

Abstract
Sphingomyelinase (SMase) activity is elevated in inflammatory states and may contribute to muscle weakness in these conditions. Exogenous SMase depresses muscle force in an oxidant-dependent manner. However, the pathway stimulated by SMase that leads to muscle weakness is unclear. In non-muscle cells, SMase activates the Nox2 isoform of NADPH oxidase, which requires the p47phox subunit for enzyme function. We targeted p47phox genetically and pharmacologically (apocynin) to examine the role of NADPH oxidase on SMase-induced increase in oxidants and diaphragm weakness. SMase increased cytosolic oxidants (arbitrary units: control 203±15, SMase 276±22; P < 0.05) and depressed maximal force in wild type mice (N/cm2: control 20±1, SMase 16±0.6; P < 0.05). However, p47phox deficient mice were protected from increased oxidants (arbitrary units: control 217±27, SMase 224±17) and loss of force elicited by SMase (N/cm2: control 20±1, SMase 19±1). Apocynin appeared to partially prevent the decrease in force caused by SMase (n = 3 mice/group). Thus, our study suggests that NADPH oxidase plays an important role on oxidant-mediated diaphragm weakness triggered by SMase. These observations provide further evidence that NADPH oxidase modulates skeletal muscle function.
Keywords: sphingolipids, force, Nox
Introduction
Diaphragm weakness is a fundamental problem in disease states such as pulmonary hypertension, heart failure, cancer, chronic obstructive pulmonary disease, and sepsis. Generally, weakness is dictated by muscle atrophy and impairments in contractile function. Contractile dysfunction, characterized by decreased force normalized for muscle cross sectional area (or specific force), can occur rapidly and in many instances ensues before the development of atrophy (Reid and Moylan, 2011). The enzyme sphingomyelinase (SMase) has been proposed as a novel component of the pathway leading to contractile dysfunction in inflammatory states seen in the diseases mentioned above (Reid and Moylan, 2011). Accordingly, we have shown that SMase diminishes specific force through increases in diaphragm oxidants (Ferreira et al., 2010; Ferreira et al., 2012). Of public health relevance, SMase activity is increased in chronic heart failure and sepsis (Claus et al., 2005; Doehner et al., 2007; Empinado et al., 2014).
SMase is a phosphodiesterase that generates phosphorylcholine and ceramide. Ceramide acts as second messenger molecule through direct interaction with proteins or after degradation to downstream sphingolipids (Gulbins and Li, 2006; Hannun and Obeid, 2008). Although the intermediate steps are unknown, it has become clear that oxidants mediate diaphragm weakness elicited by SMase and ceramide (Ferreira et al., 2010; Ferreira et al., 2012). Skeletal muscle possesses several oxidant-generating enzyme systems such as xanthine oxidase, nitric oxide synthases, NADPH oxidases, and mitochondria (Powers and Jackson, 2008). NADPH oxidase and mitochondria are candidate sources of oxidants produced in response to SMase activation (Garcia-Ruiz et al., 1997; Won and Singh, 2006; Zhang et al., 2003). In coronary arteries, NADPH oxidase is responsible for impaired vasodilation caused by SMase. Diaphragm expresses the Nox2 isoform of NADPH oxidase (Javesghani et al., 2002) and recent studies have shown that the enzyme is important for oxidant production during muscle contraction and stretch (Michaelson et al., 2010; Pal et al., 2013; Sakellariou et al., 2013). Based on the emerging role of NADPH oxidase in modulating skeletal muscle function and our preliminary data, we conducted the current study to test the hypotheses that NADPH oxidase mediates the increase in oxidants and decrease in force of skeletal muscle exposed to SMase.
Methods
Animals
We studied male C57BL6 and p47phox deficient mice from Taconic labs (B6.129S2-Ncf1tm1Shl). We used C57BL6 mice as recommended by the vendor as genetic control for p47phox(−/−) mice, which was originally developed on a B6.129 background strain (Jackson et al., 1995). Importantly, diaphragm contractile properties of B6.129 mice have been tested as part of a separate study in our laboratory (data not shown) and were similar to those shown here for C57BL6 mice. Mice were housed at the University of Florida in a specific pathogen free facility under a 12:12 hr light:dark cycle and had free access to food and water. All procedures conformed to the guiding principles for use and care of laboratory animals of the American Physiological Society and were approved by the Institute of Animal Care and Use Committee of the University of Florida.
NADPH oxidase is activated by phosphorylation of the p47phox subunit, which organizes cytosolic components and promotes assembly of the enzyme complex for oxidant production (Bedard and Krause, 2007). Accordingly, cellular responses mediated by the Nox2 isoform of NADPH oxidase are absent in neutrophils, macrophages, and skeletal muscle of p47phox(−/−) mice (Huang et al., 2000; Jackson et al., 1995; Pal et al., 2013). The lack of oxidative burst in neutrophils and macrophages renders p47phox(−/−) mice immunocompromised and increases susceptibility to infection (Huang et al., 2000). Accordingly, these mice are a model of chronic granulomatous disease in humans. None of the animals studied showed phenotypical signs of chronic granulomatous disease (CGD). We confirmed the deficiency of p47phox in the diaphragm of all mice used in this study via Western blot (see example below).
