

Abstract
Background
Recent randomized controlled trials have challenged the concept that increased high density lipoprotein cholesterol (HDL-C) levels are associated with coronary artery disease (CAD) risk reduction. The causal role of HDL-C in the development of atherosclerosis remains unclear. To increase precision and to minimize residual confounding, we exploited the cholesteryl ester transfer protein (CETP)-TaqIB polymorphism as an instrument based on Mendelian randomization.
Methods
The Mendelian randomization analysis was performed by two steps. First, we conducted a meta-analysis of 47 studies, including 23,928 cases and 27,068 controls, to quantify the relationship between the TaqIB polymorphism and the CAD risk. Next, the association between the TaqIB polymorphism and HDL-C was assessed among 5,929 Caucasians. We further employed Mendelian randomization to evaluate the causal effect of HDL-C on CAD based on the findings from the meta-analysis.
Results
The overall comparison of the B2 allele with the B1 allele yielded a significant risk reduction of CAD (P < 0.0001; OR = 0.88; 95% CI: 0.84–0.92) with substantial between-study heterogeneity (I2 = 55.2%; Pheterogeneity <0.0001). The result was not materially changed after excluding the Hardy-Weinberg Equilibrium (HWE)-violation studies. Compared with B1B1 homozygotes, Caucasian carriers of the B2 allele had a 0.25 mmol/L increase in HDL-C level (95% CI: 0.20–0.31; P <0.0001; I2 = 0; Pheterogeneity =0.87). However, a 1 standard deviation (SD) elevation in HDL-C levels due to the TaqIB polymorphism, was marginal associated with CAD risk (OR =0.79; 95% CI: 0.54–1.03; P =0.08).
Conclusions
Taken together, our results lend support to the concept that increased HDL-C cannot be translated into a reduction in CAD risk.
Electronic supplementary material
The online version of this article (doi:10.1186/s12881-014-0118-1) contains supplementary material, which is available to authorized users.
Keywords: Coronary artery disease, High density lipoprotein cholesterol, Polymorphism, Mendelian randomization
Background
Several clinical parameters are associated with common diseases and are helpful for predicting and preventing these common diseases. For instance, lipid profiles are well acknowledged to be associated with the risk of coronary artery disease (CAD) and myocardial infarction (MI) [1]. Observational and experimental studies have documented a strong positive association of low density lipoprotein cholesterol (LDL-C) and an inverse association of high density lipoprotein cholesterol (HDL-C) with the risk of CAD [2]. Nevertheless, the causal role of HDL-C in the development of atherosclerosis has not been fully clarified and the evidence available from conventional population studies is scarce and paradoxical [3-6]. Many large epidemiological studies, such as the Framingham study [7], have described an increase in HDL-C as being protective to CAD. However, it has been demonstrated that Dalcetrapib and Torcetrapib, two cholesteryl ester transfer protein (CETP) inhibitiors, were shown to elevate circulating HDL-C concentrations substantially, but do not benefit CAD patients in two large randomized controlled trials [8,9]. Both environmental exposures and genetic factors are thought to contribute to the majority of these inconsistencies. Deciphering the regulation of HDL-C metabolism via the interaction of inherited variations with environmental factors may help explore the underlying pathological mechanism of CAD.
CETP plays a key role in determining the circulating HDL levels and transfers cholesteryl esters from HDL-C to LDL-C, as well as very low density lipoprotein cholesterol (VLDL-C), in exchange for triglyceride rich lipoproteins [10]. An inverse association of CETP activity with HDL-C levels was observed [11]. Some genome-wide association studies have suggested that the correlation of the CETP locus with HDL-C concentrations is more significant than that of any other locus found across the genome [12,13]. Among these genetic variants, the TaqIB polymorphism (rs708272) in intron 1 [14] has been investigated extensively [15]. Significant associations of the TaqIB polymorphism with variations in CETP promoter activity [16] and circulating HDL-C concentrations [14] were observed, showing lower CETP concentrations and higher HDL-C levels among B2 carriers than among B1B1 homozygotes [14,17-19]. However, the definite relationship between the TaqIB polymorphism and CAD encountered a considerable dispute among the genotype-disease studies [12,19-23], most likely due to relatively small sample sizes, limitations in statistical power and interactions with ethnic descents, population classifications and environment exposures. We hypothesized that a large meta-analysis is a robust method that can reliably appraise the heterogeneity present in genetic association studies and also can expound the divergences. Given that the HDL-C is an intermediate in the causal pathway from the CETP gene to CAD, it would be sensible to conduct a meta-analysis that in some way integrates the triangle relationship: the TaqIB polymorphism-HDL-C (genotype-phenotype), the TaqIB polymorphism-CAD (genotype-disease), and HDL-C-CAD (phenotype-disease). The logic of this approach is greatly strengthened by the appeal to Mendelian randomization [24], which is according to Mendel’s second law (the law of independent assortment) [25]. Mendelian randomization means that the inheritance of an individual’s genes is independent by a seemingly random process at conception. Theoretically, the genetic polymorphism, which is causally and specifically bound up with the intermediate phenotype and the predicted risk of disease, could be exploited as an instrument to attain an un-confounded estimation of phenotype-disease association. In the present study, we examined whether the increase in circulating HDL-C concentrations due to the TaqIB polymorphism was correlated with a reduced risk of CAD, based on the rationale of Mendelian randomization [26].
Methods
Literature search strategy
Two authors (Wu and Lou) independently performed the search. The process was supervised by the third author (Qiu). Any disagreement was resolved by a consensus. The advanced search builder was used to refine the search. The Boolean operators “AND” and “OR” were used to combine the search themes. A stepwise search was conducted as recommended by the Cochrane Handbook for Systematic Reviews of Interventions (Version 5.1.0). A formal computerized literature search of electronic databases, PubMed/MEDLINE, Embase, CNKI (China Nation Knowledge Infrastructure Platform), Wanfang and CBM (China Biological Medicine), was conducted up to March 2014. A composition of the following MeSH terms and text words was used: ‘TaqIB’ or ‘rs708272’, ‘cholesteryl ester transfer protein’ or ‘CETP’, ‘high density lipoprotein cholesterol’ or ‘HDL-C’ and ‘coronary artery disease’ or ‘myocardial infarction’ or ‘atherosclerosis’. The additional studies were retrieved from the MEDLINE option ‘related articles’ and head searches were also added to the database. We also screened the bibliographies of the original research reports, reviews, and previous meta-analyses to optimize the databank. If the data was deficient or in an inappropriate form, we contacted with the original authors to obtain the raw data.
Selection criteria
All the studies that aimed for the association of the TaqIB polymorphism with HDL-C levels and CAD risk were potentially included. The following criteria were for the selection: (1) published articles of human-being genetics (full texts or abstracts) without racial or language restrictions; (2) if articles contained more than one geographic or other clinical characteristic subgroup, each subgroup was considered separately; (3) if multiple studies were derived from the same population, only the study with the largest sample size was involved to avoid overlapping data; (4) studies providing sufficient information on the TaqIB genotype by case–control status and/or on circulating HDL-C concentrations across the TaqIB genotype between CAD patients and controls.
CAD outcomes were diagnosed according to previous MI, angina pectoris, percutaneous trans luminal coronary angiography, coronary artery bypass grafting or severe angiographic stenosis ( ≥50% of ≥1 major coronary artery) [27]. Acute coronary syndrome (ACS) included unstable angina pectoris, as well as fatal and non-fatal MI [28]. MI was defined by the World Health Organization’s Multinational Monitoring of Trends and Determinants in Cardiovascular Disease (MONICA) criteria [29].
Extracted information
A standard data-collection procedure, in line with the inclusion criteria described above was used. Two investigators (Wu and Lou) independently extracted the variables from the individual eligible studies in duplicate and made the characteristics compatible in a pooled database. Any encountered disagreements were resolved by discussion in order to reach a consensus. The following information was extracted from all the eligible studies: first author’s name, publication year, ethnicity, geographic location, study design, population source, endpoints, clinical characteristics of the study subjects (such as age, gender, body mass index [BMI] and circulating lipid profiles levels), percentage of hypertension, diabetes, smoking status and the distribution of the TaqIB genotype both in patients and controls. The units of circulating lipid profiles were standardized to mmol/L. Continuous variables were expressed as mean ± standard deviation (SD) or median (5th and 95th percentiles). Standard error was converted to SD.
Hardy-Weinberg Equilibrium (HWE) testing
We tested conformity of the TaqIB polymorphism to HWE among controls in each individual study via the chi-square test or Fisher’s exact test, based on a Web program (http://ihg2.helmholtz-muenchen.de/cgi-bin/hw/hwa1.pl). In order to obtain robust evidence of estimating the association between the TaqIB polymorphism and CAD risk, sensitivity analyses were performed by excluding the HWE-deviating studies (P <0.05) [30]. We calculated the effect sizes for all the studies and then we calculated the effect sizes only for HWE-conforming studies.
Statistical analysis
Initially, we calculated the summary odd ratios (ORs) of CAD risk and the standard mean difference (SMD) of HDL-C concentrations corresponding to the 95% confidence interval (CI) to assess whether the TaqIB polymorphism was relevant to CAD risk or circulating HDL-C levels or both. Four genetic models, including allele comparison (B2 versus B1), dominant genetic model (B1B2 + B2B2 versus B1B1), recessive genetic model (B2B2 versus B1B2 + B1B1) and homozygote comparison (B2B2 versus B1B1) were used. We utilized a random-effects model based on the DerSimonian & Laird method to evaluate the effect size of each study and to modify the study weights on the basis of the in-study variance. The Mantel-Haenszel model was adopted for checking the possibility of heterogeneity [31]. Uniformity of findings across all studies was estimated by means of the inconsistency index (I2) statistic, ranging from 0 to 100%. A value of 0% indicated homogeneity; by contrast, high values of I2 implied that heterogeneity accounted for most of the between-study variation [32,33]. The between-study heterogeneity was differentiated with a Chi-square-based Q statistic test [34] where P <0.1 indicated heterogeneity across the studies. The significance level of the combined ORs which was estimated by the Z test, was P <0.05. Next, we examined pre-specified groupings of study characteristics with homogeneous effects, such as ethnicity (Asian, Caucasian and mixed-population), study design (prospective and retrospective), population source (hospital-based [H-B] and population-based [P-B]), and endpoints (CAD, MI and ACS). In addition, we conducted a meta-regression, as a complement to the estimation of the relationship between the TaqIB polymorphism and CAD risk to find the potential effect of environmental covariates on genetic heterogeneity.
Sensitivity analysis was performed by sequentially removing each individual study to identify those that likely biased the overall estimates. We used the visual funnel plot and Egger’s linear regression test to estimate publication bias. The standard error of log (OR) for each study was plotted against its OR. An asymmetric plot suggests the possible presence of publication bias, which can be verified by a T-test. P <0.05 of the I2 statistic and Egger’s test was regarded as significant [35].
Cumulative meta-analysis was used to decipher the impact of the first published study on the subsequent studies and the evolution of the synthesized effects over time was in accordance with the publications ordered by time. Data administration and statistical analysis were conducted by using Review Manager software release 5.0 (Oxford, UK) and Stata 11.0 (Stata Corporation, College Station, TX, USA). All P values were 2-sided.
