

Abstract
Diamond-Blackfan anemia is a congenital erythroid hypoplasia caused by functional haploinsufficiency of genes encoding ribosomal proteins. Mutations involving the ribosomal protein S19 gene are detected in 25% of patients. Enforced expression of ribosomal protein S19 improves the overall proliferative capacity, erythroid colony-forming potential and erythroid differentiation of hematopoietic progenitors from ribosomal protein S19-deficient patients in vitro and in vivo following xenotransplantation. However, studies using animal models are needed to assess the therapeutic efficacy and safety of the viral vectors. In the present study we have validated the therapeutic potential of gene therapy using mouse models of ribosomal protein S19-deficient Diamond-Blackfan anemia. Using lentiviral gene transfer we demonstrated that enforced expression of ribosomal protein S19 cures the anemia and lethal bone marrow failure in recipients transplanted with ribosomal protein S19-deficient cells. Furthermore, gene-corrected ribosomal protein S19-deficient cells showed an increased pan-hematopoietic contribution over time compared to untransduced cells without signs of vector-mediated toxicity. Our study provides a proof of principle for the development of clinical gene therapy to cure ribosomal protein 19-deficient Diamond-Blackfan anemia.
Introduction
Diamond-Blackfan anemia (DBA) is a congenital erythroid hypoplasia that presents early in infancy. The classic hematologic profile of DBA consists of macrocytic anemia with reticulocytopenia, normal or decreased levels of neutrophils, and a variable platelet count.1 In addition to the hematopoietic symptoms, DBA is characterized by the presence of physical abnormalities and a predisposition to cancer.2–4
Mutations in genes that encode ribosomal proteins (RP) have been identified in approximately 60–70% of DBA patients.5–13 Among these genes, RPS19 is the most common DBA gene (25% of the cases).5 All reported patients are heterozygous for the given mutation, and in most cases the mutations are predicted to result in haploinsufficiency of the respective ribosomal protein.14,15
Corticosteroids form the main therapeutic regimen in DBA.4 However, although approximately 80% of patients initially respond to corticosteroids, only half of these patients sustain the therapeutic response, while the remaining patients need chronic transfusion therapy. Twenty percent of patients undergo spontaneous remission and maintain acceptable hemoglobin levels without therapeutic intervention. The only curative treatment for DBA is allogeneic bone marrow (BM) transplantation.16
Current DBA therapies carry risks of serious side effects, and a high proportion of deaths are treatment-related underscoring the need for development of novel therapies.4 We have previously demonstrated that enforced expression of RPS19 improves the overall proliferative capacity, erythroid colony-forming potential and erythroid differentiation of hematopoietic progenitors from RPS19-deficient DBA patients.17,18 Furthermore, gene-correction of stem cells from RPS19-deficient DBA patients improves cell engraftment and erythroid differentiation following transplantation into immunocompromised mice.19 Despite these encouraging findings, it has not been clear whether gene replacement therapy using ribosomal protein genes can cure the anemia and bone marrow failure in vivo. In the current study we assessed the therapeutic efficacy and safety of gene therapy using mouse models of RPS19-deficient DBA.
Methods
Design of lentiviral vector constructs and lentivirus production
The self-inactivating lentiviral vectors used in this study were derived from the pRRL.PPT.PGK.GFPpre vector.20 A codon-optimized human RPS19 cDNA was designed and inserted downstream of the spleen focus-forming virus (SFFV) promoter. Following the RPS19 cDNA, internal ribosomal entry site (IRES), GFP, and improved post-transcriptional regulatory element (Pre*) were inserted to form the pRRL.PPT.SF.RPS19co.iresGFP.pre* vector (hereafter termed SFFV-RPS19). A similar vector, in which the RPS19 cDNA was replaced with an equally long non-coding spacer sequence, was used as a control (pRRL.PPT.SF.spacer.iresGFP.pre*; hereafter termed SFFV-GFP). Lentiviral vectors were produced by the Vector Unit at Lund University.
Mice
Generation of the transgenic Rps19 knockdown mice has been reported previously.21 Briefly, this model contains an Rps19-targeting shRNA (shRNA-D) that is expressed by a doxycycline-responsive promoter located downstream of the Collagen A1 gene. Rps19 deficiency was induced by feeding the mice with doxycycline-containing food pellets (200 mg/kg doxycycline; Bio-Serv), with the exception of the recipients transplanted with D/D BM which were given doxycycline in the drinking water (2 mg/mL doxycycline; Sigma-Aldrich) supplied with 10 mg/mL sucrose (Sigma-Aldrich) for the first 2 weeks of induction. Mice were maintained at Lund University animal facility and all animal experiments were performed with consent from the Lund University animal ethics committee.