Reagents and Solutions
Glycerol (<0.05% v/v), Apocynin (NADPH oxidase inhibitor; 1 mM), Diphenylene iodonium (DPI), and SMase (0.5 U/ml) were from Sigma-Aldrich. Apocynin and DPI were prepared in dimethylsulfoxide (DMSO). The final amount of DMSO in the tissue bath was less than 1% (v/v) in all experiments. The experimental buffer solution for all experiments was composed of (in mM) 137 NaCl, 5 KCl, 1 MgSO4, 1 NaH2PO4, 2 CaCl2, and 24 NaHCO3 and bubbled with 95%O2 (PO2 ~700 mmHg) and 5% CO2 (PCO2 ~25 mmHg) to maintain approximate pH 7.4.
Diaphragm oxidants
We employed a protocol consistent with our recent studies (Ferreira et al., 2010; Ferreira et al., 2012). Briefly, we used 2′,7′-dichlorofluorescein diacetate (DCFH-DA; Molecular Probes, Eugene, OR) to measure oxidants. We loaded diaphragm bundles with DCFH-DA (20 μM) for 15 min at 37°C, then added vehicle or SMase (0.5 U/ml) to the bath. After 15 min for in vitro exposure to vehicle or SMase, we measured fluorescence of the oxidized derivative (DCF) in diaphragm bundles (480 nm excitation, 520 nm emissions; exposure 10 ms; area 0.60 mm2) using an epifluorescence microscope (Zeiss Axio Observer.A1; Zeiss Microscopy, Jena, Germany) connected to an AxioCam MRm camera (Zeiss Microscopy) and a computer-controlled shutter driver (Uniblitz® VCM D-1; Vincent Associates, Rochester, NY) in the excitation light pathway. We quantified DCF fluorescence with Zen Pro software (Zeiss microscopy).
Western Blots
We homogenized diaphragm bundles on ice using 2× lysis buffer (20 mM Hepes; pH 7.4, 2 mM EGTA, 1% Triton-X100, 50% Glycerol, 50 mM β-Glycerophosphate, 1× Protease Inhibitor Cocktail, 1× Phosphatase Inhibitor Cocktail) and then diluted 1:1 in 2× sample loading buffer (186 mM Tris, pH 7.5, 350 mM DTT, 75% glycerol, 6% SDS, and 0.03% bromphenol blue). We loaded mouse diaphragm samples and a commercial macrophage lysate (ML-8741, ECM Biosciences, Versailles, KY) into 4–20% SDS-polyacrylamide gels, run at 200 V for ~50 min at room temperature (Criterion TGX stain-free gels; Bio-Rad Laboratories, Hercules, CA). The gels were activated and scanned for UV-induced fluorescence to determine total protein in each lane (Gel Doc™ EZ System, Bio-Rad Laboratories), and subsequently proteins were transferred to a nitrocellulose membrane at 100 mA overnight at 4°C. We blocked the membrane for 1 h at room temperature using LI-COR blocking buffer (LI-COR, Lincoln, NE), followed by incubation in p47phox (Sigma) and phospho(S370)-p47phox (Assay Biotech) antibody at 1:1000, and secondary fluorescent antibodies (IRDye, LI-COR) at 1:10,000 for ~45 minutes at room temperature. To test for potential differences in abundance of select antioxidant enzymes, we blocked the membrane for 1 hr, incubated in antibody against superoxide dismutase isoform 1 and 2 (SOD1, and SOD2,) and catalase (Ab) for 3 days at 4°C, followed by secondary fluorescent antibody at 1:20,000 for 1 hr at room temperature. We imaged the membranes using Odyssey Infrared Scanner (LI-COR) and quantified the signal using Image Studio Lite (LI-COR). The integrated intensity from infrared imaging was normalized to total protein for each lane to compare protein abundances.
Diaphragm Bundle Preparation
We anesthetized mice using isoflurane (5%, induction; 3% maintenance), excised the diaphragm, and placed immediately in buffer solution (see above) for dissection. We cut a costal diaphragm bundle maintaining a segment of the rib and central tendon for attachment to the muscle mechanics apparatus (Aurora Scientific, 300C L-R model). The rib was tied onto a metal pin located on a glass rod, while the central tendon was attached to the force transducer with 4.0 silk suture. We adjusted bundle length to achieve the highest twitch force (optimal length, l0) and allowed 15 min for equilibration at 37oC. We then added SMase to the bath and waited 45–60 min to start the isometric force-frequency protocol. In experiments with apocynin or DPI, we added the inhibitor after the thermo-equilibration period and waited another 15 min before exposing the muscle to SMase or vehicle. The protocol consisted of maximal electrical stimulations (current 600 mA, pulse frequency 1–300 Hz, pulse duration 0.25 ms, train duration 300 ms) delivered by a high-power biphasic stimulator (701C, Aurora Scientific Inc.) at 1 min intervals. At the end of the protocol, we measured peak force for each stimulus, and bundle length and weight to estimate cross-sectional area. Force-frequency data were fit by the Hill equation (Ferreira et al., 2011).
Statistical analysis
All statistical tests were performed using Prism v6.0 (GraphPad Inc, La Jolla, CA). Where appropriate, comparisons were made using either t-test or Wilcoxon rank sum, and one-way ANOVA standard or repeated measures with Bonferroni’s test for post-hoc analysis. We accepted statistical significance when P < 0.05. Data are show as mean ± SE.
