

Abstract
The tripeptide N-formyl-Met-Leu-Phe (fMLF) is a potent neutrophil chemoattractant and the reference agonist of the G protein-coupled N-formylpeptide receptor (FPR). As it plays a very important role in host defense and inflammation, there has been considerable interest in the development of fMLF analogues in the hope of identifying potential therapeutic agents. Herein we report the design, synthesis and evaluation of AApeptides that mimic the structure and function of fMLF. The lead AApeptides can effectively induce calcium mobilization and mitogen-activated protein kinase (MAPK) signal transduction pathways in FPR-transfected rat basophilic leukemic (RBL) cells. More intriguingly, at high concentrations, certain AApeptides are even more effective than fMLF in the induction of calcium mobilization. Their agonistic activity is further supported by their ability to stimulate chemotaxis and production of superoxide in HL-60 cells. Similar to fMLF, these AApeptides are much more selective towards FPR1 than FPR2. These results suggest that the fMLF-mimicking AApeptides may emerge to be a new class of therapeutic agents that target FPRs.
Keywords: fMLF, AApeptides, peptidomimetics, chemotaxis, calcium mobilization
INTRODUCTION
Neutrophils are one of the key components of innate immune system [1] as they play an essential role in host defense against bacterial and fungal infections.[2] Their responses can be modulated by the interaction between chemoattractants and cell surface receptors.[3] For instance, peptides bearing N-formyl-methionine residue, either derived from bacteria or mitochondria in eukaryotic cells, are potent chemoattractants of neutrophils.[2b, 4] They bind to a class of G protein-coupled formyl peptide receptors (FPRs) on the membrane of neutrophils, triggering intracellular signaling pathways that enhance the bactericidal activities of phagocytes including the migration of phagocytic cells towards the sites of inflammation as well as superoxide production.[2, 5] Among these peptides, N-formyl-Met-Leu-Phe (fMLF), the smallest formyl peptide that binds to FPR1 with high affinity, has been widely used as the reference agonist to study the GPCR-mediated host defense and biochemical signaling pathways in phagocytes.[5a, 6] Studies indicate that fMLF plays a critical role in the regulation of a variety of physiological functions, and show great promise for the treatment of bacterial infection, HIV, inflammation, and cancer.[7] Due to its biological interest and translational potential, there has been an extensive effort in developing fMLF analogues as potential molecular probes as well as therapeutic agents. [8]
We have recently developed a new class of peptidomimetics termed “AApeptides”, as they are based on N-acylated-N-aminoethyl amino acid residues (Figure 1) [9] derived from chiral PNA backbones. Half of the side chains in an AApeptide are derived from amino acids, and the other half of side chains come from any acylating agents including carboxylic acids,[10] acyl chlorides,[11] and sulfonyl chlorides [10]. As such, the potential for the introduction of chemically diverse functional groups into AApeptides is limitless. According to the relative positions of side chains, AApeptides are classified into two subfamilies: α-AApeptides and γ-AApeptides. Both α-AApeptides and γ-AApeptides have exhibited high resistance against proteolytic degradation. [9a, 9b, 12] By recapturing the structure and function of bioactive peptides, these AApeptides have found a wide variety of applications such as development of antimicrobial agents mimicking host defense peptides, [13] design of Tat and RGD mimetics, [12, 14] and protein/peptide recognition. [11] These endeavors have demonstrated the potential applications of AApeptides in biomedical sciences. Thus, we believe it is of interest to evaluate the ability of AApeptides for their mimicry of fMLF, so as to develop novel therapeutic agents in the future. In this study, a focused library of AApeptides was designed, synthesized and tested for their ability to activate neutrophils through FPRs.
Figure 1.
