

Abstract
Development of molecular imaging agents for fibrillar β–amyloid (Aβ) positron emission tomography (PET) during the last decade brought molecular imaging of Alzheimer’s disease (AD) pathology into the spotlight. Large cohort studies with longitudinal follow-up in cognitively normal, mild cognitive impairment and AD patients indicate that Aβ deposition can be detected many years before the onset of symptoms with molecular imaging and its progression can be followed longitudinally. The utility of Aβ PET in differential diagnosis of AD is greatest when there is no pathologic overlap between the two dementia syndromes such as in frontotemporal lobar degeneration and AD. However Aβ PET alone may be insufficient in distinguishing dementia syndromes that commonly have overlapping Aβ pathology, such as dementia with Lewy bodies and vascular dementia, which represent the two most common dementia pathologies after AD. The role of molecular imaging in AD clinical trials is growing rapidly especially in an era when preventive interventions are designed towards eradicating the pathology targeted by molecular imaging agents.
Keywords: Molecular imaging, Alzheimer’s disease, PET, PiB, amyloid, mild cognitive impairment, cognitively normal
The pathologic hallmarks of AD are neurofibrillary tangles of hyperphosphorylated tau and extracellular plaques of β-amyloid (Aβ) proteins which involve the brain many years before the emergence of symptoms. Molecular imaging with agents that bind to Aβ and tau proteins offer the promise for detecting the presence and progression of Alzheimer’s disease pathology during the preclinical stage when the disease course may be altered by early intervention. Imaging of the Aβ pathology with PET has been used in clinical research setting for almost a decade and was recently approved by US Federal Drug Administration (FDA) for clinical use. Imaging of tau pathology with PET has been investigated less, however its impact on understanding the pathophysiology of AD and on clinical practice is expected to be significant. Both imaging of Aβ and tau will likely contribute independently to early diagnosis, differential diagnosis and tracking disease progression during the preclinical, prodromal and clinical stages of AD.
Detecting preclinical and prodromal AD pathology with molecular imaging
Over the past decade, discovery of Aβ imaging with Pittsburgh compound–B (PiB)1 PET provided a window into the pathophysiology of AD in living individuals. Although autopsy studies have long suggested a high prevalence Aβ pathology with moderate to frequent plaques reaching 47% in cognitively normal older adults, imaging of Aβ pathology with PET provided an in-vivo confirmation of this observation. The prevalence of PiB positivity ranges from 20% to 34% in independent cohorts of cognitively normal individuals.2–6 The variability is likely associated with the ascertainment of participants and the cut-off used for PiB positivity as well as the median age of the cohorts. For example, in a population-based study of cognitively normal older adults that included individuals with neurological, psychiatric or systemic illnesses in order to study a representative sample of the population, the prevalence of PiB positivity was 31% with a global cortical PiB uptake cut-off of >1.5 but increased to 44% with a cut-off of >1.47, which is on a par with the autopsy studies in community based cohorts of cognitively normal elderly.8
Although Aβ pathology is common in cognitively normal individuals, the harmful effects of Aβ pathology on cognitive function are modest. 5, 9–12 The risk of cognitive decline further increases with Aβ load.6, 13, 14 High Aβ load on PET appears have subtle effects on memory, attention/executive function and visual-spatial processing5, 6, 10, 13–18. The relationship between Aβ load and cognitive domain functions does not appear to follow a specific functional-anatomical pattern but localize to the frontal, lateral temporal and parietal lobes, posterior cingulate and precuneus cortex independent of the cognitive domains that are affected5. Therefore the effects of Aβ detected on PET appear to be global and APOE ε4 status further modifies the association between Aβ load and cognition5, 19. Although cognitively normal carriers of the APOE ε4 have higher Aβ load on PET compared to non-carriers3, 5, 20, when matched on Aβ load, APOE ε4 carriers tend to perform worse on cognitive tests compared to non-carriers5 (Figure 1). Thus, APOE ε4 not only increases the risk for Aβ deposition, but also influences AD pathology by modulating the harmful effects of Aβ on cognitive function through other potentially synergistic mechanisms such as enhancing hyperphosphorylation of tau protein21 and reducing choline acetyltransferase activity.22
Figure 1. Associations between cortical PiB retention and standardized memory and global cognitive domain scores according to APOE ε4 status.
