/gut.2007.128231. [DOI] [PubMed] [Google Scholar]
- 10.Navaneethan U, Venkatesh PG, Shen B. Clostridium difficile infection and inflammatory bowel disease: understanding the evolving relationship. World J Gastroenterol. 2010;16:4892–4904. doi: 10.3748/wjg.v16.i39.4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Issa M, Ananthakrishnan AN, Binion DG. Clostridium difficile and inflammatory bowel disease. Inflamm Bowel Dis. 2008;14:1432–1442. doi: 10.1002/ibd.20500. [DOI] [PubMed] [Google Scholar]
- 12.Fekety R, Silva J, Toshniwal R, Allo M, Armstrong J, Browne R, Ebright J, Rifkin G. Antibiotic-associated colitis: effects of antibiotics on Clostridium difficile and the disease in hamsters. Rev Infect Dis. 1979;1:386–397. doi: 10.1093/clinids/1.2.386. [DOI] [PubMed] [Google Scholar]
- 13.Nell S, Suerbaum S, Josenhans C. The impact of the microbiota on the pathogenesis of IBD: lessons from mouse infection models. Nat Rev Microbiol. 2010;8:564–577. doi: 10.1038/nrmicro2403. [DOI] [PubMed] [Google Scholar]
- 14.Chen X, Katchar K, Goldsmith JD, Nanthakumar N, Cheknis A, Gerding DN, Kelly CP. A mouse model of Clostridium difficile-associated disease. Gastroenterology. 2008;135:1984–1992. doi: 10.1053/j.gastro.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 15.Kang SJ, Kim JM, Koh SJ, Kim SH, Im JP, Jung HC, Kim JS. The guggulsterone derivative GG-52 inhibits NF-κB signaling in bone marrow-derived dendritic cells and attenuates colitis in IL-10 knockout mice. Life Sci. 2013;92:1064–1071. doi: 10.1016/j.lfs.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 16.Kim JS, Jobin C. The flavonoid luteolin prevents lipopolysaccharide-induced NF-kappaB signalling and gene expression by blocking IkappaB kinase activity in intestinal epithelial cells and bone-marrow derived dendritic cells. Immunology. 2005;115:375–387. doi: 10.1111/j.1365-2567.2005.02156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kelly CP, Becker S, Linevsky JK, Joshi MA, O’Keane JC, Dickey BF, LaMont JT, Pothoulakis C. Neutrophil recruitment in Clostridium difficile toxin A enteritis in the rabbit. J Clin Invest. 1994;93:1257–1265. doi: 10.1172/JCI117080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134:577–594. doi: 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- 19.Uhlig HH, Powrie F. Mouse models of intestinal inflammation as tools to understand the pathogenesis of inflammatory bowel disease. Eur J Immunol. 2009;39:2021–2026. doi: 10.1002/eji.200939602. [DOI] [PubMed] [Google Scholar]
- 20.Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, Rennick DM, Sartor RB. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun. 1998;66:5224–5231. doi: 10.1128/iai.66.11.5224-5231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berg DJ, Davidson N, Kühn R, Müller W, Menon S, Holland G, Thompson-Snipes L, Leach MW, Rennick D. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J Clin Invest. 1996;98:1010–1020. doi: 10.1172/JCI118861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kühn R, Löhler J, Rennick D, Rajewsky K, Müller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 23.Berg DJ, Zhang J, Weinstock JV, Ismail HF, Earle KA, Alila H, Pamukcu R, Moore S, Lynch RG. Rapid development of colitis in NSAID-treated IL-10-deficient mice. Gastroenterology. 2002;123:1527–1542. doi: 10.1053/gast.2002.1231527. [DOI] [PubMed] [Google Scholar]
- 24.Strober W, Fuss IJ. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology. 2011;140:1756–1767. doi: 10.1053/j.gastro.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buonomo EL, Madan R, Pramoonjago P, Li L, Okusa MD, Petri WA. Role of interleukin 23 signaling in Clostridium difficile colitis. J Infect Dis. 2013;208:917–920. doi: 10.1093/infdis/jit277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davidson NJ, Hudak SA, Lesley RE, Menon S, Leach MW, Rennick DM. IL-12, but not IFN-gamma, plays a major role in sustaining the chronic phase of colitis in IL-10-deficient mice. J Immunol. 1998;161:3143–3149. [PubMed] [Google Scholar]