Transduction and transplantation of hematopoietic stem and progenitor cells
c-Kit+ cells were enriched from the BM of the transgenic mice (CD45.2) using CD117 MicroBeads and MACS separation columns (Miltenyi), and pre-stimulated in serum-free StemSpan®SFEM medium supplemented with penicillin/streptomycin (GIBCO), murine stem cell factor (100 ng/mL, PeproTech), human thrombopoietin (50 ng/mL, PeproTech), murine interleukin-3 (10 ng/mL, PeproTech) and human interleukin-6 (10 ng/mL, PeproTech) in six-well plates (non-tissue culture treated; BD) for 1 day (0.5×106 cells/mL). Retronectin-coated (20 ng/mL; Takara) six-well plates were preloaded with the SFFV-RPS19 or SFFV-GFP vector (100 μL/well corresponding to a MOI of 15–30), and 1×106 cells were seeded into each well in 3 mL pre-stimulation medium. After incubation for 1 day, 1.0×106 (experiment 1) or 0.5×106 (experiments 2 and 3) bulk transduced cells were transplanted in 500 μL phosphate-buffered saline into the tail veins of lethally irradiated (900 cGy) wild-type recipients (CD45.1). Secondary transplants were performed by intravenous injection of 3×106 whole BM cells into lethally irradiated wild-type recipients (CD45.1.2).
An additional experiment (experiment 4) was performed by transducing FACS-sorted lineage-Sca1+c-Kit+ (LSK) hematopoietic stem and progenitor cells. In this experiment 100×103 LSK were pre-stimulated in serum-free medium supplemented with penicillin/streptomycin, murine stem cell factor (100 ng/mL) and human thrombopoietin (100 ng/mL) in 48-well plates (tissue culture treated, BD). After 1 day, LSK were transferred into 96-well plates (tissue culture treated, BD; 20×103 LSK/well in 100 μL pre-stimulation medium), and transduced with 100 μL virus-containing pre-stimulation media, corresponding to a MOI of ~50). One day after transduction, 10×103 bulk transduced LSK, together with 250×103 fresh BM cells of the same genotype, were transplanted as above.
Results
Induction of Rps19 deficiency causes lethal bone marrow failure that is cured by enforced expression of RPS19
As the enforced expression of RPS19 improves the erythroid development in CD34+ cells from RPS19-deficient DBA patients,17–19 we asked whether gene therapy could cure the lethal BM failure in our mouse model of RPS19-deficient DBA without vector-mediated toxicity.21 Briefly, this model contains an Rps19-targeting shRNA (shRNA-D) that is expressed by a doxycycline-responsive promoter located downstream of the Collagen A1 gene (Figure 1A). Experimental animals were bred to be either heterozygous (D/+) or homozygous (D/D) for the shRNA in order to generate two models with intermediate or severe Rps19 deficiency, respectively (Figure 1B). To genetically correct the Rps19 deficiency, we developed lentiviral vectors harboring a codon-optimized human RPS19 cDNA driven by the internal SFFV promoter, followed by IRES and GFP (SFFV-RPS19) (Figure 1C).21 The codon-optimized RPS19 cDNA was further modified to prevent its recognition and downregulation by the Rps19-targeting shRNA used. A similar vector without the RPS19 cDNA was used as a control vector (SFFV-GFP). In order to assess the functionality of these vectors, we cultured transduced c-Kit-enriched BM cells from control and D/D mice in liquid cultures in the presence of doxycycline. The D/D cells transduced with the SFFV-GFP control vector failed to expand during 4 days of culture (Figure 1D). In contrast, the SFFV-RPS19 vector mediated a 6-fold increase in total cell number when compared to the SFFV-GFP vector. Next we quantified the expression of endogenous Rps19 and vector-derived RPS19 in these cultures on day 4. The SFFV-RPS19-vector-transduced control cells showed, on average, a 1.5-fold higher expression of RPS19 compared to endogenous Rps19, while the SFFV-GFP-vector-transduced cells showed no RPS19 expression (Figure 1E). The expression of RPS19 was on average more pronounced in the D/D cells transduced with SFFV-RPS19 vector (2.2-fold). This is expected since at this time-point the D/D culture consists mainly of transduced cells, and thus this value reflects the true RPS19 expression more accurately. Indeed, the SFFV-GFP-vector-transduced D/D cells could not be analyzed due to their poor proliferation and survival.