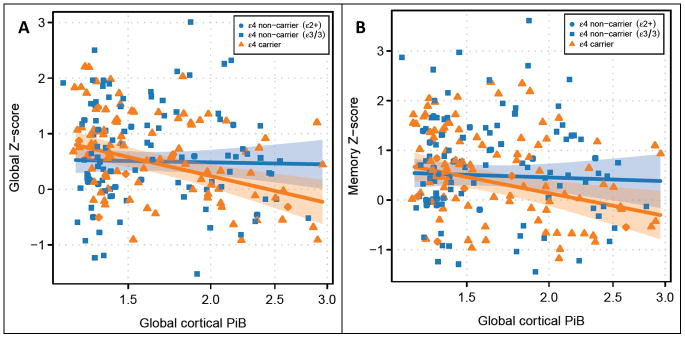
Higher Aβ load is associated with greater global cognitive impairment (partial rs = −0.18; p<0.01) and memory impairment (partial rs = −0.14; p<0.01). However global cognitive function in APOE ε4 carriers is influenced more by the Aβ load compared to APOE ε4 non-carriers matched on age, gender, education and Aβ load (sequential ANOVA interaction; p=0.01), suggesting that APOE isoforms modulate the harmful effects of Aβ on cognitive function (A). A similar trend is seen with memory function (sequential ANOVA interaction; p=0.08). With permission from Neurology 5
In 2011, the clinical diagnostic criteria for AD were revised under the auspices of the National Institutes of Aging and the Alzheimer’s Association (NIA-AA).23 These new guidelines included imaging markers in the diagnostic criteria for AD and proposed research criteria that include imaging evidence of AD for the diagnosis of preclinical AD.24 The new criteria require evidence of Aβ pathology of AD for the diagnosis of preclinical AD either through molecular imaging or cerebrospinal fluid (CSF) biomarkers. Any additional imaging or biomarker evidence of AD-related neurodegeneration measured with AD pattern of atrophy on MRI or AD pattern of hypometabolism on F-18 fluorodeoxyglucose (FDG) PET as well as presence of subtle cognitive difficulties in addition to the Aβ pathology increases the stage of preclinical AD from stage 1 to stage 3. We operationalized the preclinical AD research criteria in a population-based sample of cognitively normal older adults from the Mayo Clinic Study of Aging.25 At fixed cut-points corresponding to 90% sensitivity for diagnosing AD and the 10th percentile of cognitive scores of cognitively normal individuals, 43% of our sample was classified as stage 0, 16% stage 1 (Aβ PET positive), 12 % stage 2 (Aβ PET positive and neurodegeneration positive on MRI or FDG PET), 3% stage 3 (Aβ PET positive and neurodegeneration positive on MRI or FDG PET and subtle cognitive difficulties). 26 Furthermore, the proportion of subjects who progressed to MCI or dementia increased with advancing stage (Figure 2). 27 However, 23% of our population did not fit the preclinical AD stages because they had normal Aβ PET imaging, but abnormal neurodegeneration biomarker studies, which we classified as suspected non-AD pathophysiology (SNAP). The SNAP group is of particular interest because they progress to MCI in the short term (10% in 15 months), albeit at a rate similar to stage 1 preclinical AD subjects (11% in 15 months). The pathologic basis of positive neurodegeneration biomarker findings in the absence of Aβ pathology in this cognitively normal group is under investigation.28
Figure 2. Preclinical staging of Alzheimer’s disease and short-term progression rates.
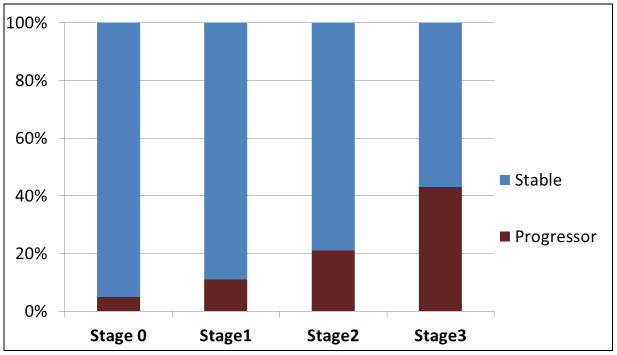
Using the preclinical staging criteria, at fixed cut-points corresponding to 90% sensitivity for diagnosing AD and the 10th percentile of cognitive scores of cognitively normal individuals, Stage 0 corresponds to low Aβ load on PET and absence of imaging markers of neuronal injury (i.e. normal hippocampal volumes on MRI and/or absence of AD-like pattern of hypometabolism on PET); Stage 1 corresponds to high Aβ load on PET and absence of imaging markers of neuronal injury; Stage 2 corresponds to high Aβ load on PET and presence of imaging markers of neuronal injury; Stage 3 corresponds to low Aβ load on PET, presence of imaging markers of neuronal injury, and subtle cognitive impairment. The proportion (%) of patients who progressed to mild cognitive impairment during a median follow-up of 15 months is demonstrated. Diagnosis of MCI was made according to Petersen criteria, 90 blinded to the imaging biomarker data used for staging.
According to the new guidelines by the NIA-AA, the prodromal stage of AD is characterized by mild cognitive impairment (MCI) and research criteria further classifies MCI patients as MCI due to AD, based on biomarker evidence of AD pathophysiology. A recent study from the Mayo Clinic Study of Aging and Alzheimer’s Disease Neuroimaging Initiative demonstrated that the NIA-AA criteria apply to most MCI subjects in both the community and clinical trials settings however, a sizeable proportion of subjects had conflicting biomarkers, which need to be explored. 29 In this population, neurodegeneration on MRI increased the rate of progression to dementia in patients with MCI due to AD and appeared to be a key factor in predicting progression relative to Aβ deposition alone.