Results
Phosphorylation of serine residues in p47phox is known to activate NADPH oxidase (Dang et al., 2006; El-Benna et al., 2009). Using a phosphorylation site-specific antibody, we found that phospho-to-total p47phox ratio was increased by 27 ± 7% after SMase exposure (Fig. 1).
Figure 1. SMase increases phosphorylation of p47phox in diaphragm bundles.

Diaphragm bundles were exposed to SMase (0.5 U/ml) for 45 min. Optical density of phosphorylated p47phox (serine 370) was divided by the total p47phox signal. Data are shown as relative to mean of vehicle group. (+) control, commercial lysate from mouse macrophages treated with lipopolysaccharide for 18 hrs. * P < 0.05 (n = 4 mice/group)
Regarding oxidants, SMase increased DCF fluorescence in WT diaphragm bundles by ~36% and genetic deficiency of p47phox, as confirmed by Western Blot, prevented the increase in oxidants (Fig. 2A). These effects occurred in the presence of diminished abundance of catalase and SOD1, but no change in SOD2 in diaphragm from p47phox(−/−) mice (Fig. 2B). SMase depressed submaximal diaphragm force (Fig. 3) and increased the frequency that elicits 50% maximal force (rightward shift of force-frequency relationship), without changing twitch characteristics (Table 1). SMase decreased the maximal rate of force development during twitch, but this effect was not present when the data were normalized to peak twitch force (Table 1). In general, diaphragm bundles from p47phox(−/−) mice were protected from the impaired contractile function caused by SMase in WT (Table 1, and Fig. 3). This protection occurred at stimulus frequencies ranging from twitch to maximal tetanic contraction.
Figure 2. SMase-induced increase in diaphragm oxidants is prevented in p47phox deficient mice.

Image illustrates expression of p47phox (~44 kDa; see Fig. 1) in whole-diaphragm lysates of wild type (WT) mice and p47phox deficient mice. Data are mean ± SE of arbitrary units of dichlorofluorescein (DCF) fluorescence in diaphragms bundles (n = 5–7 mice/group). B) Image illustrates expression of superoxide dismutase isoform 1 (SOD1; cytosolic, Cu/Zn-dependent) and isoform 2 (SOD2; mitochondrial, Mn-dependent), and catalase in whole-diaphragm lysates. We used total protein (whole lane) for normalization of western blot data. The protein gel band corresponding to ~40kDa is shown as representative of relative protein content for each lane. Catalase and SOD1 blots are normalized to same protein gel. SOD2 is normalized to a different protein gel with similar loading. Data are mean ± SE of western blot signal normalized to total protein and expressed relative to mean of WT group (fold change). * P < 0.05
Figure 3. p47phox deficient mice are protected against SMase-induced decrease in diaphragm force.

Specific force was measured after 60 min exposure to vehicle or SMase (0.5 U/ml) in wild type (WT, C57BL6; n = 8 mice/group) and p47phox deficient mice (n = 4 vehicle; 7 SMase). Lines are best fit of mean data from WT vehicle (---) and SMase (—) using Hill equation (Ferreira et al., 2009). *P < 0.05 for WT vehicle vs. WT SMase, σ P < 0.05 for p47phox(−/−) SMase vs. WT SMase.
Table 1.
Diaphragm twitch and tetanic force-frequency characteristics.
| WT | p47phox(−/−) | |||
|---|---|---|---|---|
|
| ||||
| vehicle | SMase | vehicle | SMase | |
| TPT (ms) | 15.9 ± 1.1 | 15.5 ± 0.8 | 16.3 ± 1.0 | 15.3 ± 0.5 |
| ½ RT (ms) | 13.0 ± 0.8 | 12.6 ± 0.5 | 13.5 ± 1.0 | 13.3 ± 0.5 |
| Max +dP/dt (N/cm2·s) | 284 ± 55 | 227 ± 38* | 315 ± 34 | 271 ± 48 |
| (+dP/dt)/Pt (s−1) | 67 ± 2 | 68 ± 1 | 66 ± 2 | 67 ± 1 |
| F50 (Hz) | 52 ± 2 | 60 ± 2* | 52 ± 3 | 57 ± 2 |
| nH | 3.52 ± 0.13 | 3.89 ± 0.09 | 3.30 ± 0.09 | 3.95 ± 0.17† |
TPT, time to peak tension; ½ RT, on-half relaxation time; maximal rate of specific force development during twitch; Pt, peak twitch force F50, stimulus frequency that elicits 50% maximal force; nH, Hill coefficient (~ slope of force-frequency relationship). Pt force is shown in Fig. 3. Data are mean ± SE. For WT, n = 8 mice per group. For p47phox(−/−), n = 4 (vehicle) and n = 7 mice (SMase).
P < 0.05 vs. WT vehicle;
P < 0.05 vs. p47phox(−/−) vehicle
To further examine the role of NADPH oxidase, we used the pharmacological inhibitors DPI and apocynin. DPI caused a dose-dependent decrease in diaphragm force that rendered the compound invalid for our studies (data not shown). In our preparation, apocynin increased DCF fluorescence in cell- and tissue-free experiments (not shown). Thus, we were not able to test whether apocynin prevented the increase in cytosolic oxidants elicited by SMase. Nonetheless, diaphragm function experiments in a subset of animals (n = 3 mice/group) suggest that apocynin partially protected the diaphragm from the decrease in force elicited by SMase (Fig. 4). Apocynin did not alter SMase effects on force at frequencies ≤ 50 Hz, but blunted the decrease in force for frequencies ranging from 80 to 300 Hz.