- 27.Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, Kleinschek MA, Owyang A, Mattson J, Blumenschein W, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–1316. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishida Y, Maegawa T, Kondo T, Kimura A, Iwakura Y, Nakamura S, Mukaida N. Essential involvement of IFN-gamma in Clostridium difficile toxin A-induced enteritis. J Immunol. 2004;172:3018–3025. doi: 10.4049/jimmunol.172.5.3018. [DOI] [PubMed] [Google Scholar]
- 29.Kurihara Y, Furue M. Interferon-γ enhances phorbol myristate acetate-induced cell attachment and tumor necrosis factor production via the NF-κB pathway in THP-1 human monocytic cells. Mol Med Rep. 2013;7:1739–1744. doi: 10.3892/mmr.2013.1419. [DOI] [PubMed] [Google Scholar]
- 30.Pourshafie MR, Sonnenfeld G. Treatment of an infected murine macrophage cell line (J774A.1) with interferon-gamma but not tumor necrosis factor-alpha or live Mycobacterium intracellulare alone modulates the expression of adhesion molecules. J Interferon Cytokine Res. 1997;17:69–75. doi: 10.1089/jir.1997.17.69. [DOI] [PubMed] [Google Scholar]
- 31.Tong Q, Vassilieva EV, Ivanov AI, Wang Z, Brown GT, Parkos CA, Nusrat A. Interferon-gamma inhibits T84 epithelial cell migration by redirecting transcytosis of beta1 integrin from the migrating leading edge. J Immunol. 2005;175:4030–4038. doi: 10.4049/jimmunol.175.6.4030. [DOI] [PubMed] [Google Scholar]
- 32.Utech M, Ivanov AI, Samarin SN, Bruewer M, Turner JR, Mrsny RJ, Parkos CA, Nusrat A. Mechanism of IFN-gamma-induced endocytosis of tight junction proteins: myosin II-dependent vacuolarization of the apical plasma membrane. Mol Biol Cell. 2005;16:5040–5052. doi: 10.1091/mbc.E05-03-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Günzel D, Florian P, Richter JF, Troeger H, Schulzke JD, Fromm M, Gitter AH. Restitution of single-cell defects in the mouse colon epithelium differs from that of cultured cells. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1496–R1507. doi: 10.1152/ajpregu.00470.2005. [DOI] [PubMed] [Google Scholar]
- 34.Adams RB, Planchon SM, Roche JK. IFN-gamma modulation of epithelial barrier function. Time course, reversibility, and site of cytokine binding. J Immunol. 1993;150:2356–2363. [PubMed] [Google Scholar]
- 35.Bilsborough J, Viney JL. Gastrointestinal dendritic cells play a role in immunity, tolerance, and disease. Gastroenterology. 2004;127:300–309. doi: 10.1053/j.gastro.2004.01.028. [DOI] [PubMed] [Google Scholar]
- 36.Niess JH, Reinecker HC. Lamina propria dendritic cells in the physiology and pathology of the gastrointestinal tract. Curr Opin Gastroenterol. 2005;21:687–691. doi: 10.1097/01.mog.0000181710.96904.58. [DOI] [PubMed] [Google Scholar]
- 37.Ausiello CM, Cerquetti M, Fedele G, Spensieri F, Palazzo R, Nasso M, Frezza S, Mastrantonio P. Surface layer proteins from Clostridium difficile induce inflammatory and regulatory cytokines in human monocytes and dendritic cells. Microbes Infect. 2006;8:2640–2646. doi: 10.1016/j.micinf.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 38.Lee JY, Kim H, Cha MY, Park HG, Kim YJ, Kim IY, Kim JM. Clostridium difficile toxin A promotes dendritic cell maturation and chemokine CXCL2 expression through p38, IKK, and the NF-kappaB signaling pathway. J Mol Med (Berl) 2009;87:169–180. doi: 10.1007/s00109-008-0415-2. [DOI] [PubMed] [Google Scholar]
- 39.Smith CL, Geier MS, Yazbeck R, Torres DM, Butler RN, Howarth GS. Lactobacillus fermentum BR11 and fructo-oligosaccharide partially reduce jejunal inflammation in a model of intestinal mucositis in rats. Nutr Cancer. 2008;60:757–767. doi: 10.1080/01635580802192841. [DOI] [PubMed] [Google Scholar]
- 40.Lawley TD, Clare S, Walker AW, Stares MD, Connor TR, Raisen C, Goulding D, Rad R, Schreiber F, Brandt C, et al. Targeted restoration of the intestinal microbiota with a simple, defined bacteriotherapy resolves relapsing Clostridium difficile disease in mice. PLoS Pathog. 2012;8:e1002995. doi: 10.1371/journal.ppat.1002995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yun B, Oh S, Griffiths MW. Lactobacillus acidophilus modulates the virulence of Clostridium difficile. J Dairy Sci. 2014;97:4745–4758. doi: 10.3168/jds.2014-7921. [DOI] [PubMed] [Google Scholar]