Molecular imaging studies with Aβ-binding ligands in preclinical AD indicate that approximately a third of the population of cognitively normal individuals and 71% of patients with MCI in the community have high cortical Aβ load. In cognitively normal individuals, high levels of Aβ deposition are associated with subtle cognitive deficits, cognitive decline and a higher risk of cognitive impairments in the future. However, these relationships appear to be modified by the genetic markers5, 30, life-style activities31 or cognitive reserve32.
Molecular imaging for differential diagnosis of AD
High sensitivity and specificity of PiB binding to fibrillar Aβ have been demonstrated in vitro33, in mouse models34 and human tissue35. The newer 18F agents for Aβ PET have undergone a similar validation process,36–40 and appear to show similar properties to PiB 41–45. The specificity of PiB to fibrillar Aβ is preserved even in patients with protein deposits associated with other neurodegenerative dementias such as α-synuclein in dementia with Lewy bodies (Figure 3). 46–49 However, it is important to note that there may be disagreements between the autopsy report and the PET findings because of the heterogeneity of Aβ deposits. For example, PiB labels both neuritic and diffuse plaques, although labeling of diffuse/amphorous plaques is less prominent than compact/cored plaques. 35, 50 Patients with dementia with Lewy bodies or Parkinson’s disease dementia, who typically have high loads of diffuse plaques may have a positive Aβ PET scan but would not be classified as AD because of the absence of neuritic plaques and low Braak neurofibrillary tangle stage.46, 47 Another example is cerebral amyloid angiopathy (CAA). PiB binds to vascular amyloid in patients with CAA but not all patients with CAA have parenchymal Aβ deposits for the diagnosis of AD. Thus, while PiB is highly specific to Aβ, not all Aβ deposits may be considered for the pathologic diagnosis of AD.35, 50 Furthermore, there appears to be a threshold for detection where it may not be possible to detect low levels of fibrillar Aβ deposition51, None the less the agreement between high Aβ load on PET and pathologic diagnosis of AD in clinical setting is high and demonstrated in antemortem imaging and autopsy confirmation studies and case series.52–54 The sensitivity and specificity of amyloid PET tracers to the different fibrillar Aβ deposits need further investigation.
Figure 3. Correlations of cortical PiB retention and Aβ (A) and Lewy body (B) densities in individual brain regions of a case with dementia with Lewy bodies.
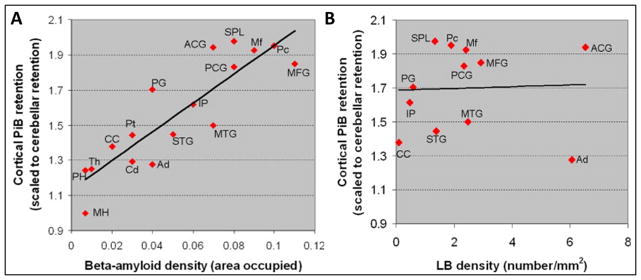
There was a strong correlation between PiB retention and Aβ density in the 17 ROIs that were analyzed on pathological examination using Spearman rank order correlation (r=0.899; p<0.0001). There was no correlation between Lewy body density and PiB retention (r=0.13; p=0.66). MH: middle hippocampus; PH: posterior hippocampus; Th: thalamus; Cd: caudate; Ad: amygdala; CC: calcarine cortex; Pt: putamen; STG: superior temporal gyrus; MTG: middle temporal gyrus; IP: inferior parietal; PG: precentral gyrus; PCG: posterior cingulate gyrus; MFG: middle frontal gyrus; Mf: midfrontal; ACG: anterior cingulate gyrus; Pc: Precuneus; SPL: superior parietal lobule. With permission from Neurobiology of Aging 45
One of the key applications of Aβ PET imaging in clinical practice is differential diagnosis of AD. The accuracy of Aβ PET in distinguishing AD and frontotemporal lobar degeneration (FTLD) is quite high 55 with overall classification accuracy of 97% in cases with histopathologic confirmation.56 On the other hand, the two most common dementia pathologies after AD are vascular disease and Lewy body pathologies, which commonly are present with additional AD pathology. In these cases, presence of intermediate to high Aβ load may be insufficient to determine the predominant pathology contributing to the dementia syndrome. In keeping with the autopsy data, 25% to 35% of patients with vascular dementia57, 58, and 60% to 80% of DLB patients 46, 54, 59–62 have high Aβ load on PET. Thus high levels of amyloid load may be insufficient in distinguishing these dementia syndromes from AD and a multi-modality imaging approach may be useful. We have shown that FDG PET, Aβ PET and structural MRI are complementary in distinguishing patients with AD and DLB54 and may be useful in predicting presence of AD pathology in patients with DLB.63 (Figure 4) Molecular imaging of the impaired nigrostriatal dopaminergic transmission in DLB with 2beta-carbomethoxy-3beta-(4-iodophenyl)-N-(3-fluoropropyl) nortropane (FP-CIT) with SPECT 64 or loss of monoaminergic terminal integrity with vesicular monoamine transporter type 2 (VMAT2) radioligands may further detect the Lewy body related pathologic features in cases with mixed dementia and be complementary to Aβ PET65.
Figure 4. Multi-modality imaging markers in distinguishing Alzheimer’s disease (AD) and dementia with Lewy bodies (DLB).

