

Abstract
The current treatment approach for severe aplastic anemia in children is based on studies performed in the 1980s, and updated evidence is required. We retrospectively compared the outcomes of children with acquired severe aplastic anemia who received immunosuppressive therapy within prospective trials conducted by the Japanese Childhood Aplastic Anemia Study Group or who underwent bone marrow transplantation from an HLA-matched family donor registered in the Japanese Society for Hematopoietic Cell Transplantation Registry. Between 1992 and 2009, 599 children (younger than 17 years) with severe aplastic anemia received a bone marrow transplant from an HLA-matched family donor (n=213) or immunosuppressive therapy (n=386) as first-line treatment. While the overall survival did not differ between patients treated with immunosuppressive therapy or bone marrow transplantation [88% (95% confidence interval: 86–90) versus 92% (90–94)], failure-free survival was significantly inferior in patients receiving immunosuppressive therapy than in those undergoing bone marrow transplantation [56% (54–59) versus 87% (85–90); P<0.0001]. There was no significant improvement in outcomes over the two time periods (1992–1999 versus 2000–2009). In multivariate analysis, age <10 years was identified as a favorable factor for overall survival (P=0.007), and choice of first-line immunosuppressive therapy was the only unfavorable factor for failure-free survival (P<0.0001). These support the current algorithm for treatment decisions, which recommends bone marrow transplantation when an HLA-matched family donor is available in pediatric severe aplastic anemia.
Introduction
Aplastic anemia is defined as peripheral blood pancytopenia caused by bone marrow failure; the pathogenesis of this disease is thought to involve autoimmune processes.1–3 The principal interventions responsible for improved survival in aplastic anemia are bone marrow transplantation (BMT) and immunosuppressive therapy (IST). In children, BMT from an HLA-matched family donor (MFD) is the treatment of choice for severe aplastic anemia (SAA).1,4–6 For children lacking an MFD, IST with a combination of antithymocyte globulin and cyclosporine has been used as a therapeutic option.6–10 However, this treatment approach is based on the results of comparative studies between these therapies that were conducted mainly in the 1980s, and there have been few recent studies that compare the outcome of BMT recipients with comparable patients receiving IST.
The largest pediatric series in previous studies was reported by the European Group for Blood and Marrow Transplantation (EBMT) and included 304 children treated from 1970 to 1988; that study indicated survival was better following first-line BMT than after first-line IST (63% versus 48%; P=0.002) but did not compare failure-free survival after the two therapies.6 Our previous analysis showed a significant advantage for patients receiving BMT from an MFD as first-line treatment in a study of 100 children with SAA who were treated between 1984 and 1998.1 In patients who received first-line IST, 10-year overall and failure-free survival rates were 55% and 40%, respectively, both of which were markedly inferior to the rates in patients who initially underwent BMT, which was associated with 10-year overall survival and failure-free survival rates greater than 90%. Since the 1980s, the outcomes of both BMT and IST have improved, likely due to better supportive care and advanced treatment and transplantation protocols. A recently published Cochrane review regarding BMT from an MFD and IST as first-line treatment also pointed out that all studies included in the analysis had a high risk of bias due to their study design and were conducted more than 10 years ago and may not be applicable to the standard of care of today.11 Updated evidence to aid treatment decisions in pediatric SAA is, therefore, required.
In children, the choice of an appropriate treatment is particularly influenced by the long-term sequelae of the disease and its therapy. Thus, failure-free survival is much more important than survival alone when analyzing the long-term outcomes of children with aplastic anemia. Lack of response, relapse, and clonal evolution are problematic in the IST setting, whereas graft failure, acute and chronic graft-versus-host disease (GVHD), and infectious complications limit the success of BMT. In the present study, we compared the outcomes of children with SAA who received IST or BMT from an MFD as first-line treatment using data from nationwide IST and BMT registries.
Methods
Patients
Between 1992 and 2009, a total of 599 consecutive children (younger than 17 years) with acquired SAA underwent BMT from an MFD or received IST as first-line treatment in Japan; 213 patients with an MFD underwent BMT and were registered in the Transplant Registry Unified Management Program (TRUMP) conducted by the Japanese Society for Hematopoietic Cell Transplantation, and 386 patients without an MFD were enrolled in two consecutive prospective multicenter trials (AA-92/97) conducted by the Japanese Childhood Aplastic Anemia Study Group and were initially treated with IST (Table 1). The disease severities were defined as previously reported.12,13 Underlying inherited marrow failure disorders were excluded clinically and by chromosome fragility testing. Marrow cytogenetic studies were performed for all patients, and patients with clonal cytogenetic abnormalities were excluded from this study. Patients with paroxysmal nocturnal hemoglobinuria with clinical symptoms and positive findings on the Ham test/sucrose test were also excluded from this analysis. All treatments were performed after obtaining written informed consent from patients or their parents in accordance with the Declaration of Helsinki.
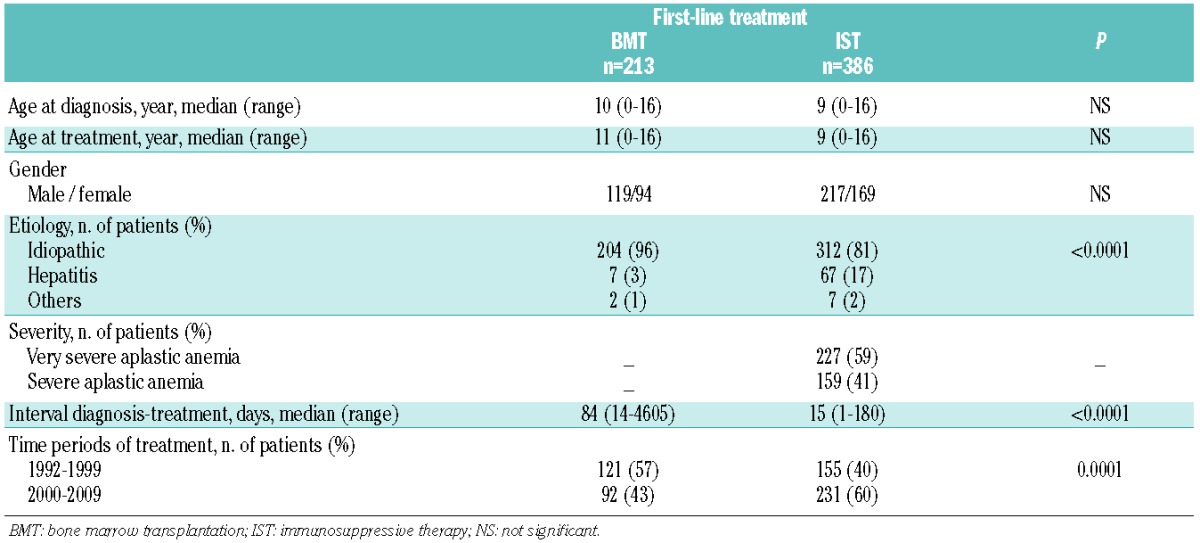
Table 1.