Figure 1.

The Rps19 knockdown mouse model and the design of lentiviral vectors. (A) Overview of the modified loci. (B) Breeding strategy to adjust the level of Rps19 downregulation. (C) Overview of the generated vectors. (D–E) c-Kit–enriched hematopoietic progenitors (0.25×106) from the BM of uninduced mice were transduced and seeded in liquid cultures in the presence of doxycycline. (D) Cell counts on day 4. (E) Expression of endogenous Rps19 and vector-derived RPS19 on day 4. Data shown in (D) and (E) represent the average of two independent experiments with two technical replicates each. SA: splice acceptor; pA: polyadenylation signal; RSV: Rous sarcoma virus; ψ:packaging signal; SD: splice donor; RRE: rev-response element; cPPt: polypurine tract. Black arrowheads in panel (A) indicate transcriptional start sites.
Next we assessed the efficacy of the generated vectors in vivo. Administration of doxycycline to the recipients with D/D BM results in acute and lethal BM failure, while recipients with D/+ BM develop a mild, chronic phenotype (Online Supplementary Figure S1).21 Since the D/D mice develop lethal BM failure shortly after doxycycline administration, we chose this model to rigorously test whether gene correction can rescue the lethal phenotype and cure the disease. The D/+ model was used in parallel to test the vectors upon Rps19 haploinsufficiency,21 a condition that is not lethal and allows long-term monitoring of experimental animals. We transduced uninduced BM cells from the control, D/+ and D/D mice with the vectors, and transplanted the transduced cells into wild-type recipient mice. We decided to use wild-type recipients since we have shown previously that the hematopoietic phenotype in Rps19-deficient mice is autonomous to the blood system.21 Initial transduction efficiencies with therapeutic and control vectors varied on average between 40% and 50% based on the percentage of GFP+ cells before transplantation (Online Supplementary Figure S2). Following engraftment and stable regeneration of the blood system, the recipient mice were administered doxycycline to down-regulate the endogenous Rps19 in order to induce the disease (Figure 2A). After 2 weeks of doxycycline administration, the recipients transplanted with SFFV-RPS19 or SFFV-GFP control cells had similar blood cellularity (Figure 2B). The recipients with the SFFV-GFP-transduced D/D BM developed lethal BM failure as exemplified by dramatic decreases in erythrocyte, reticulocyte, white blood cell and platelet counts, and died around this time-point. The reduction in the mean corpuscular volume in these mice reflects a complete lack of new erythrocytes produced under doxycycline administration. Remarkably, the recipients transplanted with the SFFV-RPS19 D/D BM had nor mal blood cellularity. In contrast to the recipients with SFFV-GFP D/D BM, the recipients with SFFV-GFP D/+ BM showed mild reductions in the number of erythrocytes and white blood cells and in hemoglobin concentration, but were able to compensate for the erythroid defect as indicated by the normal reticulocyte count. Similarly to the recipients with D/D BM, SFFV-RPS19 cured the erythroid defect and improved the number of white blood cells (Figure 2B).
Figure 2.
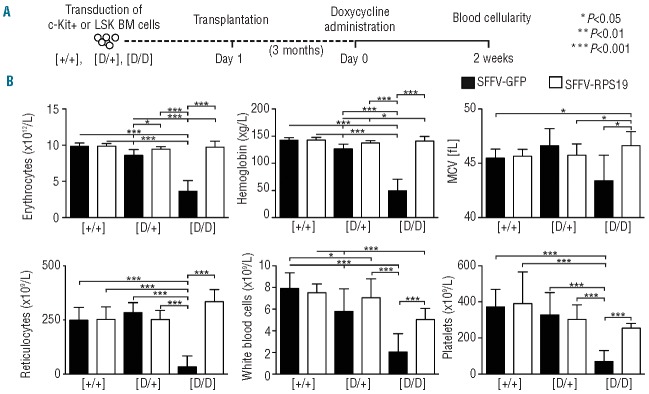
Enforced expression of RPS19 rescues the lethal bone marrow failure in Rps19-deficient mice. (A) Experimental strategy to validate the therapeutic potential of RPS19 gene correction. (B) Erythrocyte number, hemoglobin concentration, mean corpuscular volume (MCV), reticulocyte number, white blood cell number and platelet number on day 14 after doxycycline administration (n = 23, 24, 16, 16, 10 and 11 for the Control SFFV-GFP, Control SFFV-RPS19, D/+ SFFV-GFP, D/+ SFFV-RPS19, D/D SFFV-GFP and D/D SFFV-RPS19, respectively). Error bars represent standard deviation.