Results
Results of study search and characteristics
The flowchart depicting the screening process of study selection is shown in Figure 1 and the excluded articles, with the reasons for exclusion are also described. A total of 470 relevant publications were obtained by the preliminary search in PubMed, EMBASE, CNKI, Wangfang and CBM. After evaluation for our inclusion criteria, 46 articles including 47 studies with adequate information related to the association between the TaqIB polymorphism and CAD risk, were included in the final analysis [11,17,20-22,36-66]. The clinical characteristics of the eligible studies are summarized in Additional file 1: Table S1. All the eligible studies were published between 1991 and 2012.There were 20 studies performed in Asian subjects [11,38,47,51,57,58,60,63,65,66], 26 in Caucasian subjects [17,20-22,36,37,40-46,49,50,52-56,59,61,62,64,67] and one mixed- population study [48]. In consideration of the study design, 6 studies were prospective [20,40,43,45,49] and the remaining 41 studies were retrospective [11,17,21,22,36-38,41,42,44,46-48,50-67]. In addition,11 of the 47 studies were P-B [20-22,36,37,40,43,46,49,54] and the remaining studies were H-B [11,17,38,39,41,42,44,45,47,48,50-53,55-66]. CAD was regarded as the main outcome in most of the studies [11,17,21,22,36-40,42,44,45,47,49-51,53,55-61,64-66], except 8 studies analyzed MI [20,43,46,48,52,54,62,63] and 2 studies analyzed ACS [41] as the endpoint. A significant departure from HWE was found in 7 studies [21,39,48,52,63]. The article by Jensen et al. [49] contained data from two independent studies (Nurses’ Health Study [NHS] and Health Professionals Follow-up Study [HPFS]); therefore we calculated the ORs separately in our meta-analysis. The study descriptions were shown in Additional file 2.
Figure 1.
Flow diagram of the search strategy and study selection for the meta-analysis.
Overall analysis
Forty-seven studies with 23,928 cases and 27,068 controls were incorporated into the meta-analysis. The allele and genotype frequency of the TaqIB polymorphism is shown in Table 1. The results of HWE examination among the controls for all the eligible studies are also listed. The synthetic overall percentage of the B2 allele was 41.4% in cases and 43.7% in controls. The B2 allele had a similar proportion among Asians subjects (40.1% in cases versus 47.1% in controls) compared to that among Caucasian subjects (41.2% in cases versus 42.5% in controls).
Table 1.
The distribution of the Taq1B allele and genotype among CAD and controls, and P value of HWE in controls
| Sample size | B1 allele, % | B2 allele, % | B1B1 genotype | B1B2 genotype | B2B2 genotype | HWE, | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| First author | Cases | Controls | Cases | Controls | Cases | Controls | Cases | Controls | Cases | Controls | Cases | Controls | P value |
| Arca M | 415 | 403 | 60.4 | 58.1 | 39.6 | 41.9 | 153 | 134 | 187 | 184 | 68 | 71 | 0.57 |
| Bhanushali AA | 90 | 150 | 59.4 | 50.3 | 40.6 | 49.7 | 33 | 37 | 40 | 76 | 17 | 42 | 0.820 |
| Blankenberg S | 1214 | 574 | 60.8 | 57.2 | 39.3 | 42.8 | 407 | 175 | 644 | 303 | 149 | 93 | 0.047 |
| Corella D | 557 | 1180 | 62.4 | 63.6 | 37.6 | 36.4 | 224 | 482 | 247 | 537 | 86 | 161 | 0.557 |
| Dedoussis GV | 237 | 237 | 60.5 | 58.2 | 39.5 | 41.8 | 83 | 78 | 121 | 120 | 33 | 39 | 0.530 |
| Durlach A | 96 | 138 | -a | - | - | - | - | - | - | - | 13 | 39 | - |
| Eiriksdottir G | 388 | 794 | 59.1 | 52.6 | 40.9 | 47.4 | 128 | 194 | 191 | 396 | 59 | 155 | 0.072 |
| Falchi A | 100 | 100 | 58.5 | 56.0 | 41.5 | 44.0 | 30 | 30 | 57 | 52 | 13 | 18 | 0.581 |
| Freeman DJ | 498 | 1108 | 58.8 | 55.2 | 41.2 | 44.8 | 164 | 339 | 259 | 541 | 76 | 225 | 0.733 |
| Fumeron F | 608 | 724 | 60.0 | 59.5 | 40.0 | 40.5 | 209 | 258 | 312 | 346 | 87 | 120 | 0.826 |
| Horne BD | 3223 | 1588 | 57.5 | 56.0 | 42.5 | 44.0 | 1064 | 508 | 1579 | 762 | 580 | 318 | 0.293 |
| Hsieh MC | 101 | 264 | 42.1 | 29.7 | 57.9 | 70.3 | 19 | 23 | 47 | 111 | 35 | 130 | 0.920 |
| Izar MC | 386 | 604 | 40.1 | 43.0 | 59.9 | 57.0 | 32 | 66 | 238 | 374 | 107 | 145 | 0.000 |
| Jensen MK [HPFS] | 259 | 513 | 58.7 | 58.9 | 41.3 | 41.1 | 89 | 180 | 126 | 244 | 44 | 89 | 0.686 |
| Jensen MK [NHS] | 246 | 486 | 58.5 | 58.3 | 41.5 | 41.7 | 84 | 166 | 120 | 235 | 42 | 85 | 0.907 |
| Kaestner S | 204 | 35 | 53.9 | 60.0 | 46.1 | 40.0 | 53 | 13 | 114 | 16 | 37 | 6 | 0.778 |
| Kawasaki I | 24 | 361 | 79.2 | 53.6 | 20.8 | 46.4 | 15 | 101 | 8 | 185 | 1 | 75 | 0.565 |
| Keavney B | 4685 | 3460 | 57.7 | 56.9 | 42.3 | 43.1 | 1477 | 1100 | 2175 | 1527 | 790 | 646 | 0.005 |
| Kolovou G | 374 | 97 | 60.7 | 46.4 | 39.3 | 53.6 | 126 | 22 | 202 | 45 | 46 | 29 | 0.573 |
| Li J | 236 | 54 | 58.8 | 58.8 | 41.2 | 41.3 | 82 | 15 | 73 | 19 | 21 | 6 | 0.997 |
| Liu S | 384 | 384 | 58.1 | 56.9 | 41.9 | 43.1 | 125 | 122 | 196 | 193 | 63 | 69 | 0.628 |
| McCaskie PA | 556 | 2683 | 59.3 | 57.0 | 40.7 | 43.0 | 196 | 860 | 262 | 1328 | 93 | 485 | 0.482 |
| Meiner V | 577 | 659 | 57.1 | 52.6 | 42.9 | 47.4 | 173 | 166 | 282 | 320 | 95 | 134 | 0.383 |
| Mohrschladt MF | 116 | 184 | 56.9 | 54.9 | 43.1 | 45.1 | 36 | 57 | 60 | 88 | 20 | 39 | 0.642 |
| Muendlein A | 332 | 225 | 62.0 | 57.3 | 38.0 | 42.7 | 125 | 71 | 162 | 116 | 45 | 38 | 0.420 |
| Padmaja N | 504 | 338 | 58.5 | 49.3 | 41.5 | 50.7 | 163 | 86 | 264 | 161 | 77 | 91 | 0.386 |
| Park KW | 119 | 106 | - | - | - | - | 49 | 30 | - | - | - | - | - |
| Poduri A | 265 | 150 | 64.3 | 49.3 | 35.7 | 50.7 | 117 | 33 | 107 | 82 | 41 | 35 | 0.252 |
| Porchay-Balderelli I | 223 | 2901 | 63.9 | 59.5 | 36.1 | 40.5 | 95 | 1012 | 95 | 1431 | 33 | 458 | 0.198 |
| Qin Q | 249 | 167 | 58.8 | 58.4 | 41.2 | 41.6 | 81 | 49 | 131 | 97 | 37 | 21 | 0.012 |
| Rahimi Z | 207 | 92 | 62.3 | 50.0 | 37.7 | 50.0 | 57 | 20 | 144 | 52 | 6 | 20 | 0.211 |
| Rejeb J | 212 | 104 | 71.0 | 65.9 | 29.0 | 34.1 | 104 | 45 | 93 | 47 | 15 | 12 | 0.959 |
| Schierer A | 349 | 2082 | 47.0 | 49.0 | 53.0 | 51.0 | - | - | - | - | - | - | - |
| Tenkanen H | 72 | 226 | 54.2 | 55.5 | 45.8 | 44.5 | 19 | 64 | 40 | 123 | 13 | 39 | 0.125 |
| Van Acker BA | 792 | 539 | 59.4 | 58.1 | 40.6 | 41.9 | 275 | 171 | 391 | 284 | 126 | 84 | 0.06 |
| Wang SH | 111 | 75 | 58.6 | 56.0 | 41.4 | 44.0 | 38 | 22 | 54 | 41 | 19 | 12 | 0.327 |
| Wang W | 128 | 247 | 64.8 | 54.0 | 35.2 | 46.0 | 50 | 72 | 66 | 123 | 12 | 52 | 0.968 |
| Whiting BM | 3319 | 1385 | 58.1 | 56.7 | 41.9 | 43.3 | 792 | 280 | 1201 | 377 | 402 | 170 | 0.039 |
| Wu JH | 200 | 285 | 56.7 | 52.0 | 43.3 | 48.0 | 45 | 63 | 79 | 159 | 25 | 52 | 0.007 |
| Yan SK | 106 | 64 | 60.4 | 56.3 | 39.6 | 43.8 | 41 | 19 | 46 | 34 | 19 | 11 | 0.526 |
| Yang J | 83 | 163 | 62.7 | 52.1 | 37.3 | 47.9 | 31 | 47 | 42 | 76 | 10 | 40 | 0.401 |
| Yilmaz H | 173 | 111 | 59.0 | 55.5 | 41.0 | 44.5 | 66 | 39 | 72 | 46 | 35 | 26 | 0.093 |
| Zhang GB | 88 | 94 | 58.0 | 60.6 | 42.0 | 39.4 | 31 | 32 | 40 | 50 | 17 | 12 | 0.268 |
| Zhang YX | 334 | 301 | 67.7 | 65.1 | 32.3 | 34.9 | 174 | 136 | 104 | 120 | 56 | 45 | 0.034 |
| Zhao SP | 238 | 203 | 62.0 | 56.4 | 38.0 | 43.6 | 95 | 60 | 105 | 109 | 38 | 34 | 0.191 |
| Zheng KQ | 203 | 100 | 60.6 | 60.5 | 39.4 | 39.5 | 66 | 33 | 114 | 55 | 23 | 12 | 0.132 |
| Zhou DF | 47 | 330 | 57.4 | 68.0 | 42.6 | 32.0 | 17 | 157 | 20 | 135 | 10 | 38 | 0.280 |
| Total | 23928 | 27068 | 58.6 | 56.3 | 41.4 | 43.7 | 7533 | 7667 | 10910 | 11720 | 3634 | 4521 | |
HWE: Hardy-Weinberg equilibrium. The P value of HWE determined by the χ2 test or Fisher’s exact test in control groups; a: No data.
We assessed the association between the TaqIB polymorphism and CAD risk for each study under different genetic models (Table 2).The overall comparison of the B2 allele with the B1 allele demonstrated a significant risk reduction of CAD (allele comparison: P <0.0001, OR = 0.88, 95% CI: 0.84–0.92; dominant model: P <0.0001, OR = 0.85, 95% CI: 0.79–0.91) with substantial between-study heterogeneity (allele comparison: I2 = 55.2%, Pheterogeneity <0.0001; dominant model: I2 = 48.6%, Pheterogeneity <0.0001) (Figure 2).
Table 2.