Figure 4. Apocynin blunts the decrease in diaphragm force elicited by SMase.
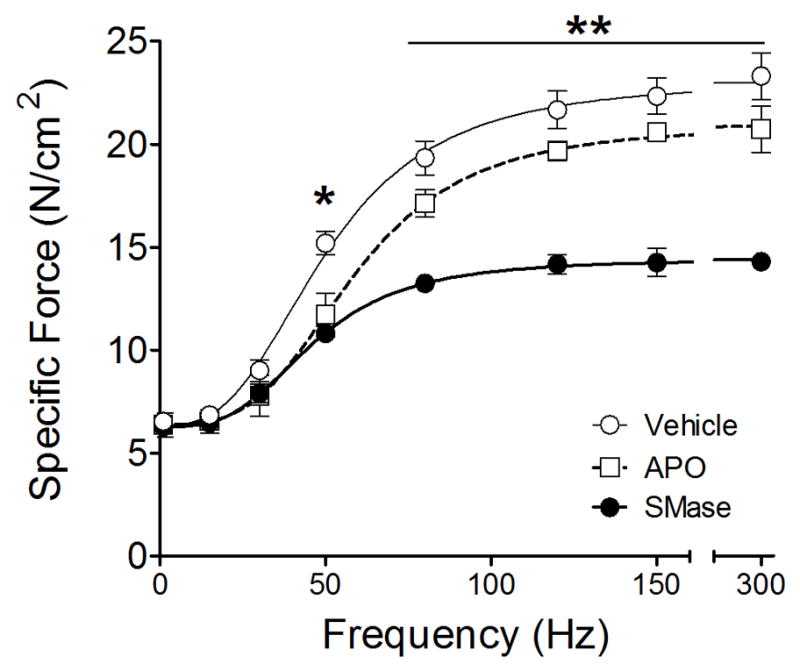
Diaphragm bundles were exposed to vehicle (control; n = 3 mice/group), SMase (0.5 U/ml, 60 min; n = 3 mice/group), and apocynin (APO, 1 mM, 15 min; SMase, 45 min; n = 3 mice/group). Lines represent best fit of mean data using Hill equation (Ferreira et al., 2009). * P < 0.05 for vehicle vs. SMase or APO. ** P < 0.05 SMase vs. APO or vehicle.
Discussion
Our main finding was that diaphragms from p47phox(−/−) mice were protected from increased cytosolic oxidants and depression of force induced by exogenous SMase. Apocynin appeared to exert partial protection of weakness caused by SMase. Altogether, our data suggest that p47phox-dependent NADPH oxidase is responsible for diaphragm dysfunction elicited by SMase.
There are several isoforms and site of action for SMases (Clarke et al., 2011; Marchesini and Hannun, 2004). Putative mediators of diaphragm dysfunction in vivo include the secretory (acid) and neutral isoforms of SMase (Reid and Moylan, 2011). Secretory SMase acts on the outer leaflet on the cell membrane, while neutral SMase exerts its effects on the inner leaflet (Hannun and Obeid, 2008). Therefore, the signaling events elicited by each isoform could be different. In strict terms, our experiments with exogenous SMase would mimic the effects of the secretory isoform. However, ceramide generated on the outer leaflet can flip-flop onto the inner leaflet and serve as a signaling molecule (Contreras et al., 2003). In this setting, the cellular events would be similar to those triggered by neutral SMase. Indeed, recombinant exogenous SMase has been widely used to mimic neutral SMase activation in cells, e.g., see (Giltiay et al., 2005). Therefore, we expect that our experiments will reflect the effects of both secretory and neutral SMases.
Skeletal muscle cells express the Nox4 and Nox2 isoforms (catalytic cores) of NADPH oxidase that are constitutively complexed with p22phox (Javesghani et al., 2002; Sakellariou et al., 2013; Sun et al., 2011). Both isoforms display post-translational regulation of activity (Lassegue et al., 2012) and may contribute to SMase effects on muscle. Activation of Nox2 requires phosphorylation of p47phox (El-Benna et al., 2009; Johnson et al., 1998; Lassegue et al., 2012). SMase activates several kinases known to phosphorylate p47phox such as p38MAPK and PKCζ (Dang et al., 2001; Dang et al., 2006). In our hands, SMase did not activate (phosphorylate) p38MAPK, while we were not able to reliably detect activated (phospho) PKCζ in diaphragm homogenates (data not shown). However, we found that SMase increased phosphorylation levels of p47phox at serine 370. This increase in p47phox phosphorylation status at serine 370 may seem modest (~27%), but should be physiologically relevant. In the inactive state, the auto-inhibitory region of p47phox prevents interaction of the protein with other subunits. Phosphorylation of serine residues in the auto-inhibitory region (aminoacids 303 to 340) leads to conformational changes in p47phox that allows interaction with other subunits and activation of NADPH oxidase. However, phosphorylation of serine residues outside the auto-inhibitory region (e.g., S359 and S370) are also important for NADPH oxidase activation as they modulate the interaction with p67phox and formation of the functional enzyme complex. In leukocytes, phosphorylation of p47phox at serine 370 is an early and required step for NADPH oxidase activation and superoxide production (Johnson et al., 1998). Thus, our findings suggest that SMase activates NADPH oxidase via phosphorylation of p47phox in diaphragm. The role of p47phox and NADPH oxidase were further examined using measurements of oxidants and contractile function ex vivo.