To determine whether long-term therapeutic benefits would be observed, the mice were monitored for 4 months (Figure 3A). Remarkably, the recipients with SFFV-RPS19 D/D BM continued to show normal blood cellularity over time, demonstrating a long-term cure of the acute lethal BM failure (Figure 3B). The recipients with SFFV-GFP D/+ BM were able to compensate for the erythroid defect over time and showed only a reduction in the number of white blood cells after 4 months. The number of white blood cells was slightly improved by SFFV-RPS19, although the observed increase was not statistically significant. Finally, at this time-point all groups showed high overall donor reconstitution confirming the absence of recipient-derived hematopoiesis (Online Supplementary Figure S3).
Figure 3.
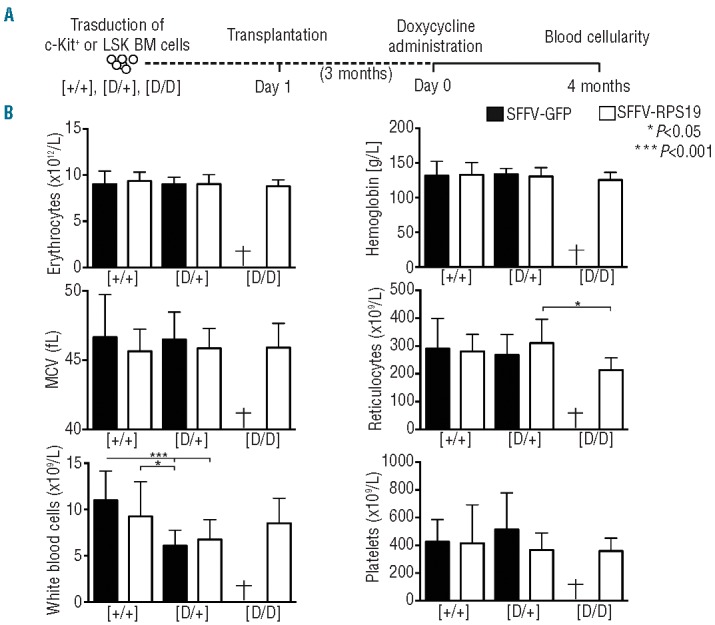
Enforced expression of RPS19 results in long-term rescue of the hematopoietic defect of Rps19-deficient mice. (A) Experimental strategy to validate the long-term therapeutic potential of RPS19 gene correction. (B) Erythrocyte number, hemoglobin concentration, mean corpuscular volume (MCV), reticulocyte number, white blood cell number and platelet number after 4 months of doxycycline administration (n = 22, 24, 14, 16 and 11 for the Control SFFV-GFP, Control SFFV-RPS19, D/+ SFFV-GFP, D/+ SFFV-RPS19 and D/D SFFV-RPS19, respectively). Error bars represent standard deviation.
Gene-corrected Rps19-deficient cells gain a competitive advantage resulting in increased contribution to hematopoiesis in vivo
To assess how gene correction affects the hematopoietic contribution of hematopoietic stem and progenitor cells over time, we monitored the percentage of GFP+ white blood cells in the peripheral blood of the recipients (Figure 4A). As expected based on the initial transduction efficiencies, the mean percentage of total GFP+ cells before the administration of doxycycline varied between 30% and 63% (Figure 4B). Following doxycycline administration, the mean percentage of GFP+ cells remained relatively stable in the recipients repopulated with SFFV-GFP and SFFV-RPS19 control BM, and SFFV-GFP D/+ BM, while it decreased in the recipients with SFFV-GFP D/D BM. Importantly, there was considerable variation in the levels of reconstitution of GFP+ cells between individual mice in these groups. By contrast, the recipients with SFFV-RPS19 D/+ BM and SFFV-RPS19 D/D BM showed clear increases in the frequency of total GFP+ cells at 4 months (46% to 64%, and 63% to 77%, respectively) with relatively small variation. As the total white blood cells include lymphoid cells with a long life span, we analyzed the percentage of GFP+ myeloid cells in the peripheral blood, which provides a more dynamic read-out for BM activity. Similarly to the total white blood cells, the percentage of GFP+ myeloid cells in the recipients with SFFV-GFP and SFFV-RPS19 control BM, and SFFV-GFP D/+ BM remained relatively stable and a notable variation was observed between individual recipients (Figure 4C). By contrast, in the recipients with SFFV-RPS19 D/+ BM and SFFV-RPS19 D/D BM, administration of doxycycline increased the mean percentage of GFP+ cells to almost 100% with minimal variation between individual recipients.