Summary estimates for ORs and 95% CI in different subgroups under various genetic contrasts
| Genotype contrasts | Study population | Study number, (case/control), n(n/n) | P heterogeneity | I 2 , % | P value a | OR | 95% CI |
|---|---|---|---|---|---|---|---|
| Total studies | |||||||
| Allele comparison | 45(23,713/26,824) | 0.000 | 55.2 | 0.000 | 0.88 | 0.84–0.92 | |
| (B2 versus B1) | |||||||
| Dominant model | 45(23,483/24,848) | 0.000 | 48.6 | 0.000 | 0.85 | 0.79–0.91 | |
| (B1B2 + B2B2 versus B1B1) | |||||||
| Recessive model | 45(23,460/24,880) | 0.000 | 53.7 | 0.000 | 0.81 | 0.74–0.88 | |
| (B2B2 versus B1B2 + B1B1) | |||||||
| Homozygote comparison | 44(23,364/24,742) | 0.000 | 58.1 | 0.000 | 0.76 | 0.68–0.84 | |
| (B2B2 versus B1B1) | |||||||
| Studies comfirming to HWE | |||||||
| Allele comparison | 38(13,326/20,048) | 0.000 | 56.6 | 0.000 | 0.86 | 0.81–0.91 | |
| Dominant model | 38(13,096/18,072) | 0.002 | 45.1 | 0.000 | 0.82 | 0.76–0.89 | |
| Recessive model | 38(13,073/18,104) | 0.000 | 55.1 | 0.000 | 0.77 | 0.69–0.86 | |
| Homozygote comparison | 37(12,977/17,966) | 0.000 | 59.2 | 0.000 | 0.72 | 0.63–0.82 | |
| Subgroups analysis after excluding HWE-deviation studies | |||||||
| Ethnicity | |||||||
| Allele comparison | Asian | 16(2,780/4,767) | 0.000 | 67.7 | 0.002 | 0.77 | 0.66–0.90 |
| Caucasian | 22(10,546/15,281) | 0.222 | 18.0 | 0.000 | 0.91 | 0.87–0.95 | |
| Dominant model | Asian | 16(2,550/2,791) | 0.022 | 46.3 | 0.000 | 0.65 | 0.54–0.79 |
| Caucasian | 22(10,546/15,281) | 0.505 | 0 | 0.001 | 0.90 | 0.85–0.96 | |
| Recessive model | Asian | 15(2,431/2,685) | 0.000 | 66.7 | 0.008 | 0.66 | 0.49–0.90 |
| Caucasian | 23(10,642/15,419) | 0.106 | 27.9 | 0.000 | 0.84 | 0.76–0.92 | |
| Homozygote comparison | Asian | 15(2,431/2,685) | 0.000 | 66.6 | 0.000 | 0.54 | 0.38–0.76 |
| Caucasian | 22(10,546/15,281) | 0.159 | 23.2 | 0.000 | 0.82 | 0.74–0.90 | |
| Study design | |||||||
| Allele comparison | prospective | 7(2,555/7,366) | 0.116 | 41.3 | 0.061 | 0.92 | 0.84–1.00 |
| retrospective | 31(10,771/12,682) | 0.000 | 59.2 | 0.000 | 0.84 | 0.78–0.90 | |
| Dominant model | prospective | 7(2,555/7,366) | 0.161 | 35 | 0.081 | 0.89 | 0.79–1.01 |
| retrospective | 31(10,518/10,738) | 0.002 | 47.3 | 0.000 | 0.79 | 0.72–0.88 | |
| Recessive model | prospective | 7(2,555/7,366) | 0.219 | 27.4 | 0.119 | 0.89 | 0.76–1.03 |
| retrospective | 31(10,518/10,738) | 0.000 | 58.1 | 0.000 | 0.73 | 0.64–0.84 | |
| Homozygote comparison | prospective | 7(2,555/7,366) | 0.106 | 42.7 | 0.080 | 0.84 | 0.70–1.02 |
| retrospective | 30(10,422/10,600) | 0.000 | 61.6 | 0.000 | 0.67 | 0.57–0.79 | |
| Population source | |||||||
| Allele comparison | P-B | 10(4,782/8,365) | 0.308 | 14.7 | 0.015 | 0.93 | 0.88–0.99 |
| H-B | 28(8,544/11,683) | 0.000 | 60.6 | 0.000 | 0.82 | 0.75–0.89 | |
| Dominant model | P-B | 10(4,782/8,365) | 0.361 | 8.8 | 0.025 | 0.91 | 0.84–0.99 |
| H-B | 28(8,314/9,707) | 0.002 | 49.5 | 0.000 | 0.76 | 0.68–0.86 | |
| Recessive model | P-B | 10(4,782/8,365) | 0.568 | 0 | 0.055 | 0.91 | 0.82–1.00 |
| H-B | 28(8,291/9,739) | 0.000 | 59.1 | 0.000 | 0.70 | 0.59–0.82 | |
| Homozygote comparison | P-B | 10(4,782/8,365) | 0.292 | 16.4 | 0.020 | 0.86 | 0.76–0.98 |
| H-B | 27(8,195/9,601) | 0.000 | 62.7 | 0.000 | 0.63 | 0.52–0.77 | |
| Endpoint | |||||||
| Allele comparison | CAD | 31(10,824/16,970) | 0.000 | 62 | 0.000 | 0.85 | 0.79–0.91 |
| MI | 5(2,029/2,787) | 0.200 | 33.2 | 0.032 | 0.89 | 0.80–0.99 | |
| ACS | 2(473/291) | 0.741 | 0 | 0.517 | 0.93 | 0.74–1.17 | |
| Dominant model | CAD | 31(10,594/14,994) | 0.001 | 49.5 | 0.000 | 0.81 | 0.73–0.89 |
| MI | 5(2,029/2,787) | 0.123 | 44.8 | 0.172 | 0.88 | 0.74–1.06 | |
| ACS | 2(473/291) | 0.493 | 0 | 0.357 | 0.86 | 0.61–1.19 | |
| Recessive model | CAD | 31(10,571/15,026) | 0.000 | 62.8 | 0.000 | 0.75 | 0.66–0.87 |
| MI | 5(2,029/2,787) | 0.775 | 0 | 0.006 | 0.80 | 0.69–0.94 | |
| ACS | 2(473/291) | 0.905 | 0 | 0.356 | 0.81 | 0.52–1.27 | |
| Homozygote comparison | CAD | 30(10,475/14,888) | 0.000 | 65.2 | 0.000 | 0.69 | 0.59–0.82 |
| MI | 5(2,029/2,787) | 0.300 | 17.9 | 0.008 | 0.76 | 0.63–0.93 | |
| ACS | 2(473/291) | 0.723 | 0 | 0.273 | 0.76 | 0.46–1.24 | |
aTest for overall effect; P-B: population-based, H-B: hospital-based.
Figure 2.
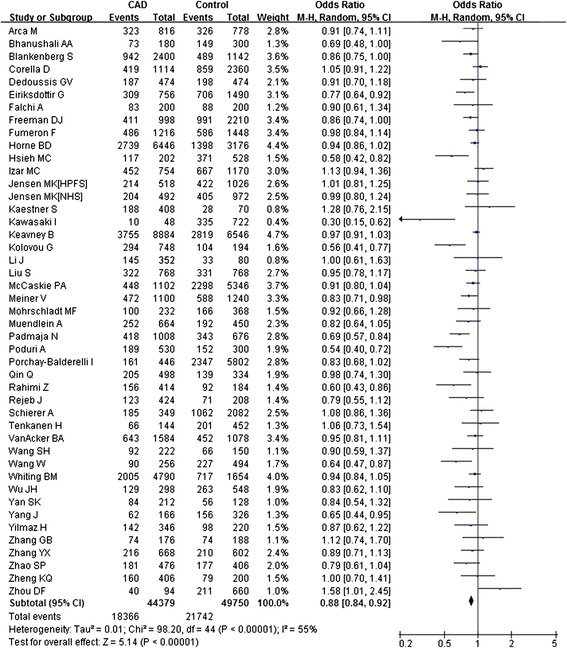
Meta-analysis for the overall association between the CETP TaqIB polymorphism and CAD under the allele comparison (B2 versus B1). ‘Events’ indicates the total number of the B2 allele. ‘Total’ indicates the total number of B2 allele plus B1 allele.
Sensitivity analysis
We hypothesized that HWE-deviating studies (7 studies containing 10,387 cases and 6,776 controls) might be partly responsible for the striking heterogeneity. Therefore, we performed a sensitivity analysis to compare the summary effects as well as the extent of between-study heterogeneity before and after excluding HWE-deviating studies. However, the summary ORs and the statistical significance of tests did not change substantially after removal of the HWE-deviating studies (allele comparison: P <0.0001, OR =0.86, 95% CI: 0.81–0.91, I2 = 56.6%, Pheterogeneity <0.0001; dominant model: P <0.0001, OR = 0.82, 95% CI: 0.76–0.89, I2 = 45.1%, Pheterogeneity <0.0001). In addition, no individual study was shown to substantially influence the overall results.
Publication bias
A remarkable publication bias of all the studies was reflected by the asymmetry of the funnel plot and verified by Egger’s regression test (t = −2.48, P =0.02 for allele comparison), but not verified by the Begg-Mazemdar test (P =0.19 for allele comparison). Nevertheless, after excluding the HWE-violating studies, the probability of publication bias was diminished and the P value for asymmetry in the funnel plot became non-significant (t = −1.71; P =0.10 for Egger’s regression test and P =0.44 for Begg-Mazemdar test). Further analysis via the trim and fill method suggested that no missing studies were needed to adjust the symmetry in the funnel plot for the TaqIB polymorphism (Figure 3).
Figure 3.
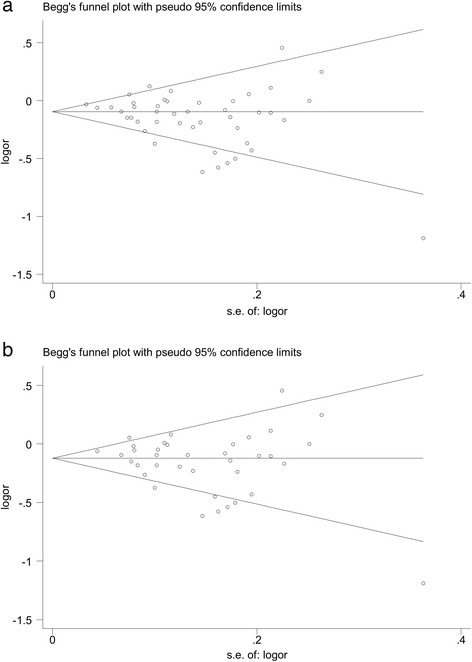
Begg’s funnel plot analysis to detect publication bias for allele comparison (B2 versus B1) of the TaqIB polymorphism (a) and after excluding the HWE-violating studies (b).
Cumulative analysis
No distinguishable evidence was found that the first published study affected the subsequent replication, as illustrated by the cumulative meta-analysis (data not shown).