Regional FDG hypometabolism and PiB uptake on PET patients with probable DLB (n=21), are compared to control subjects (n=42) and gray matter atrophy in and patients with AD (n=21), compared to DLB patients are displayed on surface rendered brain images using SPM (p<0.05; family wise error corrected for multiple comparisons). Although occipital lobe hypometabolism, global cortical PiB retention and hippocampal volumes (% of total intracranial volume ) was overlapping among probable DLB and AD patients, multi-modality imaging approach almost completely separated DLB and AD patients. Logistic regression modeling demonstrated that each imaging modality independently contributed to distinguishing the AD and DLB patients with area under the receiver operating characteristic of 0.98. With permission from Neurobiology of Aging 50
The added diagnostic value of Aβ PET imaging in differential diagnosis of dementia across different clinical settings has become a topic of significant interest with availability of 18F agents for Aβ imaging 66–70. Although added value of Aβ PET to clinical decision making is not established, 66–69 how Aβ load is measured on PET scans (i.e. visual evaluation versus various quantitative techniques) appears to make a difference in the value of this diagnostic technique in clinical setting.69
Longitudinal molecular imaging for tracking AD pathology
Longitudinal imaging of Aβ load on PET provides evidence on the progression of Aβ deposition in the preclinical to clinical AD spectrum. The hypothetical model proposed by Jack et al. 71 indicate that Aβ deposition detected with molecular imaging and CSF biomarkers follow and accelerated course early in the disease process during the preclinical and MCI stages but slowing down during the Alzheimer’s disease stage and reaching a plateau as it reaches very high levels. Many longitudinal biomarker studies on Aβ deposition agree with this model. 72–81 Cognitively normal individuals who progress to MCI and patients with MCI who progress to AD appear to have the highest rates of Aβ deposition79 correlating with cognitive decline early in the disease course76, 77. Furthermore, a higher baseline Aβ load 78, 80 and APOE ε479 are associated with higher rates of Aβ deposition. However, the association between higher baseline Aβ load measured with standardized uptake values (SUVR) and a higher rate of Aβ deposition appear to dissipate at very high levels (roughly 2.0 SUVR). After this threshold the relationship becomes an inverted U shape, gradually declining and reaching zero at highest baseline Aβ load levels (2.7 SUVR). The time estimated to start with a positive PiB scan (1.5 SUVR) to the point of plateau is approximately 15 years corresponding to a large therapeutic window for clinical trials.73
Molecular imaging in clinical trials for AD
In autosomal dominant AD, the age of symptom onset can be predicted. It is estimated that increased Aβ deposition precedes clinical symptoms for approximately 15 years providing a wide window for preventive therapies. 82, 83 The role of molecular imaging in clinical trials targeting the pathology captured with the molecular imaging agent can be two-fold: 1) To determine who has the target pathology and enrichment of trials with this information; 2) To determine whether a treatment is modifying the target pathology. Both of these applications of Aβ imaging are being used in current clinical trials of amyloid-modifying therapies for both treatment and prevention of AD.84, 85 Findings from the bapineuzumab phase 2 double-blind placebo-controlled, ascending dose study indicate that lowering of cortical fibrillar Aβ with bapineuzumab can be detected with PiB PET.86 However, even though there were reductions in Aβ load, the bapineuzumab trials were halted due to lack of improvement in clinical and functional outcomes in patients with AD dementia. Similarly, it is expected that imaging of the tau pathology of AD 87, 88 especially with agents specific to the tau pathology that are currently being developed and tested, 89 will open avenues for development of new targets for prevention.
References
- 1.Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 2.Pike KE, Ellis KA, Villemagne VL, et al. Cognition and beta-amyloid in preclinical Alzheimer’s disease: data from the AIBL study. Neuropsychologia. 2011;49:2384–2390. doi: 10.1016/j.neuropsychologia.2011.04.012. [DOI] [PubMed] [Google Scholar]