Figure 4.

Enforced expression of RPS19 confers a competitive advantage over untransduced Rps19-deficient cells. (A) Experimental strategy to validate the therapeutic potential of RPS19 gene correction. The percentage of transduced (B) total donor-derived white blood cells (CD45.2+GFP+) or (C) myeloid cells (CD45.2+Gr1+CD11b+GFP+) in the peripheral blood before doxycycline administration, 2 weeks and 4 months after doxycycline administration (n = 9–24 per group). (D) BM cellularity of the recipients 17–23 weeks after the doxycycline administration (n = 11–24 per group). (E) The percentage of transduced cells in the hematopoietic stem and progenitor compartments (n = 11–24 per group). Data in (B) are presented as box whisker plots with minimum and maximum values. Error bars in (C) represent standard deviation.
Although the recipients with D/+ BM were able to compensate for the blood cellularity over time, it is clear that enforced expression of RPS19 conferred Rps19-deficient cells a growth advantage compared to untransduced Rps19-deficient cells. Indeed, the recipients with SFFV-GFP D/+ BM showed a trend toward reduced BM cellularity that was improved by RPS19 overexpression (Figure 4D). Furthermore, using a previously described FACS strategy that allows fractionation of the myeloerythroid compartment in BM (Online Supplementary Figure S4),21,22 the mean percentage of GFP+ progenitor cells was considerably higher in the recipients with SFFV-RPS19 D/+ BM and SFFV-RPS19 D/D BM than in the other groups demonstrating the competitive advantage of gene-corrected cells already early in the hematopoietic hierarchy (Figure 4E).
Finally, in order to provide definite proof of long-term cure of the recipients with SFFV-RPS19 D/D BM, we transplanted whole BM from SFFV-RPS19 control and D/D primary recipients into lethally irradiated secondary recipients, and assessed the cellularity and GFP frequency in the peripheral blood 10 weeks after transplantation. Despite the mild reduction in the number of white blood cells in the recipients with SFFV-RPS19 D/D BM, no significant differences in blood cellularity were observed compared to that in the recipients with SFFV-RPS19 control BM (Online Supplementary Figure S5A). Furthermore, we continued to observe a high percentage of GFP+ cells in the peripheral blood of the secondary recipients with SFFV-RPS19 D/D BM (Online Supplementary Figure S5B).
Gene-corrected Rps19-deficient cells sustain polyclonal hematopoiesis and have a typical lentiviral insertion profile
In order to assess the integration profile of the SFFV-RPS19 vector as well as clonal dynamics of the transduced cells, we performed insertion site analysis on DNA of BM cells of five control and five D/+ mice obtained from recipients after 17–23 weeks of doxycycline administration (Online Supplementary Figure S6A). Integration sites were analyzed by linear amplification mediated polymerase chain reaction (LAM-PCR) and sequences retrieved by high throughput 454- sequencing. In total, 54,464 sequences, which identified 718 unique integrations (274 in the control group and 444 in the D/+ group), were collected. The mean vector copy number was similar in all analyzed animals subjected to pyrosequencing (Online Supplementary Figure S6B). According to the transduction efficiency prior to transplantation, the number of transplanted cells per mouse, the LAM-PCR protocol and the vector copy number, we did not observe a reduction in clonality but rather a stable polyclonal to oligoclonal situation in the bone marrow of all animals (for details see Online Supplementary Table S1).