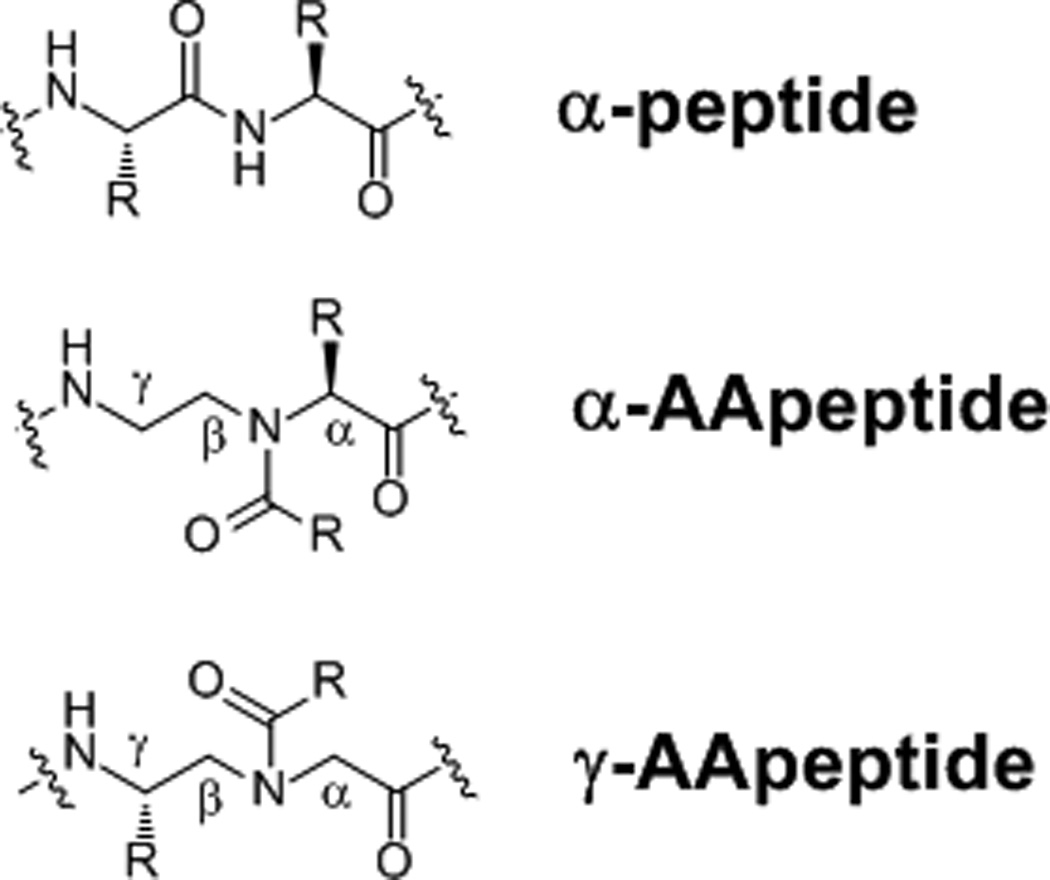
The structure of α-peptides, α-AApeptides and γ-AApeptides. In both α-AApeptides and γ-AApeptides, half of side chains are introduced through acylation, while the other half of side chains are chiral and derived from amino acids.
RESULTS AND DISCUSSION
As shown in Figure 2, AApeptides 1–7 were designed that mimic the prototype formyl peptide fMLF. These sequences contain either an N-formyl methionine or an N-formyl norleucine which is known to be critical for the agonistic activity at FPRs [5a, 8a, 15]. In addition, one AApeptide building block was also included to mimic the leucine and phenylalanine residues in fMLF. Both α-AApeptides (1 and 2) and γ-AApeptides (3–7) were designed, synthesized and tested for their activity at FPRs.
Figure 2.
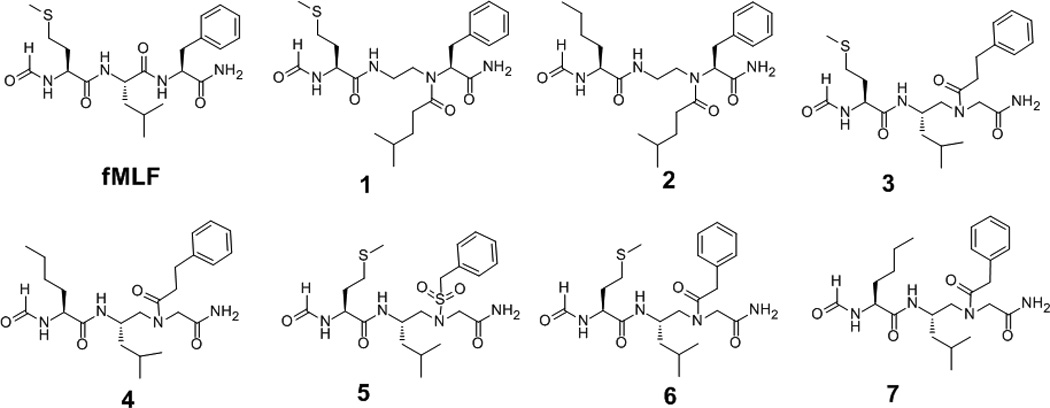
The structures of fMLF and AApeptides.
The synthesis of AApeptides was carried out following our reported procedures on the solid phase (Scheme 1). [9c, 10, 16] In brief, α-AApeptides 1 and 2 were obtained by incorporating the α-AApeptide building block directly in the synthesis. [9a] γ-AApeptides 3–7 were synthesized by using alloc protected γ-AApeptide building blocks.[11, 16b] The formyl group on the N-terminus of methionine and norleucine in the sequences was added following a published protocol. [8a] Briefly, isobutyl chloroformate and formic acid were mixed at 1:1 ratio in DCM, and then cooled down to at 0 °C, to which 1 equiv. NMM (N-methylmorpholine) was added. The resulting solution was added to resin and allowed to react for two hours. The procedure was repeated twice. The AApeptides were obtained by HPLC purification after cleavage from solid support by TFA.
Scheme 1.
