

Abstract
Multiple studies demonstrate that ubiquitination of proteins codes for regulation of cell differentiation, apoptosis, endocytosis, and many other cellular functions. There is great interest in, and considerable effort being given to defining the relationships between the structures of polyubiquitin modifications and the fates of the modified proteins. Does each ubiquitin modification achieve a specific effect, much like phosphorylation, or is ubiquitin like glycosylation, where there is heterogeneity and redundancy in the signal? The sensitive analytical tools needed to address such questions readily are not yet mature. To lay the foundation for mass spectrometry-based studies of the ubiquitin code we have assembled seven isomeric diubiquitins with all-native sequences and isopeptide linkages. Using these compounds as standards enables the development and testing of a new mass spectrometry-based strategy tailored specifically to characterize the number and sites of isopeptide linkages in polyubiquitin chains. Here we report the use of Asp-selective acid cleavage, separation by reverse phase HPLC, and characterization by tandem mass spectrometry to distinguish and characterize all seven isomeric lysine-linked ubiquitin dimers.
INTRODUCTION
Since the earliest sequence analysis of a protein conjugated by ubiquitin,[1] the sites of attachment in the target protein have been identified by locating the glycinylglycine (GG) tag left by ubiquitin when conjugates were subjected to exhaustive trypsin proteolysis. Indeed, most of our present knowledge about ubiquitination in vitro and in vivo is based on tryptic digestion and bottom up proteomics.[2–4] GG tags are also left at linkage points in ubiquitin polymers, and thus the quantitative ratios of the different linkages from mixed chains have been extracted using synthetic reference peptides carrying isotope labels.[5] This approach does not provide information about the order of the linkages and is impaired by incomplete cleavage. One alternative approach, top-down analysis of ubiquitin conjugates, is challenging for several reasons. First, the molecular mass is incremented 8.5 kDa by each ubiquitin module added, rapidly raising the requisite mass and resolution beyond the capability of many mass spectrometers. Second, fragmentation of intact polymers with multiple branches is reported not to progress to branch points even in simple dimers and cannot be used to determine the site of the linkage.[6] Thus there is a strong need to advance a technology that trims the branches to reduce the overall mass, yet preserves the branch points for characterization by LC-MS/MS,[7–9] and can eventually be incorporated into studies of biological samples.
We report here the use of time-limited proteolysis with a concentration of acetic acid that cleaves selectively at aspartic acid residues fortuitously spaced to trim the branches, and we demonstrate that this strategy allows successful application of HPLC and tandem mass spectrometry to provide information on linkage sites in dimers. We report that ubiquitin dimers linked by all-native isopeptide bonds at the seven lysine residues can be distinguished and characterized by LC-MS/MS.
With the expectation to extend the strategy to longer ubiquitin chains consisting of homogeneous and mixed linkages and to ubiquitinated proteins, we have optimized acid cleavage reactions of our reference dimers to allow the production of peptides that retain both linkage site and the carboxyl-terminus of the proximal ubiquitin moiety that would be involved in additional isopeptide linkages. This strategy is illustrated in the sequence of monoubiquitin in a MALDI spectrum of the acid cleavage products of monoubiquitin shown in Figure 1. Lysine residues in the sequence are shown in blue as potential sites of attachment, and aspartate residues are highlighted in red as potential sites for acid cleavage. Peaks are annotated in the spectrum of the mixture corresponding to peptide products, six of which contain particular attachment sites while still retaining the G76 carboxyl-terminus. Information is also obtained from cleavage products that contain unique branch sites without the carboxyl terminus.
Figure 1.

MALDI TOF mass spectrum of the product mixture of peptides from time-controlled (60 sec) acid cleavage of ubiquitin. Peaks are annotated to indicate the peptides formed. The sequence of monoubiquitin is also shown, with K linkage sites and D cleavage sites highlighted.