Subgroup analysis
In view of the considerable heterogeneity in the total analysis, subgroup analysis is an appropriate method to explore the potential sources of heterogeneity. We categorized the data in the light of the different homogeneous characteristics, such as ethnicity, study design, population source and disease type in HWE-confirming studies. The summary estimates for ORs in different subgroups under various genetic contrasts are listed in Table 2. Population stratification by ethnicity detected that the magnitude of risk reduction, given by the TaqIB polymorphism among Asian subjects, was much greater than that among Caucasian subjects, being 23% (95% CI: 0.66–0.90, P =0.002) and 9% (95% CI: 0.87–0.95, P <0.0001), respectively. Discernible heterogeneity among Asian subjects (I2 = 67.7%, Pheterogeneity <0.0001) was not observed among Caucasian subjects (I2 = 18%, Pheterogeneity =0.22). Further, the comparison of the B2 allele with the B1 allele yielded a non-significant 8% risk reduction for CAD in the prospective group (95% CI: 0.84–1.00, P = 0.06, I2 = 41.3%, Pheterogeneity =0.12), which was higher than that in the retrospective group (OR = 0.84, 95% CI: 0.78–0.90, P <0.0001, I2 = 59.2%, Pheterogeneity <0.0001). Upon stratification by population source, the ORs of CAD appeared to decrease in the H-B group (OR =0.82, 95% CI: 0.75–0.89, P <0.0001, I2 = 60.6%, Pheterogeneity <0.0001) relative to the P-B group (OR = 0.93, 95% CI: 0.88–0.99, P =0.02, I2 = 14.7%, Pheterogeneity =0.308). In addition, the risk estimate was less in the MI and ACS groups than in the CAD group (OR = 0.85, 95% CI:0.79–0.91, P <0.0001, I2 = 62%, Pheterogeneity <0.0001). Carrier status for the B2 allele carried a moderate risk reduction of MI (OR = 0.89, 95% CI: 0.80–0.99, P =0.03, I2 = 33.2%, Pheterogeneity = 0.2) and a suggestive risk reduction of ACS (OR =0.93, 95% CI: 0.74–1.17, P =0.52, I2 = 0%, Pheterogeneity =0.74).
Meta-regression analysis
The major environmental exposures were added in a series of univariate models to interpret the potential sources for between-study heterogeneity. Multiple study-level covariates, including average age, BMI, lipid fractions, percentage of male participants, smoking, hypertension and diabetes, were incorporated in our meta-regression. As a result, the CAD risk estimate for the TaqIB polymorphism was significantly influenced by smoking status (P = 0.006) and by circulating HDL-C levels (P = 0.006). This indicates that the correlation of the TaqIB polymorphism with CAD is likely to be strengthened in populations having low smoking rates or low HDL-C levels, with the reduction of CAD risk due to the B2 allele being greater in populations with low smoking rates compared with populations with high smoking rates and in populations with low HDL-C levels compared with populations with high low HDL-C levels (Figure 4).
Figure 4.
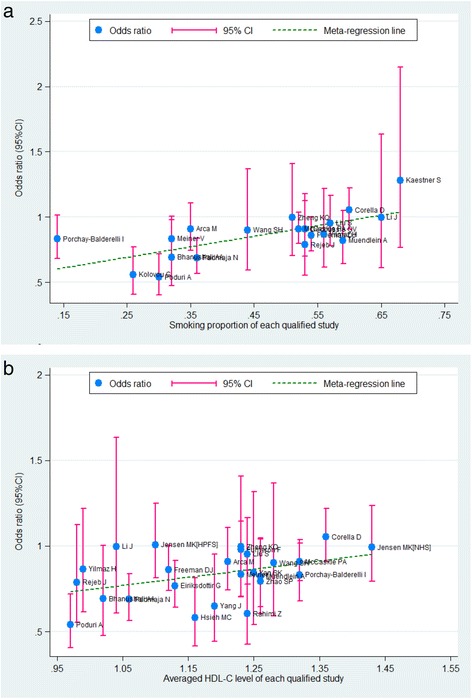
Meta-regression of overall smoking proportion (a) and averag HDL-C level (b) on in-allele risk estimates of the TaqIB polymorphism. OR is expressed as the middle of the blue solid circle whose upper and lower extremes represent the corresponding 95% CI. The green dotted line is plotted by fitting OR with overall smoking proportion (a) and averaged HDL-C level (b) for each included study.
Association of genotype-phenotype
To assess the specificity of HDL-C, we evaluated the association of the TaqIB polymorphism with HDL-C in 10 studies of Caucasian subjects, which contained 3,600 cases and 5,929 controls. Considering that some drugs such as statins probably influence circulating HDL-C levels and are generally used in treatment of CAD and MI, we calculated the SMD only among the controls. As expected, the HDL-C concentrations were dramatically increased in the B2 carriers without evidence of between-study heterogeneity. Carrier status for the B2 allele was associated with an increase of roughly 0.25 mmol/L in HDL-C (B2B1 + B2B2 versus B1B1: SMD = 0.25, 95% CI: 0.20–0.31, P <0.0001, I2 = 0, Pheterogeneity = 0.87) (Figure 5) with a low probability of publication bias, as reflected by the Egger’s test (t = 0.69, P = 0.51) and Begg-Mazemdar test (P = 0.37). None of the environmental exposures mentioned above influenced the between-study heterogeneity.
Figure 5.
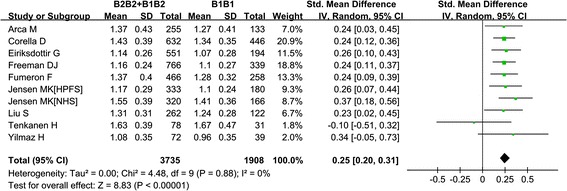
Meta-analysis for the association between the CETP TaqIB polymorphism and circulating HDL-C level among Caucasians under the dominant model (B2B2 + B1B2 versus B1B1). ‘SD’ indicates standard deviation. ‘Total’ indicates the number of measured participants.
Predicted relationship of phenotype-disease from Mendelian randomization
According to Mendelian randomization, the strength of the instrument (the TaqIB polymorphism in our study) is determined by the absolute magnitude of its association with the intermediate phenotype (HDL-C in our study). A previous individual patient data-based meta-analysis, including 13,677 Caucasian subjects, provided robust evidence that the TaqIB polymorphism is significantly associated with HDL-C among Caucasians [18]. Hence, we think the TaqIB polymorphism is a qualified predictive instrument when using a Mendelian randomization approach among Caucasians combined with our above genotype-disease results. Given most CAD and MI patients use lipid-lowering drugs, such as statins, as a primary therapy, which had a potential influence on circulating HDL-C levels, we estimated the association between HDL-C and CAD only among the controls. Among the 40 studies conforming to HWE, 14 studies (10 Caucasian studies and 4 Asian studies) provided complete information about both the TaqIB genotype -CAD association and the TaqIB genotype-HDL-C association [20,36,40,43,45,46,49,58,60,62,64,65]. The other 26 studies only estimated the association between the TaqIB genotype and CAD in detail and did not provide adequate information concerning HDL-C.
To test the reliability and robustness of our data, we did a statistical power analysis as previously described [68,69]. We assumed that OR equals 1.5 and 2.0 for differences in allele frequency, the minor allele frequency is 0.38 and a preset threshold value (α) to reject a null hypothesis is 0.05. As a result, while an allele that has an OR of 1.5 and 2, 80% and 100% of the studies achieved 80% statistical power, respectively. The statistical power of the overall comparison achieved 100% (Table 3).
Table 3.
Expected power analysis of the TaqIB polymorphism
| First author | Expected power, % a | |
|---|---|---|
| OR = 1.5 | OR = 2 | |
| Arca M | 98.2 | 100 |
| Corella D | 100 | 100 |
| Eiriksdottir G | 99.6 | 100 |
| Freeman DJ | 100 | 100 |
| Fumeron F | 99.9 | 100 |
| Jensen MK [HPFS] | 96.1 | 100 |
| Jensen MK [NHS] | 95.2 | 100 |
| Liu S | 97.5 | 100 |
| Tenkanen H | 55.8 | 95.1 |
| Yilmaz H | 64.2 | 98 |
| Total | 100 | 100 |
OR: Odds ratio. aassuming OR of 1.5 and 2.0 for differences in allele frequency, the minor allele frequency of 0.38 and Type I error probability α of 0.05.
Given the close relationship between the TaqIB polymorphism and HDL-C in these 10 studies of Caucasian subjects, we ascertained that B2 carriers had a decreased risk of CAD by 6% (95% CI: 0.86–1.03, P =0.20, I2 = 0, Pheterogeneity =0.52). There seems to be no evidence of publication bias (t = −0.11, P =0.92 for Egger’s test). Utilizing the TaqIB polymorphism as an instrument, we measured the magnitude of the association between increased HDL-C concentrations with CAD under the assumptions required for Mendelian randomization. The Mendelian randomization estimate was computed on the basis of assumed linearity of the association between HDL-C variation and OR for CAD on a log scale [70]. Considering that the National Cholesterol Education Program (NCEP) has set the guidelines for lipids and HDL-C <1.04 mmol/L (40 mg/dL) is considered low HDL-C and HDL-C >1.55 mmol/L (60 mg/dL) is high HDL-C [71], we established a concentration gradient based on increased amounts of HDL-C (mmol/L): 0.03, 0.05, 0.10, 0.20, 0.30, 0.50 and 1.00. The results suggest a marginal significant association between genetically increased circulating HDL-C levels and the reduced risk of CAD (OR = 0.79 per 1 mmol/L increase in HDL-C, 95% CI: 0.54–1.03, P =0.08). Although the estimate size remains borderline significant, we observed a tendency showing a greater increase in HDL-C to have a more significant reduction of CAD risk (Table 4).
Table 4.
Mendelian randomization analysis for the association of genetically raised HDL-C with CAD risk using CETP TaqIB polymorphism as an instrument
| Meta-analysis of included studies | OR | 95% CI | P value a |
|---|---|---|---|
| Per 0.03 mmol/L (1 mg/dL) increase in HDL-C | 0.99 | 0.98–1.00 | 0.069 |
| Per 0.05 mmol/L (2 mg/dL) increase in HDL-C | 0.98 | 0.96–1.00 | 0.069 |
| Per 0.10 mmol/L (4 mg/dL) increase in HDL-C | 0.97 | 0.93–1.00 | 0.069 |
| Per 0.20 mmol/L (8 mg/dL) increase in HDL-C | 0.94 | 0.87–1.01 | 0.069 |
| Per 0.30 mmol/L (10 mg/dL) increase in HDL-C | 0.91 | 0.81–1.01 | 0.070 |
| Per 0.50 mmol/L (20 mg/dL) increase in HDL-C | 0.87 | 0.72–1.01 | 0.071 |
| Per 1.00 mmol/L (40 mg/dL) increase in HDL-C | 0.79 | 0.54–1.03 | 0.082 |
a: Test for overall effect.
Further, weighted linear regressions of the log odds ratio of genotype-disease have been fitted, against the mean difference in HDL-C level as the explanatory variable, to test the strength of the association between HDL-C and CAD. As a result, there was a statistically significant linear relationship between the standard error of the log odds ratio of the genotype-disease and the mean difference change in HDL-C levels in controls (correlation coefficient = 5.69, P = 0.005). The results suggest that HDL-C indeed lies on the causal pathway between the CETP gene and CAD and studies carried out in populations with a large genotype-phenotype difference might be expected to show a large genotype-disease odds ratio.