Patients’ characteristics.
Immunosuppressive therapy and bone marrow transplantation procedures
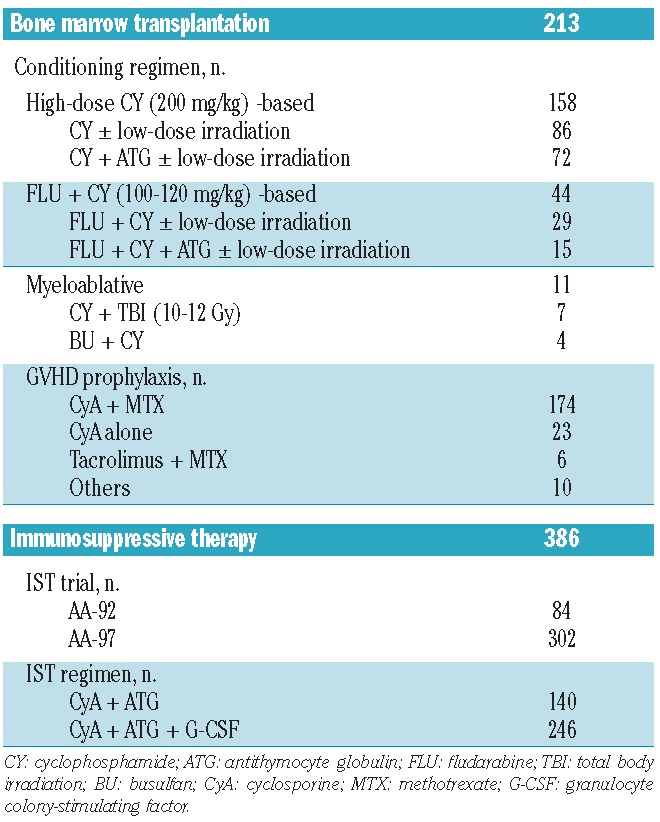
The characteristics of the treatment procedures are detailed in Table 2. Three hundred and eighty-six patients were enrolled in the AA-92 (n=84) and AA-97 (n=302) trials, and all the patients were initially treated with a combination of antithymocyte globulin and cyclosporine A. Response to IST and disease relapse were evaluated as previously reported.12 Transplantation data were collected with the use of standardized forms provided by the TRUMP. A total of 213 patients underwent BMT from an MFD as first-line treatment following the local protocols for conditioning regimens and GVHD prophylaxis. Patients who did not reach neutrophil counts >0.5×109/L for 3 consecutive days after transplantation were considered to have had primary graft failure. Patients with initial engraftment in whom absolute neutrophil counts subsequently declined to <0.5×109/L were considered to have had secondary graft failure. Acute and chronic GVHD were evaluated according to standard criteria.14–16 More details on methods are provided in the Online Supplementary Methods section.
Table 2.
Treatment characteristics.
Statistical analyses
The date of analysis was July 30, 2012. Survival probabilities were estimated by the Kaplan-Meier method and compared between different groups of patients using the log-rank test. The influence of potential risk factors on overall survival and failure-free survival was assessed according to first-line treatment (BMT or IST), time period of treatment (1992–1999 or 2000–2009), age and other variables related to each treatment. Overall survival was defined as the time from diagnosis to death or last follow-up. Failure-free survival was defined as survival with treatment response. Death, primary or secondary graft failure, and secondary malignancy in the BMT group, and death, relapse, disease progression requiring stem cell transplantation (SCT) from an alternative donor or second IST, clonal evolution and evolution to paroxysmal nocturnal hemoglobinuria in the IST group were considered treatment failures. For multivariate analyses, the Cox proportional hazard regression model was used. P values less than 0.05 were considered statistically significant. This study was approved by the institutional ethics committee of the Japanese Red Cross Nagoya First Hospital.
Results
Patients’ characteristics
The characteristics of the 599 children are detailed in Table 1. The groups treated first-line with BMT (n=213) or IST (n=386) were similar with regards to age at diagnosis, age at treatment and male/female ratio. The majority of patients in both groups had a diagnosis of idiopathic disease, although the proportion of patients with non-idiopathic disease was higher in the IST group. Seven patients (3%) in the BMT group and 67 patients (17%) in the IST group suffered from hepatitis-associated disease. Nine patients had drug-induced or virus-associated disease. Information on the proportion of very severe disease was not available for 141 patients who underwent BMT because the severity of the SAA was not a required item for the registry. The clinical features of these patients were similar to those of the remaining patients. In the IST group, details regarding the severity of disease were provided for all patients: 227 (59%) had very severe disease and 159 (41%) suffered from severe disease. As expected, the time to treatment was significantly longer in the BMT group; the median interval between diagnosis and treatment was 15 days (range, 1–180 days) and 84 days (range, 14–4605 days) for those treated with IST and BMT, respectively. In accordance with decisions taken by the patients and the parents, ten patients underwent BMT more than 5 years after diagnosis. None of the patients who received IST before BMT from an MFD were included in the BMT group.