METHODS AND MATERIALS
Assembly of diubiquitins
Diubiquitin (Ub2) was assembled either enzymatically (K48, K63) or chemically (K6, K11, K27, K29, K33) using recombinant ubiquitin (Ub) monomers. Linkage-specific ubiquitin conjugating enzymes E2-25K and the heterodimer Ubc13:Mms2 were used to make K48- and K63-linked Ub2, respectively. Enzymatic reactions were performed using wild-type ubiquitin, and quenched after 1–2 hours using 2 drops of glacial acetic acid, as described previously[10, 11] Ub2 was purified from unreacted monomers and longer chains using cation exchange chromatography on a HP SP column (GE Healthcare).
K6, K11, K27, K29 and K33-linked Ub2 were chemically assembled using a strategy employing a combination of removable, orthogonal amine protecting groups (Boc and Alloc) and a silver-mediated condensation reaction between the two ubiquitin monomers. The assembly scheme is outlined below; details of each reaction step, including reagents are reported elsewhere.[12] Linkage specificity was achieved by introducing a Lys(Boc) mutation at the target lysine of interest in ubiquitin. The Lys(Boc) ubiquitin variant was expressed in E. coli BL21(DE3) cells, together with an orthogonal tRNA synthetase/tRNA pair that incorporated the unnatural amino acid Lys(Boc).[13] After purification, all remaining amines on the Lys(Boc) ubiquitin variant were subsequently protected with the orthogonal protecting group, Alloc. The ε-NH2 of the target lysine was made available for isopeptide linkage by removal of the Boc protecting group using trifluoroacetic acid. The second ubiquitin monomer, wild-type ubiquitin, was activated with E1 to form a C-terminal thioester on ubiquitin (Ub-COSR), and all of its amines were also protected with the Alloc protecting group. Ub2 was assembled via a silver-mediated condensation reaction between Ub-COSR and the single available ε-NH2 amine on the other ubiquitin. After ligation, the Alloc protecting groups were removed, followed by protein denaturation and renaturation. Ub2 was purified from unreacted monomers as described above. Structures of the synthetic dimers were characterized by NMR and mass spectrometry.[12] All Ub2 species were exchanged into distilled water prior to microwave treatment. Diubiquitin concentrations were estimated using their extinction coefficient at 280nm (2980 cm−1M−1).

Assembly scheme for K6, K11, K27, K29 and K33-linked diubiquitins. In this report the ubiquitin moiety containing a free C-terminus (G76) is designated proximal, while the distal moiety has its G76 conjugated to a lysine on the proximal ubiquitin.
Microwave-Assisted Acid Cleavage
Diubiquitin samples were diluted to 0.1mg/ml in a 100 µL 12.5% acetic acid solution and digested at 140°C using 300W microwave energy for 30 sec or 15 min in a CEM Discover microwave (Matthews, NC). Acidic solvent was removed by lyophilization in a FreeZone 2.5 Plus from Labconco Corporation (Kansas City, MO) before further analysis. These conditions have been previously reported to hydrolyze proteins with high selectivity at Asp residues,[14] yielding peptides that produce searchable CID spectra.[15]
MALDI Analysis
A Kratos Axima CFR MALDI-TOF MS (Shimadzu Biosciences, Columbia, MD) was used in linear mode to acquire mass spectra. One hundred scans were integrated per spectrum. Laser power was 84V and the matrix used was α-cyano-4-hydroxycinnamic acid at 10mg/ml in 70/30/0.1 acetonitrile/water/TFA. The 0.1mg/ml protein sample and the matrix solution were mixed 1:1 by volume for MALDI analysis.