In agreement with our previous findings (Ferreira et al., 2010; Ferreira et al., 2012), SMase increased cytosolic oxidants in the diaphragm. The SMase exposure time may explain the differences in magnitude of change in cytosolic oxidants for the current study (15 min exposure, ~50% increase) compared to our previous studies (30 min exposure, ~100% increase; (Ferreira et al., 2010; Ferreira et al., 2012). This was established by measuring DCF fluorescence, which is a non-specific oxidant probe. Thus, we cannot determine with certainty which reactive species were increased by SMase. Our novel observation is that the increase in cytosolic oxidants was absent in diaphragms of mice deficient in p47phox, which suggests a role for NADPH oxidase. Given our findings in p47phox deficient mice, it was important to examine adaptations in select antioxidant enzymes that confound data interpretation. To our surprise, protein abundance of catalase and SOD1 (cytosolic, Cu/Zn-dependent) were decreased in p47phox(−/−) whereas SOD2 (mitochondrial, Mn-dependent) was unchanged. This suggests that oxidants derived from NADPH oxidase regulate antioxidant enzyme expression in skeletal muscle. It may also explain the similar basal levels of cytosolic oxidants in p47phox(−/−) and WT mice measured in our study and in the study by Pal et al. (2013). Overall, our data supports the recent emergence of NADPH oxidase as an important source of oxidants in skeletal muscle during “stress” conditions (Pal et al., 2013; Sakellariou et al., 2013; Semprun-Prieto et al., 2011).
The traditional view of p47phox-dependent Nox2 is that the enzyme is localized in the cell membrane (Bedard and Krause, 2007), clustering within t-tubules in skeletal muscle (Hidalgo et al., 2006; Pal et al., 2013; Sakellariou et al., 2013), and generates superoxide in the extracellular space. In this scenario, we propose two mechanisms for NADPH oxidase to increase cytosolic oxidants. 1) The presence of extracellular superoxide dismutase (SOD) that rapidly converts superoxide to hydrogen peroxide (Fattman et al., 2003), which can cross the cell membrane and reach the cytosol. 2) The existence of NADPH oxidases in membranes of intracellular compartments in muscle cells [e.g., mitochondria and sarcoplasmic reticulum (Case et al., 2013; Sakellariou et al., 2013; Xia et al., 2003)], which would generate superoxide in the cytosol. Consistent with our findings, other investigators have also measured increases in cytosolic oxidants by DCF fluorescence due to activation of NADPH oxidase in skeletal muscle (Espinosa et al., 2009; Pal et al., 2013; Sakellariou et al., 2013). The superoxide or hydrogen peroxide generated by NADPH oxidases can directly increase DCF fluorescence or be rapidly converted to hydrogen peroxide, peroxynitrite, or hydroxyl radicals that also elevate DCF fluorescence. Excess peroxynitrite (Supinski et al., 1999), hydroxyl radicals (Callahan et al., 2001), and hydrogen peroxide (Andrade et al., 1998) decrease maximal tetanic force. Thus, we explored the possibility that NADPH oxidase mediates diaphragm weakness stimulated by SMase.
The depression of force that we observed in diaphragm bundles exposed to SMase was of similar magnitude to that reported in our previous studies (Ferreira et al., 2010; Ferreira et al., 2012). This weakness originates from dysfunction in sarcomeric proteins (Ferreira et al., 2012), and it may also involve effects of oxidants on calcium release (Hidalgo et al., 2006; Wehrens et al., 2005). SMase decreased the rate of twitch force development assessed in absolute values (N/cm2·s), but not the effects were not present when the data were normalized to peak twitch force (in N/cm2). These findings suggest that the loss of force occurs mainly by a decrease in number of force-generating cross-bridges and/or force generated per cross-bridge. Importantly, muscles from mice deficient in the p47phox subunit of NADPH oxidase were protected from SMase-induced decrease in force. This shows that SMase-induced muscle weakness requires p47phox, presumably to increase muscle oxidants that are known to impair contractile function. We also tested the protective effects of apocynin for its ability to inhibit NADPH oxidase as secondary studies to confirm our observations in knockout animals. In a subset of animals (n = 3), apocynin blunted the decrease in muscle force stimulated by SMase. The partial protection with apocynin may reflect the small number of animals or incomplete inhibition of NADPH oxidase. It is important to note that apocynin can exert antioxidant effects independent of NADPH oxidase (Heumuller et al., 2008). Our measurements of muscle force were performed 45 (apocynin vs. vehicle) or 60 min after exposure to SMase (p47phox(−/−) vs. wild type). Based on the relatively similar decrease in maximal force 45 and 60 min after exposure to SMase (Ferreira et al., 2010), we consider that the difference in time would have minimal or no impact on the final outcome of our experiments. Overall, our studies using p47phox deficient mice and apocynin support the hypothesis that SMase causes diaphragm weakness via activation of NADPH oxidase.