Immunosuppressive therapy
Response to IST at 6 months was not evaluable in 11 patients for the following reasons: early death (n=7) or BMT from an alternative donor within 6 months of IST (n=4). The causes of the early deaths were sepsis (n=3), interstitial pneumonia (n=2), hemolysis of unknown cause (n=1) and accidental ingestion (n=1). Of the patients who underwent BMT from an alternative donor within 6 months, two patients died of graft failure or cardiac toxicity related to the preconditioning regimen. Overall, 238 of the 375 evaluable patients (63%) improved with first-line IST and achieved a partial response (n=151) or complete response (n=87) at 6 months. All of these patients achieved transfusion independence.
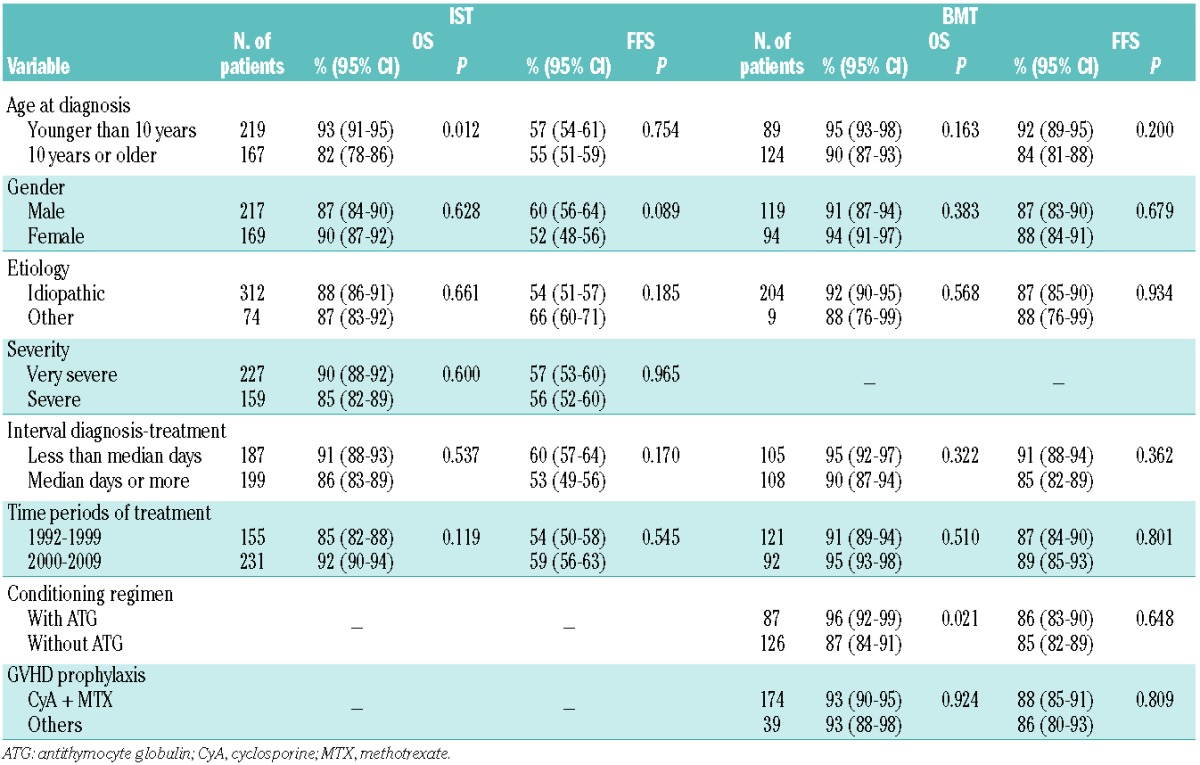
For all 386 patients who received IST initially, the 10-year overall survival rate was 88% [95% confidence interval (CI): 86–90], as shown in Figure 1A, and the median follow-up time for living patients was 106 months (range, 22–224 months). In contrast to the high rate of overall survival, the result regarding survival with response was unsatisfactory, the 10-year failure-free survival rate being 56% (95% CI: 54–59) (Figure 1B). The cause of treatment failure included death in 12 patients [due to intracranial hemorrhage (n=2), pneumonia (n=1), traffic accident (n=1) and sudden death (n=1) in addition to the seven early deaths], relapse in 23 patients, disease progression requiring second-line treatment in 109 patients, evolution to myelodysplastic syndrome in 15 patients, and appearance of paroxysmal nocturnal hemoglobinuria in two patients. After failed IST, a total of 113 patients underwent SCT from an alternative donor as second- or third-line treatment. The 10-year overall survival of these patients who received a transplant after failed IST was 79% (95% CI: 75–83) with a median of 435 days from diagnosis and SCT. We then analyzed the influence of potential risk factors for survival in the IST group. The prognostic significance of the clinical parameters is shown in Table 3. In the univariate analysis, age younger than 10 years at diagnosis was associated with a favorable overall survival rate [93% (95% CI: 91–95) versus 82% (95% CI: 78–86); P=0.012], and this was confirmed in a multivariate model. However, the rate of failure-free survival did not differ between patients in the two age groups. No other variables were significantly associated with survival after IST in either univariate or multivariate analyses.
Figure 1.