Discussion
Because HDL-C serves an important role in CAD pathogenesis, we expected that the CETP TaqIB polymorphism, which modulates HDL-C, to likewise confer the risk of disease. To our best knowledge, the current meta-analysis is one of the largest systematic reviews of studies investigating the relationship between the CETP polymorphism, HDL-C and potential risk of CAD. The synthesized estimation of the TaqIB polymorphism concerning CAD risk was presented in our meta-analysis containing 50,996 subjects. Subsequently, the possible causal inference of HDL-C on CAD risk was further assessed. The overall OR of the TaqIB polymorphism under a random-effects model was suggestive of a modest protective effect on CAD. Considering that violation of HWE might diminish the total effect of the meta-analysis, we performed a sensitivity analysis by excluding the studies demonstrating a departure from HWE. Trends in the ORs did not change substantially by estimation of the association among only the HWE-confirming studies. We provide robust evidence that the TaqIB polymorphism is associated with CAD and that the HWE-violation only slightly influences the interpretations of the observed results in our meta-analysis. Interestingly, the pre-specified subgroup analyses revealed that the B2 allele carriers had a remarkable higher risk reduction in CAD among Asians compared with Caucasians, in spite of similar frequency of the B2 allele. It remains unclear the real reasons for the divergence across the different ethnic groups, because we did not directly test the TaqIB polymorphism in different ethnic groups. We also did not estimate the association between the TaqIB polymorphism and other phenotypes. Given some GWASs have identified that numerous lipid-associated genetic variations differ with ethnicity [72-74], we expect that future investigations of the TaqIB polymorphism will uncover its genetic ancestral backgrounds and pleiotropic effects. In addition, the magnitude of association was greater in H-B studies than in P-B studies; the OR is likely overestimated, since subjects sampled from a single hospital may not reflect the real environmental exposures in the source population. In theory, participants recruited from the community, or from the general population, are more representative and reliable. Prospective studies and studies regarding MI or ACS as endpoints concerning the association between the TaqIB polymorphism and CAD did not provide robust evidence for a statistically significant association [75]. Of note, the variability of environmental exposures may be another source of between-study heterogeneity, as reflected from our meta-regression analysis. The protective effect of the B2 allele on CAD risk was potentiated in a population with low smoking rates and low circulating HDL-C levels. Although meta-regression reflects an ecological correlation, rather than a causal inference, our results imply that the underlying interactions of the TaqIB genotype with lifestyle and phenotype may influence the development of atherosclerosis. Considering a wide range of confidence intervals of the overall evaluation, further data from large-scale and well-designed studies are required to improve the precise of the study effect.
The strength of the instrument (the TaqIB polymorphism in our study) is determined by the absolute magnitude of its association with the intermediate phenotype (HDL-C in our study). A previous individual patient data-based meta-analysis including 7 large population-based studies and 3 randomized placebo-controlled trials (13,677 Caucasian subjects), have provided robust evidence that the TaqIB polymorphism is significantly associated with HDL among Caucasians [18]. Hence, the TaqIB polymorphism was certified as a qualified predictive instrument when using a Mendelian randomization approach among Caucasians because it correlates with a specific phenotype and consequently impacts the disease in the pathway intermediated by the phenotype. Our results were concordant with two previous meta-analyses including 15,704 and 19,035 participants respectively [18,19]. However, neither of these studies assessed the relationship of HDL-C and CAD after adjustment for TaqIB polymorphism and the triangulation of the TaqIB polymorphism and HDL-C associations with CAD risk remained confounded.
Mendelian randomization, a form of instrumental variable analysis [76], was applied to determine the magnitude of the causal relationship of increased HDL-C with CAD by using the TaqIB polymorphism as the instrument. Interestingly, genetically elevated HDL-C levels did not reduce CAD risk. Our results supplement the evidence challenging conventional views that raising circulating HDL-C levels may translate into protection of CAD risk. Recently, Voight et al. [77] utilized the Asn396Ser polymorphism in the endothelial lipase gene (LIPG) and a genetic score containing 14 common single nucleotide polymorphisms (solely concerning HDL-C) as two instruments for Mendelian randomization. As a result, a specific and substantial increase in HDL-C levels due to LIPG Asn396Ser and the genetic score was verified to be not related to MI. Employing various instruments may contribute to the precision of instrumental variable estimates and may facilitate examination of potential instrumental variable assumptions. Besides LIPG, CETP was acknowledged to have a profound influence on HDL-C metabolism and consequently CAD risk. The TaqIB polymorphism, a silent base change affecting the 277th nucleotide, is almost fully concordant with another promoter variants −629 C→A [16,78,79]. The data from in vivo studies have documented that the −629 C→A variant is directly functional in regulating the CETP activity [16], suggesting that the TaqIB polymorphism, as a marker of the −629 C→A polymorphism, may be vital in regulating CETP activity and HDL-C metabolism. In order to maximize statistical power, we exploited the TaqIB polymorphism, rather than the −629 C→A polymorphism, as an instrument because extensive data on the TaqIB polymorphism were available. We suggest a strong possibility that raising HDL-C via specific means of inhibiting CETP will not decrease the risk of CAD. Whether CETP efforts pro- or anti-atherogenic effects, by means of the LDL-C receptor pathway, is still unclear [80,81]. As a matter of fact, the results from animal studies and cytological studies remain paradoxical. Transgenic mice expressing CETP demonstrate accelerated atherosclerosis compared with non-expressing controls [82-84]. However, another in vivo study reported that CETP expression reduced atherosclerosis in lecithin cholesterol acyltransferase (LCAT) transgenic mice [85] and a further study revealed that CETP offset the deleterious effect of LCAT [86].
Furthermore, we restate the underlying restriction of raising circulating HDL-C levels as an effective treatment to reduce residual cardiovascular risk in patients treated by conventional statin therapy. Intense research efforts on therapeutic agents have been conducted to reduce the residual cardiovascular risk by raising circulating HDL-C levels. Statins, fibrates and niacin are regarded as effective pharmacological agents for raising HDL-C. However, the Action to Control Cardiovascular Risk in Diabetes (ACCORD) study documented there was no added clinical benefit in using combination therapy with fenofibrate plus simvastatin compared with simvastatin monotherapy, despite the fact there were significantly increased levels of HDL-C in the combination therapy group [5]. In the Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides: Impact on Global Health Outcomes (AIM-HIGH) trial, the addition of niacin to statin therapy did not reduce the rate of cardiovascular events, despite remarkable improvements in HDL-C [87]. The Investigation of Lipid Level Management to Understand its Impact in Atherosclerotic Events (ILLUMINATE) trial exhibited an excess of deaths and cardiovascular disease in the group receiving the CETP inhibitor, torcetrapib and atorvastatin compared with atorvastatin alone [8]. Subsequently, another large randomized trial performed in ACS patients showed that another CETP inhibitor, dalcetrapib, increased HDL-C levels, but did not reduce the risk of recurrent cardiovascular events [9]. Recently, a potential explanation was put forward that functional HDL-C properties may play a key role on the development of CAD rather than an indirect one [88]. Cholesterol efflux and reverse cholesterol transport is an important function of HDL-C. In vitro experiments and transgenic animal model studies have revealed that circulating HDL-C concentrations do not necessarily reflect the efficacy and anti-atherogenicity of reverse cholesterol transport [89]. It is likely that the increments of HDL-C generated by the CETP inhibition are either non-functional or pro-inflammatory rather than anti-inflammatory, which impedes their physiologic role in reverse cholesterol transport.
Another possibility is that the composition of HDL-C is altered in CAD patients and they are no longer productive at high levels or after therapeutic intervention [9]. Some studies have indicated that HDL-C and its major structural protein, apolipoprotein A1 are dysfunctional and are extensively oxidized by myeloperoxidase in human atheroma. In vitro oxidation of either apolipoprotein A1 or HDL particles by myeloperoxidase impairs their cholesterol acceptor function [90]. The precise evaluation of functional HDL-C properties and change in HDL-C sub-fractions are recommended for future studies.
Common variants in lipid-associated loci that are also associated with CAD may implicate genes at these loci as possible therapeutic targets. Recently, a large meta-analysis based on GWAS aimed to investigate the genetic markers for blood lipids [91]. A genome-wide association screen for serum lipids (including approximate 2.6 million SNPs) was conducted in 100,000 individuals of European ancestry. Ninety-five loci (of which 59 are novel) were identified to show genome-wide significant association with serum lipid traits. Further, the lead SNPs from the study were tested in 24,607 CAD patients and 66,197 individuals without CAD. They documented that common variants in lipid-associated loci are consistently related to CAD, implying that these loci may be the potential therapeutic targets. They also suggested that some causal genes in lipid-associated loci may have pleiotropic effects on non-lipid parameters that play an important role on the CAD risk reduction. They provided a foundation from which to develop a broader biological understanding of lipoprotein metabolism and to identify potential therapeutic targets for preventing CAD.
There are still limitations using Mendelian randomization analysis. Potential pleiotropic effects of genes may influence the results when a naturally occurring single nuclear polymorphism is used as an instrument to evaluate disease causality. Although single genetic variation is relative to extensive diseases, many of these associations are false positives because the gene has been more generally characterized than most in population investigations of genetic association studies. Canalization or developmental compensation also provides difficulties in interpreting the gene-disease association. These processes, which bring developmental buffering, offset the influence of the genetic variation or environmental forces [92], remain difficult to evaluate. Further experimental and subject-matter studies may provide a more precise estimation. In addition, CAD has numerous clinical features, ranging from stable coronary syndrome to ACS, which is determined by the stability of the atherosclerosis plaque. We could not completely exclude noise in our data when using CAD as the endpoint. Considering more power would be obtained from a more refined phenotype than a more global one, a more precise method that identifies the vulnerable plaque is needed and may be beneficial to estimate the effect of HDL on coronary atherosclerosis.
Conclusion
In conclusion, our results provided robust evidence that the TaqIB polymorphism, which specifically raised HDL-C concentrations, was uniformly associated with a reduction in CAD risk. However, a lack of causal inference in increased HDL-C levels with the pathogenesis of CAD was detected based on the Mendelian randomization approach among Caucasians. We highlight not only the requirements for further validation of the functional role of HDL-C in reverse cholesterol transport and in the etiology of atherosclerosis, but also as a high possibility that lifestyle interventions or pharmacotherapy, which elevates circulating HDL-C levels, cannot be assumed to benefit CAD patients in reality.
Additional files
The baseline characteristics of all eligible studies in the meta-analysis.
Supplementary material. Study descriptions.
Acknowledgments
This work was supported by the youth science and technology talents “Sail” program of Shanghai Municipal Science and Technology Commission (14YF1402700), the new hundred talents program of the Shanghai Municipal Health Bureau (XBR2013100) and the National Natural Science Foundation of China (81070177 & 81370397).
Abbreviations
- HDL-C
High density lipoprotein cholesterol
- CAD
Coronary artery disease
- CETP
Cholesteryl ester transfer protein
- SD
Standard deviation
- MI
Myocardial infarction
- LDL-C
Low density lipoprotein cholesterol
- VLDL-C
Very low density lipoprotein cholesterol
- CNKI
China nation knowledge infrastructure platform
- CBM
China biological medicine
- ACS
Acute coronary syndrome
- MONICA
Monitoring of trends and determinants in cardiovascular disease
- BMI
Body mass index
- HWE
Hardy-Weinberg equilibrium
- OR
Odd ratio
- SMD
Standard mean difference
- CI
Confidence interval
- H-B
Hospital-based
- P-B
Population-based
- NHS
Nurses’ Health Study
- HPFS
Health Professionals follow-up study
- LCAT
Lecithin cholesterol acyltransferase
- ACCORD
Action to control cardiovascular risk in diabetes
- AIM-HIGH
Atherothrombosis intervention in metabolic syndrome with low HDL/High triglycerides
- ILLUMINATE
Investigation of lipid level management to understand its impact in atherosclerotic events
- GWAS
Genome-wide association studies
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ZW designed the study, collected and selected the literature data, developed the statistical model and wrote the manuscript. YLou collected and selected the data and checked the models and results. XQ established the literature search strategy and supervised the search process. YLiu, LL and QC carried out the statistical analysis and drafted the manuscript. WJ wrote the protocol, checked the results and revised the manuscript. All authors read and approved the final manuscript.
Contributor Information
Zhijun Wu, Email: totito19822005@126.com.
Yuqing Lou, Email: louyq@hotmail.com.
Xiaochun Qiu, Email: qiuxc1969@163.com.
Yan Liu, Email: liuyan_ivy@126.com.
Lin Lu, Email: rjlulin1965@163.com.
Qiujing Chen, Email: 48049954@qq.com.
Wei Jin, Email: jinwei10724@126.com.