- 3.Morris JC, Roe CM, Xiong C, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol. 2010;67:122–131. doi: 10.1002/ana.21843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aizenstein HJ, Nebes RD, Saxton JA, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol. 2008;65:1509–1517. doi: 10.1001/archneur.65.11.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kantarci K, Lowe V, Przybelski SA, et al. APOE modifies the association between Abeta load and cognition in cognitively normal older adults. Neurology. 2012;78:232–240. doi: 10.1212/WNL.0b013e31824365ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Landau SM, Mintun MA, Joshi AD, et al. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann Neurol. 2012;72:578–586. doi: 10.1002/ana.23650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mielke MM, Wiste HJ, Weigand SD, et al. Indicators of amyloid burden in a population-based study of cognitively normal elderly. Neurology. 2012;79:1570–1577. doi: 10.1212/WNL.0b013e31826e2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sonnen JA, Larson EB, Crane PK, et al. Pathological correlates of dementia in a longitudinal, population-based sample of aging. Ann Neurol. 2007;62:406–413. doi: 10.1002/ana.21208. [DOI] [PubMed] [Google Scholar]
- 9.Pike KE, Savage G, Villemagne VL, et al. Beta-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer’s disease. Brain. 2007;130:2837–2844. doi: 10.1093/brain/awm238. [DOI] [PubMed] [Google Scholar]
- 10.Chetelat G, Villemagne VL, Pike KE, et al. Relationship between memory performance and beta-amyloid deposition at different stages of Alzheimer’s disease. Neurodegener Dis. 2012;10:141–144. doi: 10.1159/000334295. [DOI] [PubMed] [Google Scholar]
- 11.Fagan AM, Mintun MA, Shah AR, et al. Cerebrospinal fluid tau and ptau(181) increase with cortical amyloid deposition in cognitively normal individuals: implications for future clinical trials of Alzheimer’s disease. EMBO Mol Med. 2009;1:371–380. doi: 10.1002/emmm.200900048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mormino EC, Kluth JT, Madison CM, et al. Episodic memory loss is related to hippocampal-mediated beta-amyloid deposition in elderly subjects. Brain. 2009;132:1310–1323. doi: 10.1093/brain/awn320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doraiswamy PM, Sperling RA, Coleman RE, et al. Amyloid-beta assessed by florbetapir F 18 PET and 18-month cognitive decline: a multicenter study. Neurology. 2012;79:1636–1644. doi: 10.1212/WNL.0b013e3182661f74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chetelat G, Villemagne VL, Pike KE, et al. Independent contribution of temporal beta-amyloid deposition to memory decline in the pre-dementia phase of Alzheimer’s disease. Brain. 2011;134:798–807. doi: 10.1093/brain/awq383. [DOI] [PubMed] [Google Scholar]
- 15.Rentz DM, Amariglio RE, Becker JA, et al. Face-name associative memory performance is related to amyloid burden in normal elderly. Neuropsychologia. 2011;49:2776–2783. doi: 10.1016/j.neuropsychologia.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodrigue KM, Kennedy KM, Devous MD, Sr, et al. beta-Amyloid burden in healthy aging: regional distribution and cognitive consequences. Neurology. 2012;78:387–395. doi: 10.1212/WNL.0b013e318245d295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sperling RA, Johnson KA, Doraiswamy PM, et al. Amyloid deposition detected with florbetapir F 18 ((18)F-AV-45) is related to lower episodic memory performance in clinically normal older individuals. Neurobiol Aging. 2013;34:822–831. doi: 10.1016/j.neurobiolaging.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hedden T, Oh H, Younger AP, et al. Meta-analysis of amyloid-cognition relations in cognitively normal older adults. Neurology. 2013;80:1341–1348. doi: 10.1212/WNL.0b013e31828ab35d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lim YY, Ellis KA, Ames D, et al. Abeta amyloid, cognition, and APOE genotype in healthy older adults. Alzheimers Dement. 2013;9:538–545. doi: 10.1016/j.jalz.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 20.Fleisher AS, Chen K, Liu X, et al. Apolipoprotein E epsilon4 and age effects on florbetapir positron emission tomography in healthy aging and Alzheimer disease. Neurobiol Aging. 2013;34:1–12. doi: 10.1016/j.neurobiolaging.2012.04.017. [DOI] [PubMed] [Google Scholar]
- 21.Ledesma MD, Medina M, Avila J. The in vitro formation of recombinant tau polymers: effect of phosphorylation and glycation. Mol Chem Neuropathol. 1996;27:249–258. doi: 10.1007/BF02815107. [DOI] [PubMed] [Google Scholar]
- 22.Dubelaar EJ, Verwer RW, Hofman MA, et al. ApoE epsilon4 genotype is accompanied by lower metabolic activity in nucleus basalis of Meynert neurons in Alzheimer patients and controls as indicated by the size of the Golgi apparatus. J Neuropathol Exp Neurol. 2004;63:159–169. doi: 10.1093/jnen/63.2.159. [DOI] [PubMed] [Google Scholar]