Lentiviral vectors preferentially integrate inside transcription units.23 Our analysis revealed that in both groups only 65% (control group) and 50% (D/+ group) of all hits were intronic. When compared to a typical lentiviral integration pattern (70–85% intronic hits), this reduction indicates a certain selection of the transduced cells in vivo. However, we did not observe an accumulation of insertions close to the transcription start site of genes, as for gamma retroviral vectors or in the case of enhancer-mediated clonal selection (Online Supplementary Table S2). In the control group 81.5% of all insertions were present with low read counts (<10 reads), which, statistically, did not differ from the D/+ animals which showed 86.4% of low read insertions (Online Supplementary Figure S6C). High read clones (>100 reads) were found in both groups at the same level (7.5% control, 6.3% D/+ group) (Online Supplementary Figure S6D) on a polyclonal background as also indicated by the gel pictures of the LAM-PCR products (Online Supplementary Figure S7). These clones indicate either potential dominant clones within the transduced compartment or simply fluctuation of highly active clones contributing to hematopoiesis. However, the presence of high read count clones was not different between the two groups and there was no accumulation of hits near possible proto-oncogenes (Online Supplementary Table S3). We observed 20 different common insertion sites24,25 of which 17 were present in both groups (Online Supplementary Table S4). One third of the common insertion sites were also found in our previous integration site analysis of other mouse cohorts transplanted with lentiviral vectors expressing other genes.26
Discussion
In this study we demonstrated that gene therapy is feasible in a mammalian model of Diamond-Blackfan anemia. Lentiviral vectors overexpressing RPS19 corrected the anemia and lethal bone marrow failure in Rps19-deficient mice showing that pathophysiological correction of the disease is possible through gene therapy. Enforced expression of RPS19 has been shown to improve the overall proliferative capacity, erythroid colony-forming potential and erythroid differentiation of hematopoietic progenitors from RPS19-deficient DBA patients in vitro.17,18 Furthermore, gene-correction improves the engraftment and erythroid differentiation of these cells when transplanted into immunocompromised mice.19 However, the xenograft recipient mice do not develop a hematopoietic phenotype characteristic of DBA and it is, therefore, essential to determine whether the severe hematopoietic defects due to Rps19 deficiency in mouse models can be corrected with gene therapy following RPS19 gene transfer into Rps19-deficient hematopoietic stem cells. Furthermore, studies using mouse models are essential to assess the safety of the therapeutic vectors.
In the current study we decided to utilize the strong and ubiquitously expressed SFFV promoter to provide a proof of principle of the feasibility of gene therapy in the treatment of RPS19-deficient DBA. Furthermore, as codonoptimization of therapeutic genes has been shown to improve gene expression, we designed a codon-optimized human RPS19 cDNA that is not recognized by the Rps19-targeting shRNA. Indeed, the expression of RPS19 driven by the SFFV promoter was approximately 2-fold higher than that of the endogenous Rps19. Furthermore, the rescue of the proliferation of transduced c-Kit+ D/D BM cells demonstrated similar or higher levels of RPS19 compared to the endogenous Rps19 protein.
The onset of Rps19 deficiency in the recipients with shRNA-D BM resulted in a phenotype that correlated with the level of Rps19 downregulation. The recipients with D/D BM developed lethal BM failure, while the recipients with D/+ BM exhibited a mild, chronic phenotype. Remarkably, SFFV-RPS19 completely cured the BM failure generated by the D/D BM, demonstrating the potential of gene therapy to cure RPS19-deficient patients with severe transfusion-dependent DBA. Furthermore, after 4 months of doxycycline administration the recipients with SFFV-RPS19 D/D BM had, on average, almost 100% GFP+ cells in all hematopoietic BM compartments, including stem cells, suggesting a competitive advantage of gene-corrected cells already in these populations. Although the recipients with D/+ BM were able to compensate for the blood cellularity over time, they also showed an increased frequency of GFP+ progenitor cells indicating a competitive advantage of gene-corrected cells in the absence of a severe defect of blood cellularity.
Taken together, our findings demonstrate the feasibility of developing clinical gene therapy for the treatment of RPS19-deficient DBA. Importantly, by designing a codon-optimized RPS19 cDNA, driven by the SFFV promoter, we have succeeded in generating a vector system that allows high enough RPS19 expression for full functional correction of the anemia and BM failure in Rps19-deficient mice. In normal cells, ribosomal proteins are produced in excess to the needs of the ribosome assembly, and the excess protein is subjected to proteosomal degradation.27 Because of this physiological regulation, it is unlikely that the ectopic expression of RPS19 would promote uncontrolled growth. This notion is supported by findings from transgenic mice overexpressing the normal RPS19 cDNA in addition to the endogenous Rps19 gene.28 Consistent with this, we did not observe any hematologic abnormalities due to enforced expression of RPS19. Our gene therapy approach to correct Rps19-deficient hematopoiesis required relatively high levels of RPS19 expression and therefore the use of the retroviral SFFV promoter to drive the transgene cassette. In our experiments and the clonality analysis we did not find signs of overt insertional mutagenesis. However, we cannot completely exclude that the use of a strong internal promoter, such as SFFV, results in a residual risk factor. Nevertheless, studies assessing the efficacy of clinically relevant promoters, such as the human phosphoglycerate kinase promoter and the elongation factor 1α short promoter, should be performed since these promoters are less likely to cause insertional oncogenesis.29 Future studies using vectors with these mammalian promoters are needed to determine whether safer vectors can generate sufficient RPS19 expression to correct the pathophysiology of the disease.