References
- 1.Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults Executive summary of the third report of the national cholesterol education program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) JAMA. 2001;285(19):2486–2497. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- 2.Shah S, Casas JP, Gaunt TR, Cooper J, Drenos F, Zabaneh D, Swerdlow DI, Shah T, Sofat R, Palmen J, Kumari M, Kivimaki M, Ebrahim S, Smith GD, Lawlor DA, Talmud PJ, Whittaker J, Day IN, Hingorani AD, Humphries SE. Influence of common genetic variation on blood lipid levels, cardiovascular risk, and coronary events in two British prospective cohort studies. Eur Heart J. 2013;34(13):972–981. doi: 10.1093/eurheartj/ehs243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.High HDL may not protect the heart. Concentrate on lowering LDL for now, experts advise.Harv Heart Lett 2012, 23(1):6. [PubMed]
- 4.Is “good” cholesterol still good for you? New research questions the benefit of drugs to raise HDL, but lifestyle steps to boost good cholesterol are still recommended.Harv Mens Health Watch 2012, 17(2):3. [PubMed]
- 5.Ginsberg HN, Elam MB, Lovato LC, Crouse JR, 3rd, Leiter LA, Linz P, Friedewald WT, Buse JB, Gerstein HC, Probstfield J, Grimm RH, Ismail-Beigi F, Bigger JT, Goff DC, Jr, Cushman WC, Simons-Morton DG, Byington RP. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010;362(17):1563–1574. doi: 10.1056/NEJMoa1001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.The BIP Study Group Secondary prevention by raising HDL cholesterol and reducing triglycerides in patients with coronary artery disease: the Bezafibrate Infarction Prevention (BIP) study. Circulation. 2000;102(1):21–27. doi: 10.1161/01.CIR.102.1.21. [DOI] [PubMed] [Google Scholar]
- 7.Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am J Med. 1977;62(5):707–714. doi: 10.1016/0002-9343(77)90874-9. [DOI] [PubMed] [Google Scholar]
- 8.Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, Komajda M, Lopez-Sendon J, Mosca L, Tardif JC, Waters DD, Shear CL, Revkin JH, Buhr KA, Fisher MR, Tall AR, Brewer B. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357(21):2109–2122. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- 9.Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, Chaitman BR, Holme IM, Kallend D, Leiter LA, Leitersdorf E, McMurray JJ, Mundl H, Nicholls SJ, Shah PK, Tardif JC, Wright RS. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367(22):2089–2099. doi: 10.1056/NEJMoa1206797. [DOI] [PubMed] [Google Scholar]
- 10.Barter PJ, Brewer HB, Jr, Chapman MJ, Hennekens CH, Rader DJ, Tall AR. Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23(2):160–167. doi: 10.1161/01.ATV.0000054658.91146.64. [DOI] [PubMed] [Google Scholar]
- 11.Schierer A, Been LF, Ralhan S, Wander GS, Aston CE, Sanghera DK. Genetic variation in cholesterol ester transfer protein, serum CETP activity, and coronary artery disease risk in Asian Indian diabetic cohort. Pharmacogenet Genomics. 2012;22(2):95–104. doi: 10.1097/FPC.0b013e32834dc9ef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kathiresan S, Willer CJ, Peloso GM, Demissie S, Musunuru K, Schadt EE, Kaplan L, Bennett D, Li Y, Tanaka T, Voight BF, Bonnycastle LL, Jackson AU, Crawford G, Surti A, Guiducci C, Burtt NP, Parish S, Clarke R, Zelenika D, Kubalanza KA, Morken MA, Scott LJ, Stringham HM, Galan P, Swift AJ, Kuusisto J, Bergman RN, Sundvall J, Laakso M, et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet. 2009;41(1):56–65. doi: 10.1038/ng.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aulchenko YS, Ripatti S, Lindqvist I, Boomsma D, Heid IM, Pramstaller PP, Penninx BW, Janssens AC, Wilson JF, Spector T, Martin NG, Pedersen NL, Kyvik KO, Kaprio J, Hofman A, Freimer NB, Jarvelin MR, Gyllensten U, Campbell H, Rudan I, Johansson A, Marroni F, Hayward C, Vitart V, Jonasson I, Pattaro C, Wright A, Hastie N, Pichler I, Hicks AA, et al. Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat Genet. 2009;41(1):47–55. doi: 10.1038/ng.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ordovas JM, Cupples LA, Corella D, Otvos JD, Osgood D, Martinez A, Lahoz C, Coltell O, Wilson PW, Schaefer EJ. Association of cholesteryl ester transfer protein-TaqIB polymorphism with variations in lipoprotein subclasses and coronary heart disease risk: the Framingham study. Arterioscler Thromb Vasc Biol. 2000;20(5):1323–1329. doi: 10.1161/01.ATV.20.5.1323. [DOI] [PubMed] [Google Scholar]
- 15.Kondo I, Berg K, Drayna D, Lawn R. DNA polymorphism at the locus for human cholesteryl ester transfer protein (CETP) is associated with high density lipoprotein cholesterol and apolipoprotein levels. Clin Genet. 1989;35(1):49–56. doi: 10.1111/j.1399-0004.1989.tb02904.x. [DOI] [PubMed] [Google Scholar]
- 16.Dachet C, Poirier O, Cambien F, Chapman J, Rouis M. New functional promoter polymorphism, CETP/-629, in cholesteryl ester transfer protein (CETP) gene related to CETP mass and high density lipoprotein cholesterol levels: role of Sp1/Sp3 in transcriptional regulation. Arterioscler Thromb Vasc Biol. 2000;20(2):507–515. doi: 10.1161/01.ATV.20.2.507. [DOI] [PubMed] [Google Scholar]
- 17.Horne BD, Camp NJ, Anderson JL, Mower CP, Clarke JL, Kolek MJ, Carlquist JF. Multiple less common genetic variants explain the association of the cholesteryl ester transfer protein gene with coronary artery disease. J Am Coll Cardiol. 2007;49(20):2053–2060. doi: 10.1016/j.jacc.2007.02.039. [DOI] [PubMed] [Google Scholar]
- 18.Boekholdt SM, Sacks FM, Jukema JW, Shepherd J, Freeman DJ, McMahon AD, Cambien F, Nicaud V, de Grooth GJ, Talmud PJ, Humphries SE, Miller GJ, Eiriksdottir G, Gudnason V, Kauma H, Kakko S, Savolainen MJ, Arca M, Montali A, Liu S, Lanz HJ, Zwinderman AH, Kuivenhoven JA, Kastelein JJ. Cholesteryl ester transfer protein TaqIB variant, high-density lipoprotein cholesterol levels, cardiovascular risk, and efficacy of pravastatin treatment: individual patient meta-analysis of 13,677 subjects. Circulation. 2005;111(3):278–287. doi: 10.1161/01.CIR.0000153341.46271.40. [DOI] [PubMed] [Google Scholar]
- 19.Thompson A, Di Angelantonio E, Sarwar N, Erqou S, Saleheen D, Dullaart RP, Keavney B, Ye Z, Danesh J. Association of cholesteryl ester transfer protein genotypes with CETP mass and activity, lipid levels, and coronary risk. JAMA. 2008;299(23):2777–2788. doi: 10.1001/jama.299.23.2777. [DOI] [PubMed] [Google Scholar]
- 20.Liu S, Schmitz C, Stampfer MJ, Sacks F, Hennekens CH, Lindpaintner K, Ridker PM. A prospective study of TaqIB polymorphism in the gene coding for cholesteryl ester transfer protein and risk of myocardial infarction in middle-aged men. Atherosclerosis. 2002;161(2):469–474. doi: 10.1016/S0021-9150(01)00673-6. [DOI] [PubMed] [Google Scholar]
- 21.Whiting BM, Anderson JL, Muhlestein JB, Horne BD, Bair TL, Pearson RR, Carlquist JF. Candidate gene susceptibility variants predict intermediate end points but not angiographic coronary artery disease. Am Heart J. 2005;150(2):243–250. doi: 10.1016/j.ahj.2004.08.034. [DOI] [PubMed] [Google Scholar]
- 22.McCaskie PA, Beilby JP, Chapman CM, Hung J, McQuillan BM, Thompson PL, Palmer LJ. Cholesteryl ester transfer protein gene haplotypes, plasma high-density lipoprotein levels and the risk of coronary heart disease. Hum Genet. 2007;121(3–4):401–411. doi: 10.1007/s00439-007-0326-2. [DOI] [PubMed] [Google Scholar]
- 23.Borggreve SE, Hillege HL, Wolffenbuttel BH, de Jong PE, Zuurman MW, van der Steege G, van Tol A, Dullaart RP. An increased coronary risk is paradoxically associated with common cholesteryl ester transfer protein gene variations that relate to higher high-density lipoprotein cholesterol: a population-based study. J Clin Endocrinol Metab. 2006;91(9):3382–3388. doi: 10.1210/jc.2005-2322. [DOI] [PubMed] [Google Scholar]
- 24.Smith GD, Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32(1):1–22. doi: 10.1093/ije/dyg070. [DOI] [PubMed] [Google Scholar]
- 25.Castle WE. Mendel’s law of heredity. Science. 1903;18(456):396–406. doi: 10.1126/science.18.456.396. [DOI] [PubMed] [Google Scholar]
- 26.Niu W, Liu Y, Qi Y, Wu Z, Zhu D, Jin W. Association of interleukin-6 circulating levels with coronary artery disease: a meta-analysis implementing mendelian randomization approach. Int J Cardiol. 2012;157(2):243–252. doi: 10.1016/j.ijcard.2011.12.098. [DOI] [PubMed] [Google Scholar]
- 27.Wright RS, Anderson JL, Adams CD, Bridges CR, Casey DE, Jr, Ettinger SM, Fesmire FM, Ganiats TG, Jneid H, Lincoff AM, Peterson ED, Philippides GJ, Theroux P, Wenger NK, Zidar JP, Anderson JL, Adams CD, Antman EM, Bridges CR, Califf RM, Casey DE, Jr, Chavey WE, 2nd, Fesmire FM, Hochman JS, Levin TN, Lincoff AM, Peterson ED, Theroux P, Wenger NK, Zidar JP. 2011 ACCF/AHA focused update incorporated into the ACC/AHA 2007 Guidelines for the Management of Patients with Unstable Angina/Non-ST-Elevation Myocardial Infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines developed in collaboration with the American Academy of Family Physicians, Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons. J Am Coll Cardiol. 2011;57(19):e215–e367. doi: 10.1016/j.jacc.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 28.Joensen AM, Jensen MK, Overvad K, Dethlefsen C, Schmidt E, Rasmussen L, Tjonneland A, Johnsen S. Predictive values of acute coronary syndrome discharge diagnoses differed in the Danish National Patient Registry. J Clin Epidemiol. 2009;62(2):188–194. doi: 10.1016/j.jclinepi.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 29.Tunstall-Pedoe H, Kuulasmaa K, Amouyel P, Arveiler D, Rajakangas AM, Pajak A. Myocardial infarction and coronary deaths in the World Health Organization MONICA Project. Registration procedures, event rates, and case-fatality rates in 38 populations from 21 countries in four continents. Circulation. 1994;90(1):583–612. doi: 10.1161/01.CIR.90.1.583. [DOI] [PubMed] [Google Scholar]
- 30.Thakkinstian A, McElduff P, D’Este C, Duffy D, Attia J. A method for meta-analysis of molecular association studies. Stat Med. 2005;24(9):1291–1306. doi: 10.1002/sim.2010. [DOI] [PubMed] [Google Scholar]