In our previous study, the increase in oxidants and decrease in muscle force triggered by SMase were also prevented by a mitochondria-targeted antioxidant (Ferreira et al., 2012). This would appear to contradict the current data. However, oxidant production by NADPH oxidase and mitochondria are not mutually exclusive events. A cross-talk between reactive oxygen species produced by NADPH oxidase and mitochondria has been reported in several cell types (Dikalov, 2011; Doughan et al., 2008). For instance, angiotensin II infusion in mice promotes activation of NADPH oxidase and leads to mitochondrial dysfunction in limb muscle (Inoue et al., 2012; Semprun-Prieto et al., 2011).
Our experimental design has advantages and disadvantages. The main advantages of our preparation are specificity of response to SMase, determination of cause-and-effect relationship, and high throughput for screening cell signaling pathways and physiological mechanisms of dysfunction. Therefore, our results should be seen as proof-of-concept and an initial and important step toward more targeted testing under physiologically-relevant conditions. Some of the limitations of our preparation are inherent of in vitro studies in muscle tissue (Ferreira et al., 2010; Ferreira et al., 2012). We used a solution with high pressure of O2 (~700 mmHg). This could cause greater production of oxidants than under more a physiological diaphragm PO2 in vivo (~30–50 mmHg) (Poole et al., 1995). Our preparation also requires high exogenous SMase activity to compensate for the decreased cell surface area exposed to the enzyme compared to in vivo (Ferreira et al., 2010). Moreover, we used a non-specific probe to measure oxidants and cannot determine precisely which oxidant moiety was increased by SMase. Future, more extensive, experiments with a variety of inhibitors and probes, e.g., (Sakellariou et al., 2013) could help better define the specific oxidant species being produced upon SMase exposure.
Conclusion
To sum up, we employed an in vitro approach to mimic the effects of endogenous SMase activation in skeletal muscle and investigate the role of NADPH oxidase. In this setting, the p47phox subunit of NADPH oxidase stood out as being required for SMase to increased oxidants and decrease force in the diaphragm. Our findings emphasize the importance of NADPH oxidase in signaling diaphragm dysfunction in conditions where SMase activity is increased. For instance, activation of p47phox –dependent NADPH oxidase may have a role in diaphragm dysfunction in sepsis and heart failure as SMase is activated therein (Claus et al., 2005; Doehner et al., 2007; Empinado et al., 2014).
Highlights.
Sphingomyelinase increases phosphorylation of p47phox in diaphragm muscle
p47phox knockout prevents sphingomyelinase-induced increases in diaphragm oxidants
p47phox knockout prevents diaphragm weakness elicited by sphingomyelinase
Acknowledgments
We would like to thank Nikhil Patel, Victoria Williams, and Hyacinth Empinado for technical assistance with tissue processing and dissections. L. F. Ferreira was supported, in part, by grants from NIH (R00-HL098453-01) and AHA (13GRNT17160000).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andrade FH, Reid MB, Allen DG, Westerblad H. Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J Physiol. 1998;509 (Pt 2):565–575. doi: 10.1111/j.1469-7793.1998.565bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Callahan LA, She ZW, Nosek TM. Superoxide, hydroxyl radical, and hydrogen peroxide effects on single-diaphragm fiber contractile apparatus. J Appl Physiol. 2001;90:45–54. doi: 10.1152/jappl.2001.90.1.45. [DOI] [PubMed] [Google Scholar]
- Case AJ, Li S, Basu U, Tian J, Zimmerman MC. Mitochondrial-localized NADPH oxidase 4 is a source of superoxide in angiotensin II-stimulated neurons. Am J Physiol Heart Circ Physiol. 2013;305:H19–28. doi: 10.1152/ajpheart.00974.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke CJ, Wu BX, Hannun YA. The neutral sphingomyelinase family: identifying biochemical connections. Adv Enzyme Regul. 2011;51:51–58. doi: 10.1016/j.advenzreg.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claus RA, Bunck AC, Bockmeyer CL, Brunkhorst FM, Losche W, Kinscherf R, Deigner HP. Role of increased sphingomyelinase activity in apoptosis and organ failure of patients with severe sepsis. FASEB J. 2005;19:1719–1721. doi: 10.1096/fj.04-2842fje. [DOI] [PubMed] [Google Scholar]
- Contreras FX, Villar AV, Alonso A, Kolesnick RN, Goni FM. Sphingomyelinase activity causes transbilayer lipid translocation in model and cell membranes. J Biol Chem. 2003;278:37169–37174. doi: 10.1074/jbc.M303206200. [DOI] [PubMed] [Google Scholar]
- Dang PM, Fontayne A, Hakim J, El Benna J, Perianin A. Protein kinase C zeta phosphorylates a subset of selective sites of the NADPH oxidase component p47phox and participates in formyl peptide-mediated neutrophil respiratory burst. J Immunol. 2001;166:1206–1213. doi: 10.4049/jimmunol.166.2.1206. [DOI] [PubMed] [Google Scholar]