Acknowledgments
We thank Beata Lindqvist for lentivirus production. This work was supported by a Hemato-Linné grant (Swedish Research Council Linnaeus), The Swedish Cancer Society (SK), the Swedish Children’s Cancer Society (SK), the Swedish Research Council (SK), the Tobias Prize awarded by the Royal Swedish Academy of Sciences financed by the Tobias Foundation (SK) and EU project grants STEMEXPAND and PERSIST. JF was funded by the Diamond-Blackfan Anemia Foundation.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Willig TN, Niemeyer CM, Leblanc T, Tiemann C, Robert A, Budde J, et al. Identification of new prognosis factors from the clinical and epidemiologic analysis of a registry of 229 Diamond-Blackfan anemia patients. DBA group of Société d’Hématologie et d’Immunologie Pédiatrique (SHIP), Gesellshaft für Pädiatrische Onkologie und Hämatologie (GPOH), and the European Society for Pediatric Hematology and Immunology (ESPHI). Pediatr Res. 1999;46(5):553–61. [DOI] [PubMed] [Google Scholar]
- 2.Campagnoli MF, Garelli E, Quarello P, Carando A, Varotto S, Nobili B, et al. Molecular basis of Diamond-Blackfan anemia: new findings from the Italian registry and a review of the literature. Haematologica. 2004;89(4):480–9. [PubMed] [Google Scholar]
- 3.Orfali KA, Ohene-Abuakwa Y, Ball SE. Diamond Blackfan anaemia in the UK: clinical and genetic heterogeneity. Br J Haematol. 2004;125(2):243–52. [DOI] [PubMed] [Google Scholar]
- 4.Lipton JM, Atsidaftos E, Zyskind I, Vlachos A. Improving clinical care and elucidating the pathophysiology of Diamond Blackfan anemia: an update from the Diamond Blackfan Anemia Registry. Pediatr Blood Cancer. 2006;46(5):558–64. [DOI] [PubMed] [Google Scholar]
- 5.Draptchinskaia N, Gustavsson P, Andersson B, Pettersson M, Willig TN, Dianzani I, et al. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat Genet. 1999;21(2):169–75. [DOI] [PubMed] [Google Scholar]
- 6.Gazda HT, Grabowska A, Merida-Long LB, Lataqiec E, Schneider HE, Lipton JM, et al. Ribosomal protein S24 gene is mutated in Diamond-Blackfan anemia. Am J Hum Genet. 2006;79(6):1110–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cmejla R, Cmejlova J, Handrkova H, Petrak J, Pospisilova D. Ribosomal protein S17 gene (RPS17) is mutated in Diamond-Blackfan anemia. Hum Mutat. 2007;28(12):1178–82. [DOI] [PubMed] [Google Scholar]
- 8.Farrar JE, Nater M, Caywood E, McDevitt MA, Kowalski J, Takemoto CM, et al. Abnormalities of the large ribosomal subunit protein, Rpl35A, in Diamond-Blackfan anemia. Blood. 2008;112(5):1582–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gazda HT, Sheen MR, Vlachos A, Choesmel V, O’Donohue MF, Schneider H, et al. Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond-Blackfan anemia patients. Am J Hum Genet. 2008; 83(6):769–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doherty L, Sheen MR, Vlachos A, Choesmel V, O’Donohue MF, Clinton C, et al. Ribosomal protein genes RPS10 and RPS26 are commonly mutated in Diamond-Blackfan anemia. Am J Hum Genet. 2010; 86(2):222–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farrar JE, Vlachos A, Atsidaftos E, Carlson-Donohoe H, Markello TC, Arceci RJ, et al. Ribosomal protein gene deletions in Diamond-Blackfan anemia. Blood. 2011;118(26):6943–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gazda HT, Preti M, Sheen MR, O’Donohue MF, Vlachos A, Davies SM, et al. Frameshift mutation in p53 regulator RPL26 is associated with multiple physical abnormalities and a specific pre-ribosomal RNA processing defect in diamond-blackfan anemia. Hum Mutat. 2012;33(7):1037–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Landowski M, O’Donohue MF, Buros C, Ghazvinian R, Montel-Lehry N, Vlachos A, et al. Novel deletion of RPL15 identified by array-comparative genomic hybridization in Diamond-Blackfan anemia. Hum Genet. 2013;132(11):1265–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Willig TN, Draptchinskaia N, Dianzani I, Ball S, Niemeyer C, Ramenghi U, et al. Mutations in ribosomal protein S19 gene and Diamond Blackfan anemia: wide variations in phenotypic expression. Blood. 1999;94(12):4294–306. [PubMed] [Google Scholar]