- 31.Cohn LD, Becker BJ. How meta-analysis increases statistical power. Psychol Methods. 2003;8(3):243–253. doi: 10.1037/1082-989X.8.3.243. [DOI] [PubMed] [Google Scholar]
- 32.Higgins JP, Thompson SG. Quantifying heterogeneity in a meta-analysis. Stat Med. 2002;21(11):1539–1558. doi: 10.1002/sim.1186. [DOI] [PubMed] [Google Scholar]
- 33.Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ. 2003;327(7414):557–560. doi: 10.1136/bmj.327.7414.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lau J, Ioannidis JP, Schmid CH. Quantitative synthesis in systematic reviews. Ann Intern Med. 1997;127(9):820–826. doi: 10.7326/0003-4819-127-9-199711010-00008. [DOI] [PubMed] [Google Scholar]
- 35.Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta-analysis detected by a simple, graphical test. BMJ. 1997;315(7109):629–634. doi: 10.1136/bmj.315.7109.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arca M, Montali A, Ombres D, Battiloro E, Campagna F, Ricci G, Verna R. Lack of association of the common TaqIB polymorphism in the cholesteryl ester transfer protein gene with angiographically assessed coronary atherosclerosis. Clin Genet. 2001;60(5):374–380. doi: 10.1034/j.1399-0004.2001.600510.x. [DOI] [PubMed] [Google Scholar]
- 37.van Acker BA, Botma GJ, Zwinderman AH, Kuivenhoven JA, Dallinga-Thie GM, Sijbrands EJ, Boer JM, Seidell JC, Jukema JW, Kastelein JJ, Jansen H, Verhoeven AJ. High HDL cholesterol does not protect against coronary artery disease when associated with combined cholesteryl ester transfer protein and hepatic lipase gene variants. Atherosclerosis. 2008;200(1):161–167. doi: 10.1016/j.atherosclerosis.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 38.Bhanushali AA, Das BR. Genetic variants at the APOE, lipoprotein lipase (LpL), cholesteryl ester transfer protein (CETP), and endothelial nitric oxide (eNOS) genes and coronary artery disease (CAD): CETP Taq1 B2B2 associates with lower risk of CAD in Asian Indians. J Community Genet. 2010;1(2):55–62. doi: 10.1007/s12687-010-0005-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blankenberg S, Tiret L, Bickel C, Schlitt A, Jungmair W, Genth-Zotz S, Lubos E, Espinola-Klein C, Rupprecht HJ. [Genetic variation of the cholesterol ester transfer protein gene and the prevalence of coronary artery disease. The AtheroGene case control study] Z Kardiol. 2004;93(Suppl 4):IV16–IV23. doi: 10.1007/s00392-004-1404-2. [DOI] [PubMed] [Google Scholar]
- 40.Corella D, Carrasco P, Amiano P, Arriola L, Chirlaque MD, Huerta JM, Martinez C, Martinez-Camblor P, Molina E, Navarro C, Quirós JR, Rodríguez L, Sánchez MJ, Ortega-Azorín C, Ros E, Sala N, González CA, Moreno C. Common cholesteryl ester transfer protein gene variation related to high-density lipoprotein cholesterol is not associated with decreased coronary heart disease risk after a 10-year follow-up in a Mediterranean cohort: Modulation by alcohol consumption. Atherosclerosis. 2010;211(2):531–538. doi: 10.1016/j.atherosclerosis.2010.03.026. [DOI] [PubMed] [Google Scholar]
- 41.Dedoussis GV, Panagiotakos DB, Louizou E, Mantoglou I, Chrysohoou C, Lamnisou K, Pitsavos C, Stefanadis C. Cholesteryl ester-transfer protein (CETP) polymorphism and the association of acute coronary syndromes by obesity status in Greek subjects: the CARDIO2000-GENE study. Hum Hered. 2007;63(3–4):155–161. doi: 10.1159/000099827. [DOI] [PubMed] [Google Scholar]
- 42.Durlach A, Clavel C, Girard-Globa A, Durlach V. Sex-dependent association of a genetic polymorphism of cholesteryl ester transfer protein with high-density lipoprotein cholesterol and macrovascular pathology in type II diabetic patients. J Clin Endocrinol Metab. 1999;84(10):3656–3659. doi: 10.1210/jcem.84.10.6064. [DOI] [PubMed] [Google Scholar]
- 43.Eiriksdottir G, Bolla MK, Thorsson B, Sigurdsson G, Humphries SE, Gudnason V. The -629C > A polymorphism in the CETP gene does not explain the association of TaqIB polymorphism with risk and age of myocardial infarction in Icelandic men. Atherosclerosis. 2001;159(1):187–192. doi: 10.1016/S0021-9150(01)00489-0. [DOI] [PubMed] [Google Scholar]
- 44.Falchi A, Giovannoni L, Piras IS, Calo CM, Moral P, Vona G, Varesi L. Prevalence of genetic risk factors for coronary artery disease in Corsica island (France) Exp Mol Pathol. 2005;79(3):210–213. doi: 10.1016/j.yexmp.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 45.Freeman DJ, Samani NJ, Wilson V, McMahon AD, Braund PS, Cheng S, Caslake MJ, Packard CJ, Gaffney D. A polymorphism of the cholesteryl ester transfer protein gene predicts cardiovascular events in non-smokers in the West of Scotland Coronary Prevention Study. Eur Heart J. 2003;24(20):1833–1842. doi: 10.1016/j.ehj.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 46.Fumeron F, Betoulle D, Luc G, Behague I, Ricard S, Poirier O, Jemaa R, Evans A, Arveiler D, Marques-Vidal P. Alcohol intake modulates the effect of a polymorphism of the cholesteryl ester transfer protein gene on plasma high density lipoprotein and the risk of myocardial infarction. J Clin Invest. 1995;96(3):1664–1671. doi: 10.1172/JCI118207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hsieh MC, Tien KJ, Chang SJ, Lo CS, Hsin SC, Hsiao JY, Hsu SC, Liang HT, Chen HC, Shin SJ, Lin SR. Cholesteryl ester transfer protein B1B1 genotype as a predictor of coronary artery disease in Taiwanese with type 2 diabetes mellitus. Metabolism. 2007;56(6):745–750. doi: 10.1016/j.metabol.2006.12.023. [DOI] [PubMed] [Google Scholar]
- 48.Izar MC, Helfenstein T, Ihara SS, Relvas WG, Santos AO, Fischer SC, Pinto LE, Lopes IE, Pomaro DR, Fonseca MI, Bodanese LC, Moriguchi EH, Saraiva JF, Introcaso L, Souza AD, Scartezini M, Torres KP, Zagury L, Jardim PC, Costa EA, Tacito LH, Forti A, Magalhaes ME, Chacra AR, Bertolami MC, Loures-Vale AA, Barros MA, Xavier HT, Lyra R, Argamanijan D, et al. Association of lipoprotein lipase D9N polymorphism with myocardial infarction in type 2 diabetes: the genetics, outcomes, and lipids in type 2 diabetes (GOLD) study. Atherosclerosis. 2009;204(1):165–170. doi: 10.1016/j.atherosclerosis.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 49.Jensen MK, Mukamal KJ, Overvad K, Rimm EB. Alcohol consumption, TaqIB polymorphism of cholesteryl ester transfer protein, high-density lipoprotein cholesterol, and risk of coronary heart disease in men and women. Eur Heart J. 2008;29(1):104–112. doi: 10.1093/eurheartj/ehm517. [DOI] [PubMed] [Google Scholar]
- 50.Kaestner S, Patsouras N, Spathas DH, Flordellis CS, Manolis AS. Lack of association between the cholesteryl ester transfer protein gene–TaqIB polymorphism and coronary restenosis following percutaneous transluminal coronary angioplasty and stenting: a pilot study. Angiology. 2010;61(4):338–343. doi: 10.1177/0003319709348297. [DOI] [PubMed] [Google Scholar]
- 51.Kawasaki I, Tahara H, Emoto M, Shoji T, Nishizawa Y. Relationship between TaqIB cholesteryl ester transfer protein gene polymorphism and macrovascular complications in Japanese patients with type 2 diabetes. Diabetes. 2002;51(3):871–874. doi: 10.2337/diabetes.51.3.871. [DOI] [PubMed] [Google Scholar]
- 52.Keavney B, Palmer A, Parish S, Clark S, Youngman L, Danesh J, McKenzie C, Delepine M, Lathrop M, Peto R, Collins R. Lipid-related genes and myocardial infarction in 4685 cases and 3460 controls: discrepancies between genotype, blood lipid concentrations, and coronary disease risk. Int J Epidemiol. 2004;33(5):1002–1013. doi: 10.1093/ije/dyh275. [DOI] [PubMed] [Google Scholar]
- 53.Kolovou G, Vasiliadis I, Kolovou V, Karakosta A, Mavrogeni S, Papadopoulou E, Papamentzelopoulos S, Giannakopoulou V, Marvaki A, Degiannis D, Bilianou H. The role of common variants of the cholesteryl ester transfer protein gene in left main coronary artery disease. Lipids Health Dis. 2011;10:156. doi: 10.1186/1476-511X-10-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meiner V, Friedlander Y, Milo H, Sharon N, Ben-Avi L, Shpitzen S, Leitersdorf E, Siscovick DS, Schwartz SM. Cholesteryl ester transfer protein (CETP) genetic variation and early onset of non-fatal myocardial infarction. Ann Hum Genet. 2008;72(Pt 6):732–741. doi: 10.1111/j.1469-1809.2008.00464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mohrschladt MF, van der Sman-de BF, Hofman MK, van der Krabben M, Westendorp RG, Smelt AH. TaqIB polymorphism in CETP gene: the influence on incidence of cardiovascular disease in statin-treated patients with familial hypercholesterolemia. Eur J Hum Genet. 2005;13(7):877–882. doi: 10.1038/sj.ejhg.5201426. [DOI] [PubMed] [Google Scholar]
- 56.Muendlein A, Saely CH, Marte T, Schmid F, Koch L, Rein P, Langer P, Aczel S, Drexel H. Synergistic effects of the apolipoprotein E epsilon3/epsilon2/epsilon4, the cholesteryl ester transfer protein TaqIB, and the apolipoprotein C3–482 C > T polymorphisms on their association with coronary artery disease. Atherosclerosis. 2008;199(1):179–186. doi: 10.1016/j.atherosclerosis.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 57.Padmaja N, Kumar RM, Balachander J, Adithan C. Cholesteryl ester transfer protein TaqIB, −629C > A and I405V polymorphisms and risk of coronary heart disease in an Indian population. Clin Chim Acta. 2009;402(1–2):139–145. doi: 10.1016/j.cca.2008.12.041. [DOI] [PubMed] [Google Scholar]
- 58.Poduri A, Khullar M, Bahl A, Sharma YP, Talwar KK. A combination of proatherogenic single-nucleotide polymorphisms is associated with increased risk of coronary artery disease and myocardial infarction in Asian Indians. DNA Cell Biol. 2009;28(9):451–460. doi: 10.1089/dna.2009.0887. [DOI] [PubMed] [Google Scholar]
- 59.Porchay-Balderelli I, Pean F, Bellili N, Jaziri R, Marre M, Fumeron F. The CETP TaqIB polymorphism is associated with the risk of sudden death in type 2 diabetic patients. Diabetes Care. 2007;30(11):2863–2867. doi: 10.2337/dc07-0869. [DOI] [PubMed] [Google Scholar]
- 60.Rahimi Z, Nourozi-Rad R, Vaisi-Raygani A, Saidi MR, Ahmadi R, Yarani R, Hamzehee K, Parsian A. Association between cholesteryl ester transfer protein TaqIB variants and risk of coronary artery disease and diabetes mellitus in the population of western Iran. Genet Test Mol Biomarkers. 2011;15(11):813–819. doi: 10.1089/gtmb.2011.0037. [DOI] [PubMed] [Google Scholar]