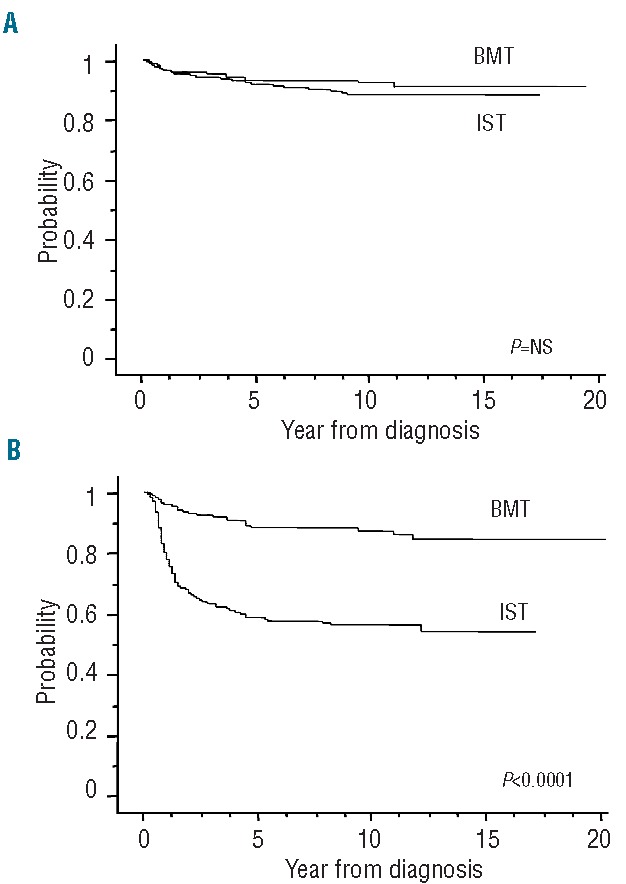
Survival of 599 children with severe aplastic anemia according to first-line treatments with immunosuppressive therapy (IST) (n=386) or bone marrow transplantation (BMT) (n=213). (A) Overall survival. The 10-year overall survival was 88% (95% CI: 86–90) in the IST group and 92% (95% CI: 90–94) in MFD BMT recipients (P=NS). (B) Failure-free survival. The 10-year failure-free survival was 56% (95% CI: 54–59) in the IST group and 87% (95% CI: 85–90) in the BMT group (P<0.0001).
Table 3.
Univariate analysis of 10-year overall survival (OS) and failure-free survival (FFS), according to first-line treatment.
Bone marrow transplantation
In the BMT group, 209 patients (98%) achieved primary engraftment at a median of 16 days after transplantation. As shown in Figure 1A and 1B, the 10-year overall survival and failure-free survival rates for all 213 patients who were treated initially with BMT from an MFD were 92% (95% CI: 90–94) and 87% (95% CI: 85–90), respectively. When the analysis was applied to the patients who underwent BMT within 180 days from diagnosis, similar results were observed; the 10-year overall survival and failure-free survival rates were 94% (95% CI: 92–96) and 89% (95% CI: 86–92), respectively. The median follow-up time for living patients was 101 months (range, 18–213 months). The cause of treatment failure included primary graft failure in two patients, secondary graft failure in ten patients, second malignancy in one patient, and death due to other complications in 12 patients. Although both patients without primary engraftment died, nine of the ten patients with secondary graft failure remain alive; eight were saved by a second transplant, and one recovered spontaneously. Twenty-five of 209 patients (12%) who had achieved primary engraftment developed grade II to IV acute GVHD, and extensive chronic GVHD was observed in 13 of 209 patients (6%) alive 100 days after BMT.
The prognostic significance of the clinical parameters, including variables related to transplantation, was then assessed. We found no association between age, gender, etiology, interval between diagnosis and BMT, or time period of treatment and treatment outcome (Table 3). Of particular interest with regards to conditioning regimens is the fact that the addition of antithymocyte globulin produced an improvement of overall survival [96% (95% CI: 92–99) versus 87% (95% CI: 84–91); P=0.021], whereas the rate of failure-free survival was comparable. A fludarabine-based regimen did not affect outcome after BMT from an MFD, although the number of patients treated with such regimens was too small to draw any conclusions. Multivariate analysis showed that none of the variables significantly influenced survival.
Survival and prognostic factors
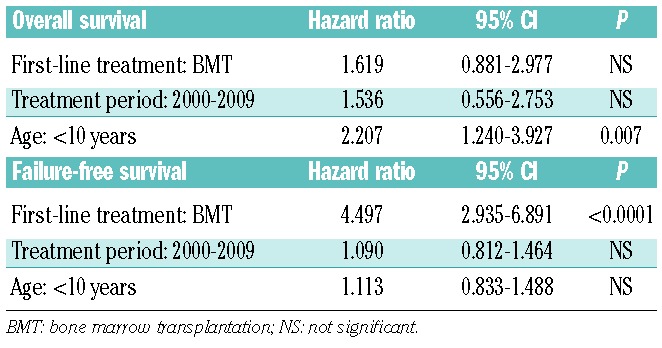
The overall outcomes of the 599 children with SAA, stratified according to their first-line treatment, are shown in Figure 1A and 1B. Our data clearly showed a significant advantage for children receiving BMT from an MFD as first-line treatment; the failure-free survival was significantly superior in patients treated with BMT than in those in whom IST was used (P<0.0001), whereas the overall survival of patients in these two treatment groups did not differ. Figure 2A and 2B show survival curves in all patients treated in the two sequential time periods, 1992–1999 and 2000–2009: results were comparable over time [10-year overall survival: 88% (95% CI: 86–90) versus 93% (95% CI: 91–95); 10-year failure-free survival: 67% (95% CI: 65–70) versus 68% (95% CI: 66–71)], indicating no significant improvement in the last two decades. When age groups were considered, overall survival at 10 years in the younger group (<10 years old) was significantly better than that in the other age groups [93% (95% CI: 92–95) versus 85% (95% CI: 83–88); P=0.007], although no difference in failure-free survival was observed (Figure 3A and 3B). The favorable overall survival in the younger group may be mostly due to that observed in the first-line IST group. In multivariate analysis, age younger than 10 years at diagnosis was identified as a favorable factor for overall survival (P=0.007), and choice of first-line BMT from an MFD was confirmed as an independent favorable factor for failure-free survival (P<0.0001), as shown in Table 4.
Figure 2.