- 23.Jack CR, Jr, Albert MS, Knopman DS, et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:257–262. doi: 10.1016/j.jalz.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roberts RO, Geda YE, Knopman DS, et al. The Mayo Clinic Study of Aging: design and sampling, participation, baseline measures and sample characteristics. Neuroepidemiology. 2008;30:58–69. doi: 10.1159/000115751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jack CR, Jr, Knopman DS, Weigand SD, et al. An operational approach to National Institute on Aging-Alzheimer’s Association criteria for preclinical Alzheimer disease. Ann Neurol. 2012;71:765–775. doi: 10.1002/ana.22628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knopman DS, Jack CR, Jr, Wiste HJ, et al. Short-term clinical outcomes for stages of NIA-AA preclinical Alzheimer disease. Neurology. 2012;78:1576–1582. doi: 10.1212/WNL.0b013e3182563bbe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knopman DS, Jack CR, Jr, Wiste HJ, et al. Brain injury biomarkers are not dependent on beta-amyloid in normal elderly. Ann Neurol. 2012 doi: 10.1002/ana.23816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Petersen RC, Aisen P, Boeve BF, et al. Criteria for mild cognitive impairment due to alzheimer’s disease in the community. Ann Neurol. 2013 doi: 10.1002/ana.23931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mosconi L, Andrews RD, Matthews DC. Comparing brain amyloid deposition, glucose metabolism, and atrophy in mild cognitive impairment with and without a family history of dementia. J Alzheimers Dis. 2013;35:509–524. doi: 10.3233/JAD-121867. [DOI] [PubMed] [Google Scholar]
- 31.Vemuri P, Lesnick TG, Przybelski SA, et al. Effect of lifestyle activities on Alzheimer disease biomarkers and cognition. Ann Neurol. 2012;72:730–738. doi: 10.1002/ana.23665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kemppainen NM, Aalto S, Karrasch M, et al. Cognitive reserve hypothesis: Pittsburgh Compound B and fluorodeoxyglucose positron emission tomography in relation to education in mild Alzheimer’s disease. Ann Neurol. 2008;63:112–118. doi: 10.1002/ana.21212. [DOI] [PubMed] [Google Scholar]
- 33.Mathis CA, Wang Y, Holt DP, et al. Synthesis and evaluation of 11C-labeled 6-substituted 2-arylbenzothiazoles as amyloid imaging agents. J Med Chem. 2003;46:2740–2754. doi: 10.1021/jm030026b. [DOI] [PubMed] [Google Scholar]
- 34.Manook A, Yousefi BH, Willuweit A, et al. Small-animal PET imaging of amyloid-beta plaques with [11C]PiB and its multi-modal validation in an APP/PS1 mouse model of Alzheimer’s disease. PLoS One. 2012;7:e31310. doi: 10.1371/journal.pone.0031310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ikonomovic MD, Klunk WE, Abrahamson EE, et al. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain. 2008;131:1630–1645. doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choi SR, Schneider JA, Bennett DA, et al. Correlation of amyloid PET ligand florbetapir F 18 binding with Abeta aggregation and neuritic plaque deposition in postmortem brain tissue. Alzheimer Dis Assoc Disord. 2012;26:8–16. doi: 10.1097/WAD.0b013e31821300bc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin KJ, Hsu WC, Hsiao IT, et al. Whole-body biodistribution and brain PET imaging with [18F]AV-45, a novel amyloid imaging agent--a pilot study. Nucl Med Biol. 2010;37:497–508. doi: 10.1016/j.nucmedbio.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 38.Wong DF, Rosenberg PB, Zhou Y, et al. In vivo imaging of amyloid deposition in Alzheimer disease using the radioligand 18F-AV-45 (florbetapir [corrected] F 18) J Nucl Med. 2010;51:913–920. doi: 10.2967/jnumed.109.069088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poisnel G, Dhilly M, Moustie O, et al. PET imaging with [18F]AV-45 in an APP/PS1–21 murine model of amyloid plaque deposition. Neurobiol Aging. 2012;33:2561–2571. doi: 10.1016/j.neurobiolaging.2011.12.024. [DOI] [PubMed] [Google Scholar]
- 40.Cselenyi Z, Jonhagen ME, Forsberg A, et al. Clinical validation of 18F-AZD4694, an amyloid-beta-specific PET radioligand. J Nucl Med. 2012;53:415–424. doi: 10.2967/jnumed.111.094029. [DOI] [PubMed] [Google Scholar]
- 41.Rowe CC, Pejoska S, Mulligan RS, et al. Head-to-head comparison of 11C-PiB and 18F-AZD4694 (NAV4694) for beta-amyloid imaging in aging and dementia. J Nucl Med. 2013;54:880–886. doi: 10.2967/jnumed.112.114785. [DOI] [PubMed] [Google Scholar]
- 42.Villemagne VL, Mulligan RS, Pejoska S, et al. Comparison of 11C-PiB and 18F-florbetaben for Abeta imaging in ageing and Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2012;39:983–989. doi: 10.1007/s00259-012-2088-x. [DOI] [PubMed] [Google Scholar]
- 43.Wolk DA, Zhang Z, Boudhar S, et al. Amyloid imaging in Alzheimer’s disease: comparison of florbetapir and Pittsburgh compound-B positron emission tomography. J Neurol Neurosurg Psychiatry. 2012;83:923–926. doi: 10.1136/jnnp-2012-302548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Becker GA, Ichise M, Barthel H, et al. PET quantification of 18F-florbetaben binding to beta-amyloid deposits in human brains. J Nucl Med. 2013;54:723–731. doi: 10.2967/jnumed.112.107185. [DOI] [PubMed] [Google Scholar]