LC-MS/MS with Electrospray Ionization
After lyophilization, samples were diluted with HPLC grade water with 0.1% formic acid back to 100 µL.5 µL were injected, concentrated and desalted on a C8 trapping column (0.5×3 mm, Agilent Technologies) for 5 minutes before being separated through a pepSil C8 column (Column Technology Inc., Fremont, CA) with a flow rate of 300 nL/min and a gradient of 18–22% in 120 min (Solvent A: 97.5% HPLC grade water, 2.5% ACN, and 0.1% formic acid; Solvent B: 97.5% ACN, 2.5% HPLC grade water and 0.1% formic). Analysis was performed in reverse-phase using a 2D nano HPLC system (Shimadzu BioSciences, Columbia, MD) interfaced to an LTQ-Orbitrap XL mass spectrometer (Thermo-Fisher, San Jose, CA). Precursor masses were acquired with a resolution of 60000 while fragment ions were acquired with a resolution of 30000 all within the orbitrap mass analyzer. Fragmentation was accomplished by CID with collision energy normalized at 35. The 4 most intense ions were isolated and fragmented in each cycle. All spectra were interpreted manually using both raw and deconvoluted spectra.
RESULTS
Microwave-supported acid hydrolysis was optimized at 140 °C for 30 sec to produce the branched target peptides from each dimer. The sequences and observed and theoretical molecular masses of the truncated products of interest are shown in Table 1. All theoretical and observed masses match within 15 ppm. Each retains the linkage point and the carboxyl terminal prospectively attached to a target protein or another ubiquitin. Cleavage at Asp produces unique products for dimers linked via K63, K48, and K33, and isomeric products with linkages at K27/K29 or K6/K11. Other peptides are produced in each proteolysis reaction including the fully truncated branched peptides (Table 2), which provide confirming structural information about the linkage point.
Table 1.
Sequences and the theoretical and observed monoisotopic masses of the truncated branched peptides analyzed from the seven all-native K-linked ubiquitin dimers
| Linkage | Sequences of the Truncated Target Peptides | Mass (Da) | |
|---|---|---|---|
| Theor. | Obser. | ||
| K63 | 5432.98 | 5433.04 | |
| K48 | 6942.77 | 6942.83 | |
| K33 | 7696.17 | 7696.23 | |
| K29 | 8935.85 | 8935.89 | |
| K27 | |||
| K11 | 11267.09 | 11267.03 | |
| K6 | 11267.12 | ||
Table 2.
Sequences and the theoretical and observed monoisotopic masses of the fully truncated branched peptides analyzed from the seven all native K-linked diubiquitins.
| Linkage | Sequences of the Fully Truncated Target Peptides | Mass (Da) | |
|---|---|---|---|
| Theor. | Obser. | ||
| K63 | 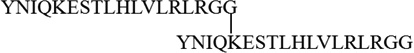 |
4174.35 | 4174.36 |
| K48 | 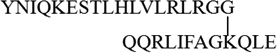 |
3490.95 | 3490.96 |
| K33 | 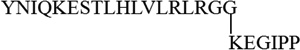 |
2717.52 | 2717.53 |
| K29 | 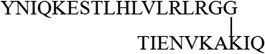 |
3220.83 | 3220.85 |
| K27 |  |
||
| K11 | 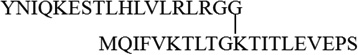 |
4312.39 | 4312.41 |
| K6 | 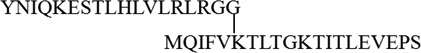 |
||
As suggested by the MALDI spectrum in Figure 1, this strategy produces a mixture of peptide products from a purified synthetic dimer, which we expect to extend to a conjugated dimers enriched from a cell lysate by immunoprecipitation.[15] Small peptides released from the ends of both branches can be removed from the larger branched peptides of interest by fractionation through a small ion exchange column prior to fractionation by HPLC interfaced to the mass spectrometer. Figure 2 summarizes chromatographic separation on a C8 reverse phase column of the seven target peptides (shown in Table 1) formed by acid cleavage of a mixture of all seven isomeric dimers. Reconstructed ion chromatograms of the protonated molecular ions indicate that separation is achieved of most of the truncated peptides of interest and indicate that relative retention times can contribute to characterization of the branched peptides. Ascertained using hydrolysis products from individual dimers and fragmentation patterns from the mixture of all seven dimers, the product from K63 elutes in a peak centered at 32 min; K27 and K29 coelute at 42 min; K33 at 48 min; K48 at 51 min; K6 and K11 coelute around 97 min. Retention times do not correlate completely with molecular masses, suggesting that molecular shapes imparted by branching are also influential. The molecular masses of the target hydrolysis products are characteristic; however four of the traces contain extra peaks. These are contributed by non-targeted hydrolysis products with the same masses formed from other dimers in the mixture. As discussed in the Introduction, fragmentation and tandem MS experiments together are required to distinguish the truncated K6/K11 and K27/K29 pairs and to provide definitive support for assignment of dimers with other linkage points.