- Dang PM, Stensballe A, Boussetta T, Raad H, Dewas C, Kroviarski Y, Hayem G, Jensen ON, Gougerot-Pocidalo MA, El-Benna J. A specific p47phox -serine phosphorylated by convergent MAPKs mediates neutrophil NADPH oxidase priming at inflammatory sites. J Clin Invest. 2006;116:2033–2043. doi: 10.1172/JCI27544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free Radic Biol Med. 2011;51:1289–1301. doi: 10.1016/j.freeradbiomed.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doehner W, Bunck AC, Rauchhaus M, von Haehling S, Brunkhorst FM, Cicoira M, Tschope C, Ponikowski P, Claus RA, Anker SD. Secretory sphingomyelinase is upregulated in chronic heart failure: a second messenger system of immune activation relates to body composition, muscular functional capacity, and peripheral blood flow. Eur Heart J. 2007;28:821–828. doi: 10.1093/eurheartj/ehl541. [DOI] [PubMed] [Google Scholar]
- Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. 2008;102:488–496. doi: 10.1161/CIRCRESAHA.107.162800. [DOI] [PubMed] [Google Scholar]
- El-Benna J, Dang PM, Gougerot-Pocidalo MA, Marie JC, Braut-Boucher F. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: structure, phosphorylation and implication in diseases. Exp Mol Med. 2009;41:217–225. doi: 10.3858/emm.2009.41.4.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Empinado HM, Deevska GM, Nikolova-Karakashian M, Yoo JK, Christou DD, Ferreira LF. Diaphragm dysfunction in heart failure is accompanied by increases in neutral sphingomyelinase activity and ceramide content. Eur J Heart Fail. 2014;16:519–525. doi: 10.1002/ejhf.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa A, Garcia A, Hartel S, Hidalgo C, Jaimovich E. NADPH oxidase and hydrogen peroxide mediate insulin-induced calcium increase in skeletal muscle cells. J Biol Chem. 2009;284:2568–2575. doi: 10.1074/jbc.M804249200. [DOI] [PubMed] [Google Scholar]
- Fattman CL, Schaefer LM, Oury TD. Extracellular superoxide dismutase in biology and medicine. Free Radic Biol Med. 2003;35:236–256. doi: 10.1016/s0891-5849(03)00275-2. [DOI] [PubMed] [Google Scholar]
- Ferreira LF, Campbell KS, Reid MB. Effectiveness of sulfur-containing antioxidants in delaying skeletal muscle fatigue. Med Sci Sports Exerc. 2011;43:1025–1031. doi: 10.1249/MSS.0b013e3182019a78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira LF, Gilliam LA, Reid MB. L-2-oxothiazolidine-4-carboxylate reverses glutathione oxidation and delays fatigue of skeletal muscle in vitro. J Appl Physiol. 2009;107:211–216. doi: 10.1152/japplphysiol.00001.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira LF, Moylan JS, Gilliam LA, Smith JD, Nikolova-Karakashian M, Reid MB. Sphingomyelinase stimulates oxidant signaling to weaken skeletal muscle and promote fatigue. Am J Physiol Cell Physiol. 2010;299:C552–560. doi: 10.1152/ajpcell.00065.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira LF, Moylan JS, Stasko S, Smith JD, Campbell KS, Reid MB. Sphingomyelinase depresses force and calcium sensitivity of the contractile apparatus in mouse diaphragm muscle fibers. J Appl Physiol. 2012;112:1538–1545. doi: 10.1152/japplphysiol.01269.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Ruiz C, Colell A, Mari M, Morales A, Fernandez-Checa JC. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione. J Biol Chem. 1997;272:11369–11377. doi: 10.1074/jbc.272.17.11369. [DOI] [PubMed] [Google Scholar]
- Giltiay NV, Karakashian AA, Alimov AP, Ligthle S, Nikolova-Karakashian MN. Ceramide- and ERK-dependent pathway for the activation of CCAAT/enhancer binding protein by interleukin-1beta in hepatocytes. J Lipid Res. 2005;46:2497–2505. doi: 10.1194/jlr.M500337-JLR200. [DOI] [PubMed] [Google Scholar]
- Gulbins E, Li PL. Physiological and pathophysiological aspects of ceramide. Am J Physiol Regul Integr Comp Physiol. 2006;290:R11–26. doi: 10.1152/ajpregu.00416.2005. [DOI] [PubMed] [Google Scholar]
- Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- Heumuller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schroder K, Brandes RP. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51:211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- Hidalgo C, Sanchez G, Barrientos G, racena-Parks P. A transverse tubule NADPH oxidase activity stimulates calcium release from isolated triads via ryanodine receptor type 1 S -glutathionylation. J Biol Chem. 2006;281:26473–26482. doi: 10.1074/jbc.M600451200. [DOI] [PubMed] [Google Scholar]
- Huang CK, Zhan L, Hannigan MO, Ai Y, Leto TL. P47(phox)-deficient NADPH oxidase defect in neutrophils of diabetic mouse strains, C57BL/6J-m db/db and db/+ J Leukoc Biol. 2000;67:210–215. doi: 10.1002/jlb.67.2.210. [DOI] [PubMed] [Google Scholar]