- 45.Landau SM, Breault C, Joshi AD, et al. Amyloid-beta imaging with Pittsburgh compound B and florbetapir: comparing radiotracers and quantification methods. J Nucl Med. 2013;54:70–77. doi: 10.2967/jnumed.112.109009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burack MA, Hartlein J, Flores HP, et al. In vivo amyloid imaging in autopsy-confirmed Parkinson disease with dementia. Neurology. 2010;74:77–84. doi: 10.1212/WNL.0b013e3181c7da8e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kantarci K, Yang C, Schneider JA, et al. Antemortem amyloid imaging and beta-amyloid pathology in a case with dementia with Lewy bodies. Neurobiol Aging. 2012;33:878–885. doi: 10.1016/j.neurobiolaging.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fodero-Tavoletti MT, Smith DP, McLean CA, et al. In vitro characterization of Pittsburgh compound-B binding to Lewy bodies. J Neurosci. 2007;27:10365–10371. doi: 10.1523/JNEUROSCI.0630-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ye L, Velasco A, Fraser G, et al. In vitro high affinity alpha-synuclein binding sites for the amyloid imaging agent PIB are not matched by binding to Lewy bodies in postmortem human brain. Journal of neurochemistry. 2008;105:1428–1437. doi: 10.1111/j.1471-4159.2008.05245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lockhart A, Lamb JR, Osredkar T, et al. PIB is a non-specific imaging marker of amyloid-beta (Abeta) peptide-related cerebral amyloidosis. Brain. 2007;130:2607–2615. doi: 10.1093/brain/awm191. [DOI] [PubMed] [Google Scholar]
- 51.Ikonomovic MD, Abrahamson EE, Price JC, et al. Early AD pathology in a [C-11]PiB-negative case: a PiB-amyloid imaging, biochemical, and immunohistochemical study. Acta Neuropathol. 2012;123:433–447. doi: 10.1007/s00401-012-0943-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Clark CM, Schneider JA, Bedell BJ, et al. Use of florbetapir-PET for imaging beta-amyloid pathology. Jama. 2011;305:275–283. doi: 10.1001/jama.2010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sojkova J, Driscoll I, Iacono D, et al. In vivo fibrillar beta-amyloid detected using [11C]PiB positron emission tomography and neuropathologic assessment in older adults. Arch Neurol. 2011;68:232–240. doi: 10.1001/archneurol.2010.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kantarci K, Lowe VJ, Boeve BF, et al. Multimodality imaging characteristics of dementia with Lewy bodies. Neurobiol Aging. 2012;33:2091–2105. doi: 10.1016/j.neurobiolaging.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Villemagne VL, Ong K, Mulligan RS, et al. Amyloid imaging with (18)F-florbetaben in Alzheimer disease and other dementias. J Nucl Med. 2011;52:1210–1217. doi: 10.2967/jnumed.111.089730. [DOI] [PubMed] [Google Scholar]
- 56.Rabinovici GD, Rosen HJ, Alkalay A, et al. Amyloid vs FDG-PET in the differential diagnosis of AD and FTLD. Neurology. 2011;77:2034–2042. doi: 10.1212/WNL.0b013e31823b9c5e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schneider JA, Arvanitakis Z, Bang W, et al. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007;69:2197–2204. doi: 10.1212/01.wnl.0000271090.28148.24. [DOI] [PubMed] [Google Scholar]
- 58.Sonnen JA, Santa Cruz K, Hemmy LS, et al. Ecology of the aging human brain. Arch Neurol. 2011;68:1049–1056. doi: 10.1001/archneurol.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Edison P, Rowe CC, Rinne JO, et al. Amyloid load in Parkinson’s disease dementia and Lewy body dementia measured with [11C]PIB positron emission tomography. J Neurol Neurosurg Psychiatry. 2008;79:1331–1338. doi: 10.1136/jnnp.2007.127878. [DOI] [PubMed] [Google Scholar]
- 60.Foster ER, Campbell MC, Burack MA, et al. Amyloid imaging of Lewy body-associated disorders. Mov Disord. 2010;25:2516–2523. doi: 10.1002/mds.23393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gomperts SN, Rentz DM, Moran E, et al. Imaging amyloid deposition in Lewy body diseases. Neurology. 2008;71:903–910. doi: 10.1212/01.wnl.0000326146.60732.d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maetzler W, Liepelt I, Reimold M, et al. Cortical PIB binding in Lewy body disease is associated with Alzheimer-like characteristics. Neurobiology of disease. 2009;34:107–112. doi: 10.1016/j.nbd.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 63.Kantarci K, Ferman TJ, Boeve BF, et al. Focal atrophy on MRI and neuropathologic classification of dementia with Lewy bodies. Neurology. 2012;79:553–560. doi: 10.1212/WNL.0b013e31826357a5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.O’Brien JT, Colloby S, Fenwick J, et al. Dopamine transporter loss visualized with FP-CIT SPECT in the differential diagnosis of dementia with Lewy bodies. Arch Neurol. 2004;61:919–925. doi: 10.1001/archneur.61.6.919. [DOI] [PubMed] [Google Scholar]
- 65.Villemagne VL, Okamura N, Pejoska S, et al. Differential diagnosis in Alzheimer’s disease and dementia with Lewy bodies via VMAT2 and amyloid imaging. Neurodegener Dis. 2012;10:161–165. doi: 10.1159/000334535. [DOI] [PubMed] [Google Scholar]