Figure 2.

Ion chromatograms of the target masses listed in Table 1 reconstructed from the LC-MS/MS analysis of the products of a 30 sec hydrolysis of a mixture of all seven isomeric dimers
Collisionally induced dissociation provides class characteristic fragment ions that will allow the investigator to recognize the truncated target peptides shown in Table 1 and are obtained from diubiquitins and potentially ubiquitin chains conjugated to peptides as they elute from the HPLC. In an earlier report[9] we noted that low mass singly-charged b ions are formed reliably from the C-terminal tail remaining from the distal ubiquitin of truncated K63-linked diubiquitin. Some or all of these ions are observed in all the truncated dimers studied here. Reconstructed ion chromatograms in Figure 3 demonstrate how the suite of b6, b7, b8, b9, and b10 ions can be used to recognize or detect candidate truncated diubiquitins eluting from the HPLC.
Figure 3.

Ion chromatograms reconstructed from the same LC-MS/MS analysis seen in Figure 2 of the masses of b6, b7, b8, b9, and b10 produced by CID on the C-terminus tail remaining on the distal ubiquitin (pictured above) present on all seven K-linked diubiquitins. At the top is a reconstructed ion chromatogram comprising the five m/z values reconstructed from Figure 2.
Figure 4 summarizes the fragment ions observed in the truncated branched peptides obtained from the K33-, K48-, and K63-linked diubiquitins. In all three cases, acid cleavage and CID combine to define the linkage site and differentiate it from all others. Note also the formation of suites of b ions, consistently observed in all target peptides (Table 1) and presented in Figure 3. Figure 5 summarizes fragmentation in the truncated products from the K6 and K11-linked isomers, and Figure 6 presents the fragmentation in the truncated products from K27- and K29-linked diubiquitins. Among these two sets of isomers, only the linkage at K6 can be unambiguously characterized from the product ions (Figure 7). The other three CID spectra do eliminate most possibilities and bracket the linkage site to one of two positions. In our hands neither ETD nor HCD produced more definitive fragmentation in these longer branched peptides, and similarly the combination of in-source fragmentation and high energy CID in a MALDI TOF instrument did not produce the fragmentation required, e.g., to distinguish K27 from K29 or K11 from K6. The CID spectra in Figures 6 and 7 eliminate all other potential branch sites and bracket the linkage sites.
Figure 4.

Fragmentation observed in truncated K33-, K48-, and K63-linked diubiquitins activated by CID in a linear ion trap. b-ions are indicated by left facing carots and y-ions are indicated by right facing carots.
Figure 5.

Fragmentation observed in truncated K6- and K11-linked diubiquitins activated by CID in a linear ion trap. Carots as indicated in Figure 4.
Figure 6.

Fragmentation observed in truncated K27- and K29-linked diubiquitins activated by CID in a linear ion trap. Carots as indicated in Figure 4.
Figure 7.

a. NanoESI charge distribution of the target product from K6-linked diubiquitin with selected precursor ion circled. b. Product ion mass spectrum with the b- and y-ions labeled that distinguishes the K6-linkage from all other linkages. The branched peptide sequence is shown with those b- and y-ions starred.