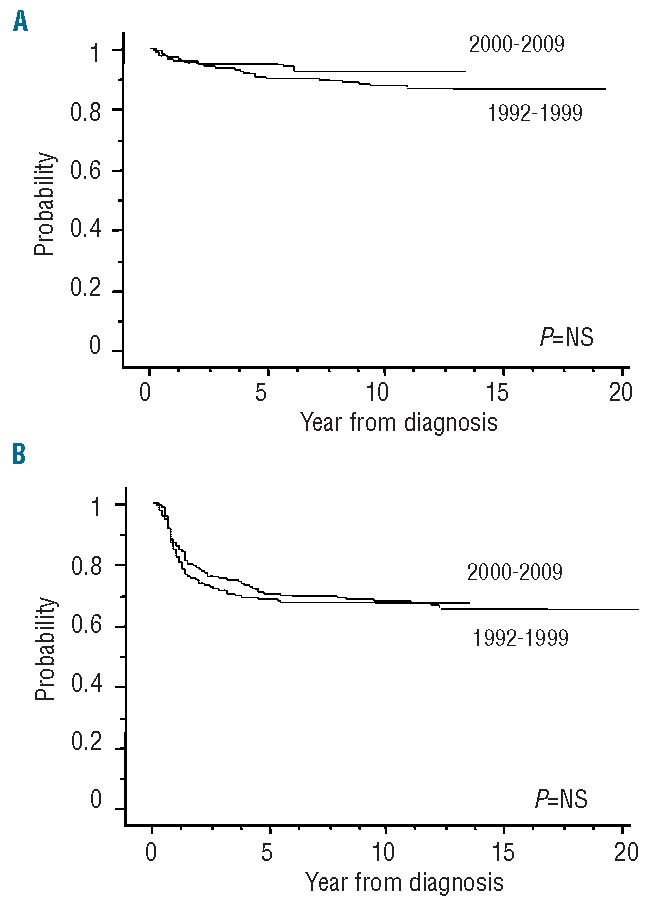
Survival of patients according to time periods of treatment: 1992–1999 (n=276) or 2000–2009 (n=323). (A) Overall survival. The 10-year overall survival was 88% (95% CI: 86–90) in 1992–1999 vs. 93% (95% CI: 91–95) in 2000–2009. (B) Failure-free survival. The 10-year failure-free survival was 67% (95% CI: 65–70) in 1992–1999 vs. 68% (95% CI: 66–71) in 2000–2009.
Figure 3.
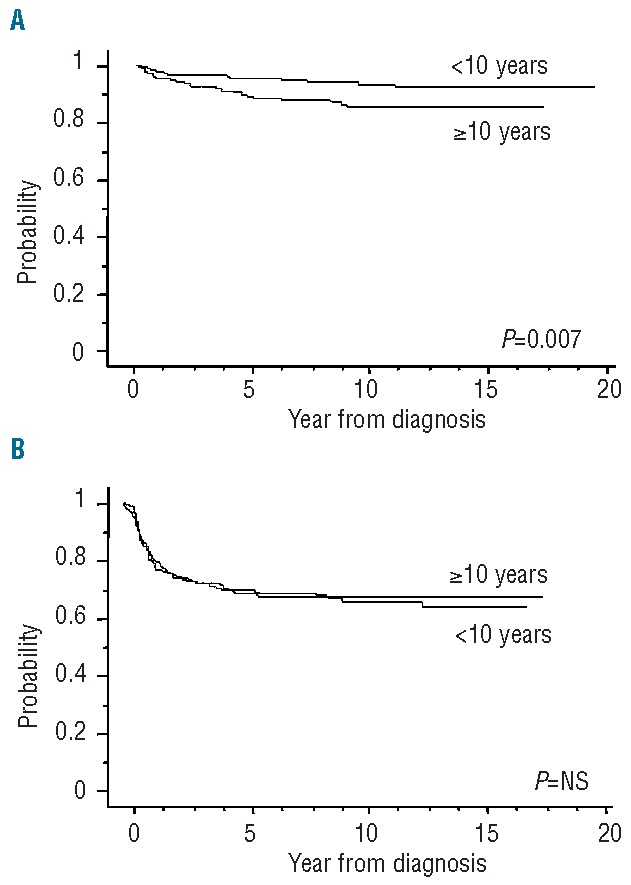
Survival of patients according to age at diagnosis: <10 years (n=308) or ≥10 years (n=291). (A) Overall survival. The 10-year overall survival in the younger group (<10 years) was significantly better than that in the other group [93% (95% CI: 92–95) vs. 85% (95% CI: 83–88); P=0.007]. (B) Failure-free survival. No difference in failure-free survival at 10 years was observed [67% (95% CI: 65–70) vs. 63% (95% CI: 59–67)].
Table 4.
Multivariate analysis of favorable factors for survival in all 599 patients with SAA.
Discussion
For children with SAA, BMT and IST have been accepted as standard treatments during the past three decades. The current guideline recommends BMT from an MFD as the treatment of choice for pediatric SAA17–19 based on the results of comparative studies performed in the 1980s.1,5,6,20,21 On the other hand, recent prospective studies with intensified IST for pediatric SAA have resulted in dramatic improvements in survival.22,23 For example, a study from the EBMT showed a 100% overall survival rate at 6 years after first-line IST in 31 SAA patients younger than 20 years treated from 2002 to 2008.22 These excellent overall survival results after IST have led to discussion about the first-line treatment in children with SAA. To obtain solid evidence on which to base treatment decisions, ideally, a randomized controlled trial is required. However, because of the rarity of the disease, no randomized controlled trials comparing IST with BMT from an MFD as first-line treatment for SAA exist, and only retrospective studies using data from registries or relatively small cohorts of patients are available. Following the previous report of 304 children treated from 1970 to 1988,6 the EBMT SAA Working Party (SAAWP) reported a consecutive study of 911 children younger than 16 years initially treated with IST (n=304) or BMT (n=607) between 1991 and 2002, which indicated that first-line IST gave an overall survival rate comparable to that of first-line BMT (81% versus 79%).10 Unfortunately, the analyses had several limitations, because the drugs used for IST varied (e.g., antithymocyte globulin only, cyclosporine A only, or a combination of antithymocyte globulin and cyclosporine A) and the donor types used for BMT were not consistent (15% of the donors were mismatched family donors or matched/mismatched unrelated donors, although the majority of those were MFD). In addition, neither EBMT study provided results on failure-free survival,6,10 which seems to be much more important than survival alone. Recent advances in supportive care and salvage therapies have effectively rescued non-responders to IST.24 On the other hand, relapse, clonal evolution in the IST group and secondary graft failure and late malignancy in the BMT group are serious problems in long-term survivors. That is the reason why overall survival is no longer the only endpoint to determine optimal first-line treatment in children with SAA. In Japan, we have conducted consecutive prospective trials with a unified IST regimen consisting of antithymocyte globulin and cyclosporine A since 1992, enrolling 386 SAA patients younger than 17 years. During the same period, 213 SAA patients younger than 17 years underwent BMT from an MFD and were registered into the TRUMP, which provided a unique opportunity to investigate updated evidence for treatment decisions in pediatric SAA, although this study also had limitations due to its retrospective nature.