- 66.Camus V, Payoux P, Barre L, et al. Using PET with 18F-AV-45 (florbetapir) to quantify brain amyloid load in a clinical environment. Eur J Nucl Med Mol Imaging. 2012;39:621–631. doi: 10.1007/s00259-011-2021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Frederiksen KS, Hasselbalch SG, Hejl AM, et al. Added Diagnostic Value of (11)C-PiB-PET in Memory Clinic Patients with Uncertain Diagnosis. Dement Geriatr Cogn Dis Extra. 2012;2:610–621. doi: 10.1159/000345783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ossenkoppele R, Prins ND, Pijnenburg YA, et al. Impact of molecular imaging on the diagnostic process in a memory clinic. Alzheimers Dement. 2013;9:414–421. doi: 10.1016/j.jalz.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 69.Frisoni GB, Bocchetta M, Chetelat G, et al. Imaging markers for Alzheimer disease: Which vs how. Neurology. 2013;81:487–500. doi: 10.1212/WNL.0b013e31829d86e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mitka M. PET imaging for Alzheimer disease: are its benefits worth the cost? Jama. 2013;309:1099–1100. doi: 10.1001/jama.2013.2101. [DOI] [PubMed] [Google Scholar]
- 71.Jack CR, Jr, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9:119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jack CR, Jr, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207–216. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jack CR, Jr, Wiste HJ, Lesnick TG, et al. Brain beta-amyloid load approaches a plateau. Neurology. 2013;80:890–896. doi: 10.1212/WNL.0b013e3182840bbe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Villemagne VL, Burnham S, Bourgeat P, et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 2013;12:357–367. doi: 10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]
- 75.Kadir A, Almkvist O, Forsberg A, et al. Dynamic changes in PET amyloid and FDG imaging at different stages of Alzheimer’s disease. Neurobiol Aging. 2012;33:198 e191–114. doi: 10.1016/j.neurobiolaging.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 76.Koivunen J, Scheinin N, Virta JR, et al. Amyloid PET imaging in patients with mild cognitive impairment: a 2-year follow-up study. Neurology. 2011;76:1085–1090. doi: 10.1212/WNL.0b013e318212015e. [DOI] [PubMed] [Google Scholar]
- 77.Ossenkoppele R, Tolboom N, Foster-Dingley JC, et al. Longitudinal imaging of Alzheimer pathology using [11C]PIB, [18F]FDDNP and [18F]FDG PET. Eur J Nucl Med Mol Imaging. 2012;39:990–1000. doi: 10.1007/s00259-012-2102-3. [DOI] [PubMed] [Google Scholar]
- 78.Sojkova J, Zhou Y, An Y, et al. Longitudinal patterns of beta-amyloid deposition in nondemented older adults. Arch Neurol. 2011;68:644–649. doi: 10.1001/archneurol.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Villemagne VL, Pike KE, Chetelat G, et al. Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease. Ann Neurol. 2011;69:181–192. doi: 10.1002/ana.22248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Villain N, Chetelat G, Grassiot B, et al. Regional dynamics of amyloid-beta deposition in healthy elderly, mild cognitive impairment and Alzheimer’s disease: a voxelwise PiB-PET longitudinal study. Brain. 2012;135:2126–2139. doi: 10.1093/brain/aws125. [DOI] [PubMed] [Google Scholar]
- 81.Jack CR, Jr, Lowe VJ, Weigand SD, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer’s disease: implications for sequence of pathological events in Alzheimer’s disease. Brain. 2009;132:1355–1365. doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fleisher AS, Chen K, Quiroz YT, et al. Florbetapir PET analysis of amyloid-beta deposition in the presenilin 1 E280A autosomal dominant Alzheimer’s disease kindred: a cross-sectional study. Lancet Neurol. 2012;11:1057–1065. doi: 10.1016/S1474-4422(12)70227-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sperling RA, Jack CR, Jr, Aisen PS. Testing the right target and right drug at the right stage. Science translational medicine. 2011;3:111cm133. doi: 10.1126/scitranslmed.3002609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Barthel H, Luthardt J, Becker G, et al. Individualized quantification of brain beta-amyloid burden: results of a proof of mechanism phase 0 florbetaben PET trial in patients with Alzheimer’s disease and healthy controls. Eur J Nucl Med Mol Imaging. 2011;38:1702–1714. doi: 10.1007/s00259-011-1821-1. [DOI] [PubMed] [Google Scholar]
- 86.Rinne JO, Brooks DJ, Rossor MN, et al. 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 2010;9:363–372. doi: 10.1016/S1474-4422(10)70043-0. [DOI] [PubMed] [Google Scholar]
- 87.Small GW, Siddarth P, Kepe V, et al. Prediction of cognitive decline by positron emission tomography of brain amyloid and tau. Arch Neurol. 2012;69:215–222. doi: 10.1001/archneurol.2011.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Small GW, Kepe V, Ercoli LM, et al. PET of brain amyloid and tau in mild cognitive impairment. N Engl J Med. 2006;355:2652–2663. doi: 10.1056/NEJMoa054625. [DOI] [PubMed] [Google Scholar]
- 89.Okamura N, Furumoto S, Harada R, et al. Novel 18F-Labeled Arylquinoline Derivatives for Noninvasive Imaging of Tau Pathology in Alzheimer Disease. J Nucl Med. 2013;54:1420–1427. doi: 10.2967/jnumed.112.117341. [DOI] [PubMed] [Google Scholar]
- 90.Petersen RC. Mild cognitive impairment as a diagnostic entity. Journal of internal medicine. 2004;256:183–194. doi: 10.1111/j.1365-2796.2004.01388.x. [DOI] [PubMed] [Google Scholar]