Table 2 shows the structures of completely truncated minor products formed from all seven diubiquitins in the 30 sec hydrolysis and favored products in a 15 min reaction. Note that each peptide contains the branch site, however the C-terminus, and thus its potential conjugated peptide, has been lost. The branch points are clearly defined by CID in all seven of these smaller branched peptides. Fragmentation is shown in Figure 8 for all completely truncated K-linked peptide products. The potential branching sites are clearly distinguished in these tandem spectra, and we expect that the smaller products will be useful to elucidate more complex structures, e.g. in immunoprecipitates from cell lysates.
Figure 8.

Fragmentation by CID observed in all truncated K-linked dimers.
It has been pointed out previously that several of the ubiquitin-like proteins from the SUMO family leave a 32 residue tag when digested with trypsin, often longer than the modified peptide.[16,17] The ubiquitin-like protein Nedd8 leaves a GG tag when cleaved with trypsin, which cannot be distinguished from ubiquitin.[18] The use of tags resulting from acid cleavage, as proposed by this laboratory[15,19] alleviates these issues with SUMO and Nedd8.
In conclusion, the middle out strategy reported here allows the seven all natural K-linked diubiquitins to be characterized readily by LC-MS/MS. Truncated branched peptides are produced in 30 sec, which carry linkage points as well as the proximal carboxyl terminus that is the potential site for further conjugation. This feature potentially allows determination of consecutive linkages in a polyubiquitin chain, which is not feasible with the current complete proteolytic methods. The shortened branched peptides undergo extensive CID fragmentation, which allows reliable identification or bracketing of conjugation sites. A set of class characteristic b ions is reliably observed in CID spectra and can be used to identify elution times of ubiquitin conjugates. A current limitation is the need for manual interpretation of product ion spectra of the branched peptides. We are working to automate spectral interpretation and allow database searching. Building on the foundation of experience with isomeric diubiquitins, we are now extending our strategy to longer polyubiquitin conjugates, with the longer term objective of applying it to characterize linkage order and composition of polyubiquitins and their conjugates in cell lysates.
Acknowledgements
This work is supported by National Institutes of Health (NIH) grants GM065334 and GM021248.
References
- 1.Thorne AW, Sautiere P, Briand G, Crane-Robinson C. The structure of ubiquitinated histone H2B. The EMBO Journal. 1987;6:1005. doi: 10.1002/j.1460-2075.1987.tb04852.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G, Roelofs J, Finley D, Gygi SP. A proteomics approach to understanding protein ubiquitination. Nat. Biotech. 2003;21:921. doi: 10.1038/nbt849. [DOI] [PubMed] [Google Scholar]
- 3.Vasilescu J, Smith JC, Etheir M, Figeys D. Proteomic analysis of ubiqutinated proteins from human MCF-7 breast cancer cells by immunoaffinity purification and mass spectrometry. J. Prot. Res. 2005;4:2192. doi: 10.1021/pr050265i. [DOI] [PubMed] [Google Scholar]
- 4.Low TY, Magliozzi R, Guardavaccaro D, Heck AJR. Unraveling the ubiquitin-regulated signaling networks by mass spectrometry-based proteomics. Proteomics. 2013;13:526. doi: 10.1002/pmic.201200244. [DOI] [PubMed] [Google Scholar]
- 5.Kirkpatrick DS, Hathaway NA, Hanna J, Elsasser S, Rush J, Finley D, King RW, Gygi SP. Quantitative analysis of in vitro ubiquitinated cyclin B1 reveals complex chain topology. Nat. Cell Biol. 2006;8:700. doi: 10.1038/ncb1436. [DOI] [PubMed] [Google Scholar]