- 61.Rejeb J, Omezzine A, Rebhi L, Naffeti I, Kchok K, Belkahla R, Bel Hadjmbarek I, Ben Rejeb N, Nabli N, Boujelbene A, Ben Abdelaziz A, Boughzala E, Bouslama A. Association of the cholesteryl ester transfer protein Taq1 B2B2 genotype with higher high-density lipoprotein cholesterol concentrations and lower risk of coronary artery disease in a Tunisian population. Arch Cardiovasc Dis. 2008;101(10):629–636. doi: 10.1016/j.acvd.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 62.Tenkanen H, Koshinen P, Kontula K, Aalto-Setala K, Manttari M, Manninen V, Runeberg SL, Taskinen MR, Ehnholm C. Polymorphisms of the gene encoding cholesterol ester transfer protein and serum lipoprotein levels in subjects with and without coronary heart disease. Hum Genet. 1991;87(5):574–578. doi: 10.1007/BF00209015. [DOI] [PubMed] [Google Scholar]
- 63.Wu JH, Lee YT, Hsu HC, Hsieh LL. Influence of CETP gene variation on plasma lipid levels and coronary heart disease: a survey in Taiwan. Atherosclerosis. 2001;159(2):451–458. doi: 10.1016/S0021-9150(01)00524-X. [DOI] [PubMed] [Google Scholar]
- 64.Yilmaz H, Isbir T, Agachan B, Karaali ZE. Effects of cholesterol ester transfer protein Taq1B gene polymorphism on serum lipoprotein levels in Turkish coronary artery disease patients. Cell Biochem Funct. 2005;23(1):23–28. doi: 10.1002/cbf.1124. [DOI] [PubMed] [Google Scholar]
- 65.Zheng KQ, Zhang SZ, He Y, Zhang L, Zhang KL, Huang DJ, Sun Y. Association between cholesteryl ester transfer protein gene polymorphisms and variations in lipid levels in patients with coronary heart disease. Chin Med J. 2004;117(9):1288–1292. [PubMed] [Google Scholar]
- 66.Park KW, Choi JH, Kim HK, Oh S, Chae IH, Kim HS, Oh BH, Lee MM, Park YB, Choi YS. The association of cholesteryl ester transfer protein polymorphism with high-density lipoprotein cholesterol and coronary artery disease in Koreans. Clin Genet. 2003;63(1):31–38. doi: 10.1034/j.1399-0004.2003.630105.x. [DOI] [PubMed] [Google Scholar]
- 67.Blankenberg S, Rupprecht HJ, Bickel C, Jiang XC, Poirier O, Lackner KJ, Meyer J, Cambien F, Tiret L. Common genetic variation of the cholesteryl ester transfer protein gene strongly predicts future cardiovascular death in patients with coronary artery disease. J Am Coll Cardiol. 2003;41(11):1983–1989. doi: 10.1016/S0735-1097(03)00408-X. [DOI] [PubMed] [Google Scholar]
- 68.Wu Z, Lou Y, Jin W, Liu Y, Lu L, Lu G. The C161T polymorphism in the peroxisome proliferator-activated receptor gamma gene (PPARgamma) is associated with risk of coronary artery disease: a meta-analysis. Mol Biol Rep. 2013;40(4):3101–3112. doi: 10.1007/s11033-012-2384-3. [DOI] [PubMed] [Google Scholar]
- 69.Xing G, Xing C. Adjusting for covariates in logistic regression models. Genet Epidemiol. 2010;34(7):769–771. doi: 10.1002/gepi.20526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Minelli C, Thompson JR, Tobin MD, Abrams KR. An integrated approach to the meta-analysis of genetic association studies using Mendelian randomization. Am J Epidemiol. 2004;160(5):445–452. doi: 10.1093/aje/kwh228. [DOI] [PubMed] [Google Scholar]
- 71.National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) Third report of the national cholesterol education program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report. Circulation. 2002;106(25):3143–3421. [PubMed] [Google Scholar]
- 72.Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, Beckmann JS, Bragg-Gresham JL, Chang HY, Demirkan A, Den Hertog HM, Do R, Donnelly LA, Ehret GB, Esko T, Feitosa MF, Ferreira T, Fischer K, Fontanillas P, Fraser RM, Freitag DF, Gurdasani D, Heikkilä K, Hyppönen E, Isaacs A, Jackson AU, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45(11):1274–1283. doi: 10.1038/ng.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Frazier-Wood AC, Manichaikul A, Aslibekyan S, Borecki IB, Goff DC, Hopkins PN, Lai CQ, Ordovas JM, Post WS, Rich SS, Sale MM, Siscovick D, Straka RJ, Tiwari HK, Tsai MY, Rotter JI, Arnett DK. Genetic variants associated with VLDL, LDL and HDL particle size differ with race/ethnicity. Hum Genet. 2013;132(4):405–413. doi: 10.1007/s00439-012-1256-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Deo RC, Wilson JG, Xing C, Lawson K, Kao WH, Reich D, Tandon A, Akylbekova E, Patterson N, Mosley TH, Jr, Boerwinkle E, Taylor HA., Jr Single-nucleotide polymorphisms in LPA explain most of the ancestry-specific variation in Lp(a) levels in African Americans. PLoS ONE. 2011;6(1):e14581. doi: 10.1371/journal.pone.0014581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Colhoun HM, McKeigue PM, Davey Smith G. Problems of reporting genetic associations with complex outcomes. Lancet. 2003;361(9360):865–872. doi: 10.1016/S0140-6736(03)12715-8. [DOI] [PubMed] [Google Scholar]
- 76.Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27(8):1133–1163. doi: 10.1002/sim.3034. [DOI] [PubMed] [Google Scholar]
- 77.Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, Hindy G, Holm H, Ding EL, Johnson T, Schunkert H, Samani NJ, Clarke R, Hopewell JC, Thompson JF, Li M, Thorleifsson G, Newton-Cheh C, Musunuru K, Pirruccello JP, Saleheen D, Chen L, Stewart A, Schillert A, Thorsteinsdottir U, Thorgeirsson G, Anand S, Engert JC, Morgan T, Spertus J, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380(9841):572–580. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Klerkx AH, Tanck MW, Kastelein JJ, Molhuizen HO, Jukema JW, Zwinderman AH, Kuivenhoven JA. Haplotype analysis of the CETP gene: not TaqIB, but the closely linked -629C– > A polymorphism and a novel promoter variant are independently associated with CETP concentration. Hum Mol Genet. 2003;12(2):111–123. doi: 10.1093/hmg/ddg013. [DOI] [PubMed] [Google Scholar]
- 79.Corbex M, Poirier O, Fumeron F, Betoulle D, Evans A, Ruidavets JB, Arveiler D, Luc G, Tiret L, Cambien F. Extensive association analysis between the CETP gene and coronary heart disease phenotypes reveals several putative functional polymorphisms and gene-environment interaction. Genet Epidemiol. 2000;19(1):64–80. doi: 10.1002/1098-2272(200007)19:1<64::AID-GEPI5>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 80.Van Eck M, Ye D, Hildebrand RB, Kar Kruijt J, de Haan W, Hoekstra M, Rensen PC, Ehnholm C, Jauhiainen M, Van Berkel TJ. Important role for bone marrow-derived cholesteryl ester transfer protein in lipoprotein cholesterol redistribution and atherosclerotic lesion development in LDL receptor knockout mice. Circ Res. 2007;100(5):678–685. doi: 10.1161/01.RES.0000260202.79927.4f. [DOI] [PubMed] [Google Scholar]
- 81.Okamoto H, Yonemori F, Wakitani K, Minowa T, Maeda K, Shinkai H. A cholesteryl ester transfer protein inhibitor attenuates atherosclerosis in rabbits. Nature. 2000;406(6792):203–207. doi: 10.1038/35018119. [DOI] [PubMed] [Google Scholar]
- 82.Marotti KR, Castle CK, Boyle TP, Lin AH, Murray RW, Melchior GW. Severe atherosclerosis in transgenic mice expressing simian cholesteryl ester transfer protein. Nature. 1993;364(6432):73–75. doi: 10.1038/364073a0. [DOI] [PubMed] [Google Scholar]
- 83.Plump AS, Masucci-Magoulas L, Bruce C, Bisgaier CL, Breslow JL, Tall AR. Increased atherosclerosis in ApoE and LDL receptor gene knock-out mice as a result of human cholesteryl ester transfer protein transgene expression. Arterioscler Thromb Vasc Biol. 1999;19(4):1105–1110. doi: 10.1161/01.ATV.19.4.1105. [DOI] [PubMed] [Google Scholar]
- 84.Herrera VL, Makrides SC, Xie HX, Adari H, Krauss RM, Ryan US, Ruiz-Opazo N. Spontaneous combined hyperlipidemia, coronary heart disease and decreased survival in Dahl salt-sensitive hypertensive rats transgenic for human cholesteryl ester transfer protein. Nat Med. 1999;5(12):1383–1389. doi: 10.1038/70956. [DOI] [PubMed] [Google Scholar]
- 85.Foger B, Chase M, Amar MJ, Vaisman BL, Shamburek RD, Paigen B, Fruchart-Najib J, Paiz JA, Koch CA, Hoyt RF, Brewer HB, Jr, Santamarina-Fojo S. Cholesteryl ester transfer protein corrects dysfunctional high density lipoproteins and reduces aortic atherosclerosis in lecithin cholesterol acyltransferase transgenic mice. J Biol Chem. 1999;274(52):36912–36920. doi: 10.1074/jbc.274.52.36912. [DOI] [PubMed] [Google Scholar]
- 86.Berti JA, de Faria EC, Oliveira HC. Atherosclerosis in aged mice over-expressing the reverse cholesterol transport genes. Braz J Med Biol Res. 2005;38(3):391–398. doi: 10.1590/S0100-879X2005000300010. [DOI] [PubMed] [Google Scholar]
- 87.Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365(24):2255–2267. doi: 10.1056/NEJMoa1107579. [DOI] [PubMed] [Google Scholar]
- 88.Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, Mohler ER, Rothblat GH, Rader DJ. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364(2):127–135. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.von Eckardstein A, Nofer JR, Assmann G. High density lipoproteins and arteriosclerosis. Role of cholesterol efflux and reverse cholesterol transport. Arterioscler Thromb Vasc Biol. 2001;21(1):13–27. doi: 10.1161/01.ATV.21.1.13. [DOI] [PubMed] [Google Scholar]
- 90.Huang Y, DiDonato JA, Levison BS, Schmitt D, Li L, Wu Y, Buffa J, Kim T, Gerstenecker GS, Gu X, Kadiyala CS, Wang Z, Culley MK, Hazen JE, Didonato AJ, Fu X, Berisha SZ, Peng D, Nguyen TT, Liang S, Chuang CC, Cho L, Plow EF, Fox PL, Gogonea V, Tang WH, Parks JS, Fisher EA, Smith JD, Hazen SL. An abundant dysfunctional apolipoprotein A1 in human atheroma. Nat Med. 2014;20(2):193–203. doi: 10.1038/nm.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, Johansen CT, Fouchier SW, Isaacs A, Peloso GM, Barbalic M, Ricketts SL, Bis JC, Aulchenko YS, Thorleifsson G, Feitosa MF, Chambers J, Orho-Melander M, Melander O, Johnson T, Li X, Guo X, Li M, Shin Cho Y, Jin Go M, Jin Kim Y, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466(7307):707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Smith GD, Ebrahim S. Mendelian randomization: prospects, potentials, and limitations. Int J Epidemiol. 2004;33(1):30–42. doi: 10.1093/ije/dyh132. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The baseline characteristics of all eligible studies in the meta-analysis.
Supplementary material. Study descriptions.