This study confirmed the excellent outcomes obtained in Japanese children with SAA treated with BMT from an MFD or IST. Consistent with the EBMT studies,10,22 the survival of children with SAA initially treated with IST has improved markedly since the 1980s, when first-line IST gave greatly inferior survival (with overall survival rates of around 40–50%) when compared with first-line BMT1,5,6,20,21; in the current analyses, the probability of overall survival at 10 years in the patients treated first-line with IST reached 88%, which was comparable to that of the group treated first-line with BMT. Recent significant advances in second-line SCT, especially with a matched unrelated donor, may contribute to this marked improvement in survival after first-line IST.25–27 In our series, a certain number of patients underwent SCT from an alternative donor after failed IST as a second- or third-line treatment. When patients were subdivided into three groups (first-line BMT from an MFD, IST only, and SCT after failed IST groups), the 10-year overall survival rates in these groups were 91%, 93% and 79%, respectively (P<0.0001), confirming that, in the case of failure of IST, SCT from an alternative donor is a very good salvage option, whereas MFD BMT and IST are excellent first-line treatments for children with SAA.
Regarding survival with response after first-line treatment, we found that the failure-free survival rate in patients treated with IST plateaued over the past two decades after having slightly improved since the 1980s (from 40% in the 1980s to 56% currently).1 Thus, unlike the overall survival results, failure-free survival in the IST group was significantly inferior to that in the MFD BMT group. Consistent with our observations, the EBMT group also demonstrated no significant improvement in outcomes in response to IST since the 1990s.10 This may suggest that the IST regimen has not improved over time. Over the past decade, with the hypothesis that more intense IST might produce better outcomes, the addition of newer immunosuppressive agents, such as mycophenolate mofetil and sirolimus to antithymocyte globulin and cyclosporine A, has been tested, but has failed to improve responses.28–31 The combination of antithymocyte globulin and cyclosporine A is, therefore, still regarded as the standard IST regimen. Another possibility is that we have reached a ceiling in the percentage of patients with the capacity to respond to IST.18 In patients refractory to IST, the pathophysiology of the disease may be different from that in patients responsive to IST, which is thought to involve autoimmune processes, although there are no good markers to routinely or reliably distinguish non-responders from responders.13,32–34 Further studies are needed to identify patients refractory to IST, because these patients might benefit from prompt alternative donor SCT.
Importantly, all patients in the current analyses were treated with horse antithymocyte globulin (Lymphoglobulin), which has recently been withdrawn from Asian and European markets and replaced by rabbit antithymocyte globulin. To date, there are only limited studies using rabbit antithymocyte globulin as first-line IST for pediatric aplastic anemia, and thus, the effectiveness of this form of antithymocyte globulin for pediatric patients remains controversial.35–38 The change of product might result in different outcomes in response to IST for children with SAA.
Survival after BMT from an MFD in children with SAA has exceeded 90% for the past two decades, and this has remained unchanged when compared with our previous observation in the 1980s. In this study, the major causes of treatment failure were primary and secondary graft failure, but notably, most patients with secondary graft failure were rescued by second transplantation or careful observation. In addition to short-term complications, long-term sequelae, such as chronic GVHD and late malignancy, should be taken into consideration to make optimal treatment decisions, especially in children. Our results showed that acute and chronic GVHD were relatively uncommon in the setting of BMT from an MFD for pediatric SAA, which is consistent with recently reported results from the EBMT SAAWP, with 11% of grade II to IV acute GVHD and 4% of extensive chronic GVHD after BMT from an MFD for SAA in all age groups.39 Regarding late malignancy, Kikuchi et al. recently published data from 329 Japanese children with SAA from the nationwide registry, confirming a low incidence of late malignancy after BMT from an MFD; the cumulative incidence of late malignancy was 0.8% at 10 years and 2.5% at 20 years, respectively, which was much lower than the cumulative incidences in reports from western countries.40 In the present series, only one patient developed a late malignancy (myelodysplastic syndrome), and was saved by second BMT. These observations suggest that this approach has been already established as first-line treatment for children with SAA.
In conclusion, our updated data clearly demonstrate that children receiving BMT from an MFD as first-line treatment have a significant advantage over children managed with first-line IST, given the dramatically better failure-free survival and the lower incidence of associated long-term sequelae in the BMT group, which supports the current algorithm for treatment decisions that recommends BMT for pediatric SAA when an MFD is available. On the other hand, IST using the combination of antithymocyte globulin and cyclosporine A is the treatment of choice for children with SAA without an MFD considering the comparable overall survival with BMT from an MFD, which could possibly be ascribed to recent improvements in outcomes after SCT from an alternative donor. In other words, patients have an excellent chance of survival, even after failed first-line IST, when they undergo second-line SCT from an alternative donor.