- Inoue N, Kinugawa S, Suga T, Yokota T, Hirabayashi K, Kuroda S, Okita K, Tsutsui H. Angiotensin II-induced reduction in exercise capacity is associated with increased oxidative stress in skeletal muscle. Am J Physiol Heart Circ Physiol. 2012;302:H1202–1210. doi: 10.1152/ajpheart.00534.2011. [DOI] [PubMed] [Google Scholar]
- Jackson SH, Gallin JI, Holland SM. The p47phox mouse knock-out model of chronic granulomatous disease. J Exp Med. 1995;182:751–758. doi: 10.1084/jem.182.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javesghani D, Magder SA, Barreiro E, Quinn MT, Hussain SN. Molecular characterization of a superoxide-generating NAD(P)H oxidase in the ventilatory muscles. Am J Respir Crit Care Med. 2002;165:412–418. doi: 10.1164/ajrccm.165.3.2103028. [DOI] [PubMed] [Google Scholar]
- Johnson JL, Park JW, Benna JE, Faust LP, Inanami O, Babior BM. Activation of p47(PHOX), a cytosolic subunit of the leukocyte NADPH oxidase. Phosphorylation of ser-359 or ser-370 precedes phosphorylation at other sites and is required for activity. J Biol Chem. 1998;273:35147–35152. doi: 10.1074/jbc.273.52.35147. [DOI] [PubMed] [Google Scholar]
- Lassegue B, San Martin A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res. 2012;110:1364–1390. doi: 10.1161/CIRCRESAHA.111.243972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesini N, Hannun YA. Acid and neutral sphingomyelinases: roles and mechanisms of regulation. Biochem Cell Biol. 2004;82:27–44. doi: 10.1139/o03-091. [DOI] [PubMed] [Google Scholar]
- Michaelson LP, Shi G, Ward CW, Rodney GG. Mitochondrial redox potential during contraction in single intact muscle fibers. Muscle Nerve. 2010;42:522–529. doi: 10.1002/mus.21724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal R, Basu Thakur P, Li S, Minard C, Rodney GG. Real-time imaging of NADPH oxidase activity in living cells using a novel fluorescent protein reporter. PLoS One. 2013;8:e63989. doi: 10.1371/journal.pone.0063989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole DC, Wagner PD, Wilson DF. Diaphragm microvascular plasma PO2 measured in vivo. J Appl Physiol. 1995;79:2050–2057. doi: 10.1152/jappl.1995.79.6.2050. [DOI] [PubMed] [Google Scholar]
- Powers SK, Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev. 2008;88:1243–1276. doi: 10.1152/physrev.00031.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid MB, Moylan JS. Beyond atrophy: redox mechanisms of muscle dysfunction in chronic inflammatory disease. J Physiol. 2011;589:2171–2179. doi: 10.1113/jphysiol.2010.203356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakellariou GK, Vasilaki A, Palomero J, Kayani A, Zibrik L, McArdle A, Jackson MJ. Studies of mitochondrial and nonmitochondrial sources implicate nicotinamide adenine dinucleotide phosphate oxidase(s) in the increased skeletal muscle superoxide generation that occurs during contractile activity. Antioxid Redox Signal. 2013;18:603–621. doi: 10.1089/ars.2012.4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semprun-Prieto LC, Sukhanov S, Yoshida T, Rezk BM, Gonzalez-Villalobos RA, Vaughn C, Michael Tabony A, Delafontaine P. Angiotensin II induced catabolic effect and muscle atrophy are redox dependent. Biochem Biophys Res Commun. 2011;409:217–221. doi: 10.1016/j.bbrc.2011.04.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun QA, Hess DT, Nogueira L, Yong S, Bowles DE, Eu J, Laurita KR, Meissner G, Stamler JS. Oxygen-coupled redox regulation of the skeletal muscle ryanodine receptor-Ca2+ release channel by NADPH oxidase 4. Proc Natl Acad Sci U S A. 2011;108:16098–16103. doi: 10.1073/pnas.1109546108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supinski G, Stofan D, Callahan LA, Nethery D, Nosek TM, DiMarco A. Peroxynitrite induces contractile dysfunction and lipid peroxidation in the diaphragm. J Appl Physiol. 1999;87:783–791. doi: 10.1152/jappl.1999.87.2.783. [DOI] [PubMed] [Google Scholar]
- Wehrens XH, Lehnart SE, Reiken S, van der Nagel R, Morales R, Sun J, Cheng Z, Deng SX, de Windt LJ, Landry DW, Marks AR. Enhancing calstabin binding to ryanodine receptors improves cardiac and skeletal muscle function in heart failure. Proc Natl Acad Sci U S A. 2005;102:9607–9612. doi: 10.1073/pnas.0500353102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won JS, Singh I. Sphingolipid signaling and redox regulation. Free Radic Biol Med. 2006;40:1875–1888. doi: 10.1016/j.freeradbiomed.2006.01.035. [DOI] [PubMed] [Google Scholar]
- Xia R, Webb JA, Gnall LL, Cutler K, Abramson JJ. Skeletal muscle sarcoplasmic reticulum contains a NADH-dependent oxidase that generates superoxide. Am J Physiol Cell Physiol. 2003;285:C215–C221. doi: 10.1152/ajpcell.00034.2002. [DOI] [PubMed] [Google Scholar]
- Zhang DX, Zou AP, Li PL. Ceramide-induced activation of NADPH oxidase and endothelial dysfunction in small coronary arteries. Am J Physiol Heart Circ Physiol. 2003;284:H605–612. doi: 10.1152/ajpheart.00697.2002. [DOI] [PubMed] [Google Scholar]