- 6.Strachan J, Roach L, Sokratous K, Tooth D, Long J, Garner TP, Searle MS, Oldham NJ, Layfield R. Insights into the molecular composition of endogenous unanchored polyubiquitin chains. J. Prot. Res. 2012;11:1969. doi: 10.1021/pr201167n. [DOI] [PubMed] [Google Scholar]
- 7.Warren MR, Parker CE, Mocanu V, Klappers D, Borchers CH. Electrospray ionization tandem mass spectrometry of model peptides reveals diagnostic fragment ions for protein ubiquitination. Rapid Commun. Mass Spectrom. 2005;19:429. doi: 10.1002/rcm.1798. [DOI] [PubMed] [Google Scholar]
- 8.Xu P, Peng J. Characterization of polyubiquitin chain structure by middle-down mass spectrometry. Anal. Chem. 2008;80:3438. doi: 10.1021/ac800016w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cannon J, Nakasone M, Fushman D, Fenselau C. Proteomic identification and analysis of K63-linked ubiquitin conjugates. Anal. Chem. 2012;84:10121. doi: 10.1021/ac302675y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Varadan R, Walker O, Pickart C, Fushman D. Structural properties of polyubiquitin chains in solution. J. Mol. Biol. 2002;324:637. doi: 10.1016/s0022-2836(02)01198-1. [DOI] [PubMed] [Google Scholar]
- 11.Varadan R, Assfalg M, Haririnia A, Raasi S, Pickart C, Fushman D. Solution conformation of Lys63-linked di-ubiquitin chain provides clues to functional diversity of polyubiquitin signaling. J. Biol. Chem. 2004;279:7055. doi: 10.1074/jbc.M309184200. [DOI] [PubMed] [Google Scholar]
- 12.Castaneda CA, Liu J, Chaturvedi A, Nowicka U, Cropp TA, Fushman D. Nonenzymatic assembly of natural polyubiquitin chains of any linkage composition and isotopic labeling scheme. J. Am. Chem. Soc. 2011;133:17855. doi: 10.1021/ja207220g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castaneda CA, Liu J, Kashyap TR, Singh RK, Fushman D, Cropp TA. Controlled enzymatic synthesis of natural-linkage, defined-length polyubiquitin chains using lysines with removable protecting groups. Chem. Commun. 2011;47:2026. doi: 10.1039/c0cc04868b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Swatkoski S, Gutierrez P, Wynne C, Petrov A, Dinman JD, Edwards N, Fenselau C. Evaluation of microwave-accelerated residue-specific acid cleavage for proteomic applications . J. Proteome Res. 2008;7:579. doi: 10.1021/pr070502c. [DOI] [PubMed] [Google Scholar]
- 15.Cannon J, Lohnes K, Wynne C, Wang Y, Edwards N, Fenselau C. High-throughput Middle-down analysis using an orbitrap. J. Proteome Res. 2010;9:3886. doi: 10.1021/pr1000994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Osula O, Swatkoski S, Cotter R. Identification of protein SUMOylation sites by mass spectrometry using combined microwave-assisted aspartic acid cleavage and trypic digestion. J. Mass Spectrom. 2012;47:644. doi: 10.1002/jms.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chicooree N, Griffiths JR, Connolly Y, Tan CT, Malliri A, Eyers CE, Smith DL. A novel approach to the analysis of SUMOylation with the independent use of trypsin and elastase digestion followed by database searching utilizing consecutive residue addition to lysine. Rapid Commun. Mass Spectrom. 2012;27:127. doi: 10.1002/rcm.6425. [DOI] [PubMed] [Google Scholar]
- 18.Leidecker O, Matic I, Mahata B, Pion E, Xirodimas DP. The ubiquitin E1 enzyme Ube1 mediates NEDD8 activation under diverse stress conditions. Cell Cycle. 2012;11:1142. doi: 10.4161/cc.11.6.19559. [DOI] [PubMed] [Google Scholar]
- 19.Lee A, Castaneda C, Singh R, Wang Y, Edwards N, Fushman D, Fenselau C. Characterization of polyubiquitin chains: building a foundation for reading the ubiquitin code; Proceedings of the 62nd Annual Conference of the American Society for Mass Spectrometry; June 15–19; Baltimore MD. 2014. [Google Scholar]