Acknowledgments
The authors would like to thank Ms. Hiroe Namizaki for secretarial assistance and Prof. Akira Kikuchi for his support of this study. We also thank all of the patients, families, and referring physicians who provided precise data.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Kojima S, Horibe K, Inaba J, Yoshimi A, Takahashi Y, Kudo K, et al. Long-term outcome of acquired aplastic anaemia in children: comparison between immunosuppressive therapy and bone marrow transplantation. Br J Haematol. 2000;111(1):321–8. [DOI] [PubMed] [Google Scholar]
- 2.Mathe G, Amiel JL, Schwarzenberg L, Choay J, Trolard P, Schneider M, et al. Bone marrow graft in man after conditioning by antilymphocytic serum. Transplant Proc. 1971;3(1):325–32. [PubMed] [Google Scholar]
- 3.Young NS. Acquired aplastic anemia. JAMA. 1999;282(3):271–8. [DOI] [PubMed] [Google Scholar]
- 4.Bacigalupo A, Brand R, Oneto R, Bruno B, Socie G, Passweg J, et al. Treatment of acquired severe aplastic anemia: bone marrow transplantation compared with immunosuppressive therapy--The European Group for Blood and Marrow Transplantation experience. Semin Hematol. 2000;37(1):69–80. [DOI] [PubMed] [Google Scholar]
- 5.Doney K, Leisenring W, Storb R, Appelbaum FR. Primary treatment of acquired aplastic anemia: outcomes with bone marrow transplantation and immunosuppressive therapy. Seattle Bone Marrow Transplant Team. Ann Intern Med. 1997;126(2):107–15. [DOI] [PubMed] [Google Scholar]
- 6.Locasciulli A, van’t Veer L, Bacigalupo A, Hows J, Van Lint MT, Gluckman E, et al. Treatment with marrow transplantation or immunosuppression of childhood acquired severe aplastic anemia: a report from the EBMT SAA Working Party. Bone Marrow Transplant. 1990;6(3):211–7. [PubMed] [Google Scholar]
- 7.Bacigalupo A, Broccia G, Corda G, Arcese W, Carotenuto M, Gallamini A, et al. Antilymphocyte globulin, cyclosporin, and granulocyte colony-stimulating factor in patients with acquired severe aplastic anemia (SAA): a pilot study of the EBMT SAA Working Party. Blood. 1995;85(5):1348–53. [PubMed] [Google Scholar]
- 8.Fuhrer M, Rampf U, Baumann I, Faldum A, Niemeyer C, Janka-Schaub G, et al. Immunosuppressive therapy for aplastic anemia in children: a more severe disease predicts better survival. Blood. 2005;106(6):2102–4. [DOI] [PubMed] [Google Scholar]
- 9.Kojima S, Hibi S, Kosaka Y, Yamamoto M, Tsuchida M, Mugishima H, et al. Immunosuppressive therapy using antithymocyte globulin, cyclosporine, and danazol with or without human granulocyte colony-stimulating factor in children with acquired aplastic anemia. Blood. 2000;96(6):2049–54. [PubMed] [Google Scholar]
- 10.Locasciulli A, Oneto R, Bacigalupo A, Socie G, Korthof E, Bekassy A, et al. Outcome of patients with acquired aplastic anemia given first line bone marrow transplantation or immunosuppressive treatment in the last decade: a report from the European Group for Blood and Marrow Transplantation (EBMT). Haematologica. 2007;92(1):11–8. [DOI] [PubMed] [Google Scholar]
- 11.Peinemann F, Bartel C, Grouven U. First-line allogeneic hematopoietic stem cell transplantation of HLA-matched sibling donors compared with first-line ciclosporin and/or antithymocyte or antilymphocyte globulin for acquired severe aplastic anemia. Cochrane Database Syst Rev. 2013;7:CD006407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kosaka Y, Yagasaki H, Sano K, Kobayashi R, Ayukawa H, Kaneko T, et al. Prospective multicenter trial comparing repeated immunosuppressive therapy with stem-cell transplantation from an alternative donor as second-line treatment for children with severe and very severe aplastic anemia. Blood. 2008;111(3):1054–9. [DOI] [PubMed] [Google Scholar]
- 13.Yoshida N, Yagasaki H, Hama A, Takahashi Y, Kosaka Y, Kobayashi R, et al. Predicting response to immunosuppressive therapy in childhood aplastic anemia. Haematologica. 2011;96(5):771–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deeg HJ. Prophylaxis and treatment of acute graft-versus-host disease: current state, implications of new immunopharmacologic compounds and future strategies to prevent and treat acute GVHD in high-risk patients. Bone Marrow Transplant. 1994;14(Suppl 4):S56–60. [PubMed] [Google Scholar]
- 15.Glucksberg H, Storb R, Fefer A, Buckner CD, Neiman PE, Clift RA, et al. Clinical manifestations of graft-versus-host disease in human recipients of marrow from HL-A-matched sibling donors. Transplantation. 1974;18(4):295–304. [DOI] [PubMed] [Google Scholar]
- 16.Shulman HM, Sullivan KM, Weiden PL, McDonald GB, Striker GE, Sale GE, et al. Chronic graft-versus-host syndrome in man. A long-term clinicopathologic study of 20 Seattle patients. Am J Med. 1980; 69(2):204–17. [DOI] [PubMed] [Google Scholar]
- 17.Marsh JC, Ball SE, Cavenagh J, Darbyshire P, Dokal I, Gordon-Smith EC, et al. Guidelines for the diagnosis and management of aplastic anaemia. Br J Haematol. 2009;147(1):43–70. [DOI] [PubMed] [Google Scholar]
- 18.Passweg JR, Marsh JC. Aplastic anemia: first-line treatment by immunosuppression and sibling marrow transplantation. Hematology Am Soc Hematol Educ Program. 2010:36–42. [DOI] [PubMed] [Google Scholar]
- 19.Scheinberg P. Aplastic anemia: therapeutic updates in immunosuppression and transplantation. Hematology Am Soc Hematol Educ Program. 2012:292–300. [DOI] [PubMed] [Google Scholar]
- 20.Bayever E, Champlin R, Ho W, Lenarsky C, Storch S, Ladisch S, et al. Comparison between bone marrow transplantation and antithymocyte globulin in treatment of young patients with severe aplastic anemia. J Pediatr. 1984;105(6):920–5. [DOI] [PubMed] [Google Scholar]