- 15.Angelini M, Cannata S, Mercaldo V, Gibello L, Santoro C, Dianzani I, et al. Missense mutations associated with Diamond-Blackfan anemia affect the assembly of ribosomal protein S19 into the ribosome. Hum Mol Genet. 2007;16(14):1720–7. [DOI] [PubMed] [Google Scholar]
- 16.Vlachos A, Federman N, Reyes-Haley C, Abramson J, Lipton JM. Hematopoietic stem cell transplantation for Diamond Blackfan anemia: a report from the Diamond Blackfan Anemia Registry. Bone Marrow Transplant. 2001;27(4):381–6. [DOI] [PubMed] [Google Scholar]
- 17.Hamaguchi I, Ooka A, Brun A, Richter J, Dahl N, Karlsson S. Gene transfer improves erythroid development in ribosomal protein S19-deficient Diamond-Blackfan anemia. Blood. 2002;100(8):2724–31. [DOI] [PubMed] [Google Scholar]
- 18.Hamaguchi I, Flygare J, Nishiura H, Brun AC, Ooka A, Kiefer T, et al. Proliferation deficiency of multipotent hematopoietic progenitors in ribosomal protein S19 (RPS19)-deficient diamond-Blackfan anemia improves following RPS19 gene transfer. Mol Ther. 2003;7(5):613–22. [DOI] [PubMed] [Google Scholar]
- 19.Flygare J, Olsson K, Richter J, Karlsson S. Gene therapy of Diamond Blackfan anemia CD34(+) cells leads to improved erythroid development and engraftment following transplantation. Exp Hematol. 2008;36(11):1428–35. [DOI] [PubMed] [Google Scholar]
- 20.Dull T, Zufferey R, Kelly M, et al. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72(11):8463–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaako P, Flygare J, Olsson K, Quere R, Ehinger M, Henson A, et al. Mice with ribosomal protein S19 deficiency develop bone marrow failure and symptoms like patients with Diamond-Blackfan anemia. Blood. 2011;118(23):6087–96. [DOI] [PubMed] [Google Scholar]
- 22.Jaako P, Debnath S, Olsson K, Bryder D, Flygare J, Karlsson S. Dietary L-leucine improves the anemia in a mouse model for Diamond-Blackfan anemia. Blood. 2012;120(11):2225–8. [DOI] [PubMed] [Google Scholar]
- 23.Mitchell RS, Beitzel BF, Schroder AR, Shinn P, Chen H, Berry CC, et al. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004;2(8):E234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suzuki T, Shen H, Agaki K, Morse HC, Malley JD, Naiman DQ, et al. New genes involved in cancer identified by retroviral tagging. Nat Genet. 2002;32(1):166–74. [DOI] [PubMed] [Google Scholar]
- 25.Wu X, Luke BT, Burgess SM. Redefining the common insertion site. Virology. 2006;344(2):292–305. [DOI] [PubMed] [Google Scholar]
- 26.Rittelmeyer I, Rothe M, Brugman MH, Iken M, Schambach A, Manns MP, et al. Hepatic lentiviral gene transfer is associated with clonal selection, but not with tumor formation in serially transplanted mice. Hepatology. 2012;58(1):397–408. [DOI] [PubMed] [Google Scholar]
- 27.Lam YW, Lamond AI, Mann M, Andersen JS. Analysis of nucleolar dynamics reveals the nuclear degradation of ribosomal proteins. Curr Biol. 2007;17(9):749–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Devlin EE, Dacosta L, Mohandas N, Elliott G, Bodine DM. A transgenic mouse model demonstrates a dominant negative effect of a point mutation in the RPS19 gene associated with Diamond-Blackfan anemia. Blood. 2010;116(15):2826–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zychlinski D, Schambach A, Modlich U, Maetzig T, Meyer J, Grassman E, et al. Physiological promoters reduce the geno-toxic risk for integrating gene vectors. Mol Ther. 2008;16(4):718–25. [DOI] [PubMed] [Google Scholar]