- 21.Bacigalupo A, Hows J, Gluckman E, Nissen C, Marsh J, Van Lint MT, et al. Bone marrow transplantation (BMT) versus immunosuppression for the treatment of severe aplastic anaemia (SAA): a report of the EBMT SAA Working Party. Br J Haematol. 1988;70(2):177–82. [DOI] [PubMed] [Google Scholar]
- 22.Tichelli A, Schrezenmeier H, Socie G, Marsh J, Bacigalupo A, Duhrsen U, et al. A randomized controlled study in patients with newly diagnosed severe aplastic anemia receiving antithymocyte globulin (ATG), cyclosporine, with or without G-CSF: a study of the SAA Working Party of the European Group for Blood and Marrow Transplantation. Blood. 2011;117(17):4434–41. [DOI] [PubMed] [Google Scholar]
- 23.Nair V, Sondhi V, Sharma A, Das S, Sharma S. Survival after immunosuppressive therapy in children with aplastic anemia. Indian Pediatr. 2012;49(5):371–6. [DOI] [PubMed] [Google Scholar]
- 24.Valdez JM, Scheinberg P, Nunez O, Wu CO, Young NS, Walsh TJ. Decreased infection-related mortality and improved survival in severe aplastic anemia in the past two decades. Clin Infect Dis. 2011;52(6):726–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bacigalupo A, Socie G, Lanino E, Prete A, Locatelli F, Locasciulli A, et al. Fludarabine, cyclophosphamide, antithymocyte globulin, with or without low dose total body irradiation, for alternative donor transplants, in acquired severe aplastic anemia: a retrospective study from the EBMT-SAA Working Party. Haematologica. 2010;95(6):976–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kennedy-Nasser AA, Leung KS, Mahajan A, Weiss HL, Arce JA, Gottschalk S, et al. Comparable outcomes of matched-related and alternative donor stem cell transplantation for pediatric severe aplastic anemia. Biol Blood Marrow Transplant. 2006;12(12):1277–84. [DOI] [PubMed] [Google Scholar]
- 27.Yagasaki H, Takahashi Y, Hama A, Kudo K, Nishio N, Muramatsu H, et al. Comparison of matched-sibling donor BMT and unrelated donor BMT in children and adolescent with acquired severe aplastic anemia. Bone Marrow Transplant. 2010;45(10):1508–13. [DOI] [PubMed] [Google Scholar]
- 28.Alsultan A, Goldenberg NA, Kaiser N, Graham DK, Hays T. Tacrolimus as an alternative to cyclosporine in the maintenance phase of immunosuppressive therapy for severe aplastic anemia in children. Pediatr Blood Cancer. 2009;52(5):626–30. [DOI] [PubMed] [Google Scholar]
- 29.Scheinberg P, Nunez O, Weinstein B, Wu CO, Young NS. Activity of alemtuzumab monotherapy in treatment-naive, relapsed, and refractory severe acquired aplastic anemia. Blood. 2012;119(2):345–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scheinberg P, Nunez O, Wu C, Young NS. Treatment of severe aplastic anaemia with combined immunosuppression: anti-thymocyte globulin, ciclosporin and mycophe-nolate mofetil. Br J Haematol. 2006;133(6):606–11. [DOI] [PubMed] [Google Scholar]
- 31.Scheinberg P, Wu CO, Nunez O, Boss C, Sloand EM, Young NS. Treatment of severe aplastic anemia with a combination of horse antithymocyte globulin and cyclosporine, with or without sirolimus: a prospective randomized study. Haematologica. 2009;94(3):348–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang MH, Kim KH, Kim HS, Jun HJ, Kim DH, Jang JH, et al. Predictors of response to immunosuppressive therapy with antithymocyte globulin and cyclosporine and prognostic factors for survival in patients with severe aplastic anemia. Eur J Haematol. 2009;84(2):154–9. [DOI] [PubMed] [Google Scholar]
- 33.Scheinberg P, Wu CO, Nunez O, Young NS. Long-term outcome of pediatric patients with severe aplastic anemia treated with antithymocyte globulin and cyclosporine. J Pediatr. 2008;153(6):814–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoshida N, Yagasaki H, Takahashi Y, Yamamoto T, Liang J, Wang Y, et al. Clinical impact of HLA-DR15, a minor population of paroxysmal nocturnal haemoglobinuria-type cells, and an aplastic anaemia-associated autoantibody in children with acquired aplastic anaemia. Br J Haematol. 2008;142(3):427–35. [DOI] [PubMed] [Google Scholar]
- 35.Scheinberg P, Nunez O, Weinstein B, Biancotto A, Wu CO, Young NS. Horse versus rabbit antithymocyte globulin in acquired aplastic anemia. N Engl J Med. 365(5):430–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marsh JC, Bacigalupo A, Schrezenmeier H, Tichelli A, Risitano AM, Passweg JR, et al. Prospective study of rabbit antithymocyte globulin and cyclosporine for aplastic anemia from the EBMT Severe Aplastic Anaemia Working Party. Blood. 2012;119(23):5391–6. [DOI] [PubMed] [Google Scholar]
- 37.Afable MG, 2nd, Shaik M, Sugimoto Y, Elson P, Clemente M, Makishima H, et al. Efficacy of rabbit anti-thymocyte globulin in severe aplastic anemia. Haematologica. 2011;96(9):1269–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jeong D-C, Chung NG, Lee JW, Jang P-S, Cho B, Kim H-K. Long-term outcome of immunosuppressive therapy with rabbit antithymocyte globulin (rATG) for childhood severe aplastic anemia for 15 years. ASH Annual Meeting Abstracts. 2011;118(21):1346. [Google Scholar]
- 39.Bacigalupo A, Socie G, Schrezenmeier H, Tichelli A, Locasciulli A, Fuehrer M, et al. Bone marrow versus peripheral blood as the stem cell source for sibling transplants in acquired aplastic anemia: survival advantage for bone marrow in all age groups. Haematologica. 2012;97(8):1142–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kikuchi A, Yabe H, Kato K, Koh K, Inagaki J, Sasahara Y, et al. Long-term outcome of childhood aplastic anemia patients who underwent allogeneic hematopoietic SCT from an HLA-matched sibling donor in Japan. Bone Marrow Transplant. 2012;48(5):657–60. [DOI] [PubMed] [Google Scholar]