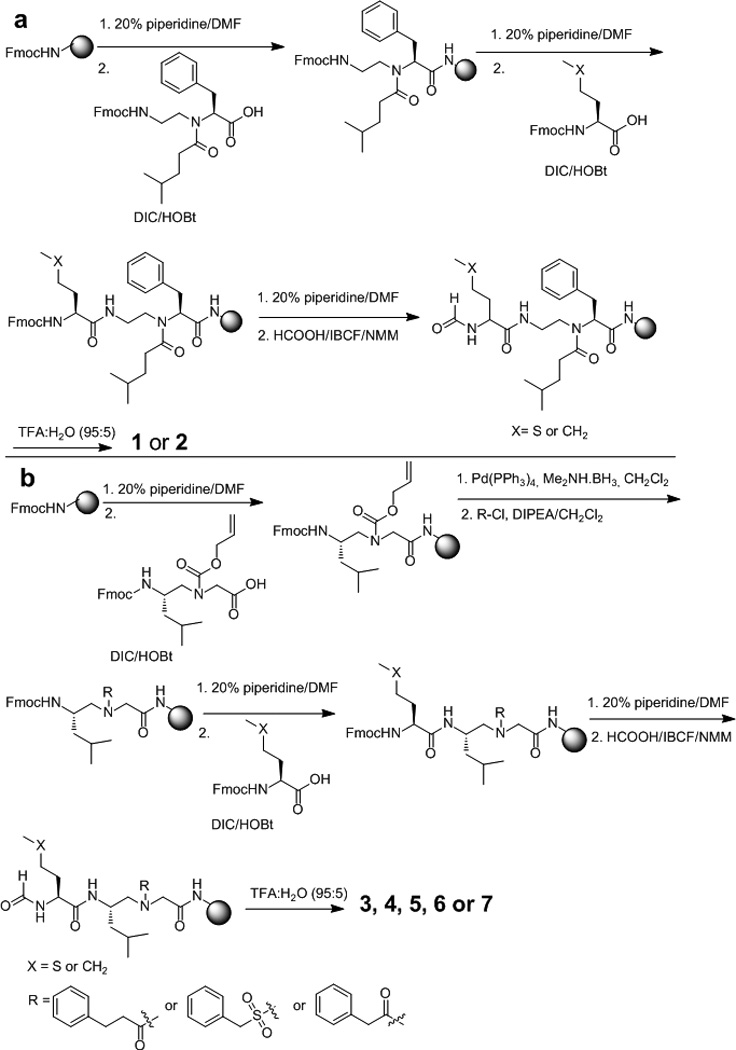
a, synthesis of α-AApeptides 1 and 2; b, synthesis of γ-AApeptides 3–7.
To assess the ability of these peptidomimetics to mimic the function of the fMLF peptide, we first tested their efficacy in the induction of calcium mobilization in human FPR1 transfected RBL-2H3 cells. As shown in Figure 3, fMLF is a potent activator of calcium mobilization, and it takes effect at the concentrations as low as 1 nM. It seems that α-AApeptides cannot mimic fMLF because both 1 and 2 do not exhibit capability to induce calcium mobilization. This may be explained by the relative positions of side chains on α-AApeptides. The aliphatic side chains in 1 and 2 are introduced via acylation, and they are next to the chiral aromatic side chains. They were designed to mimic the Leu-Phe dipeptide residues. However, since these side chains are too close to each other, their spatial relationship is quite different from that of the Leu-Phe dipeptide residues in the model peptide. As a result, they failed to mimic the functions of Leu-Phe effectively and in fact were not active at all. However, a few γ-AApeptides showed good activity in calcium mobilization assays. It is possible that in γ-AApeptides, the spatial orientation of side chains is more similar to regular peptides than in α-AApeptides. Among them, 1, 2, 5 and 6 did not display any activity under tested condition. However, 3, 4 and 7 exhibited dose-dependent function in calcium mobilization assays. 4 became effective at the concentration of 50 µM, while 3 and 7 started to stimulate calcium mobilization at 2 µM. At 50 µM, 3 had efficacy similar to fMLF at 10 µM. Surprisingly, at the concentration of 10 µM, 7 was almost 2-fold as effective as fMLF at the same concentration, although at lower concentrations it was less potent. Although the mechanism is elusive and a more detailed study is needed in the future, this unusual activity-boosting maybe explained by the multivalency theory. It is known that multivalent ligands are much more effective in receptor signaling than monomeric ligands because these ligands increase avidity and induce positive cooperativity when interacting with receptors.[17] We hypothesize that, although 7 is less effective than fMLF at low concentrations, it may aggregate at cell membranes at higher concentrations. Such aggregation, even non-covalent, may lead to multivalent effect, thereby boosting its activity dramatically. The structure-activity relationship of these γ-AApeptides is also somewhat different from peptide-based fMLF mimetics. It is known that replacement of the methionine with norleucine in fMLF does not reduce bioactivity of the peptide;[15b] nevertheless, we observed that 4 is less effective than 3. Interestingly, 6 is not active, but 7 is the most potent molecule identified so far in this group. These results suggest that all three side chains in AApeptides are important for their activity, and any one of them has to have correct orientation for all side chains in order to be active. Since these AApeptides are short and presumably do not have any secondary structures, a small change in one side chain may affect the overall structure dramatically. These findings may also provide insight into the development of more potent fMLF mimetics in the future.
Figure 3.
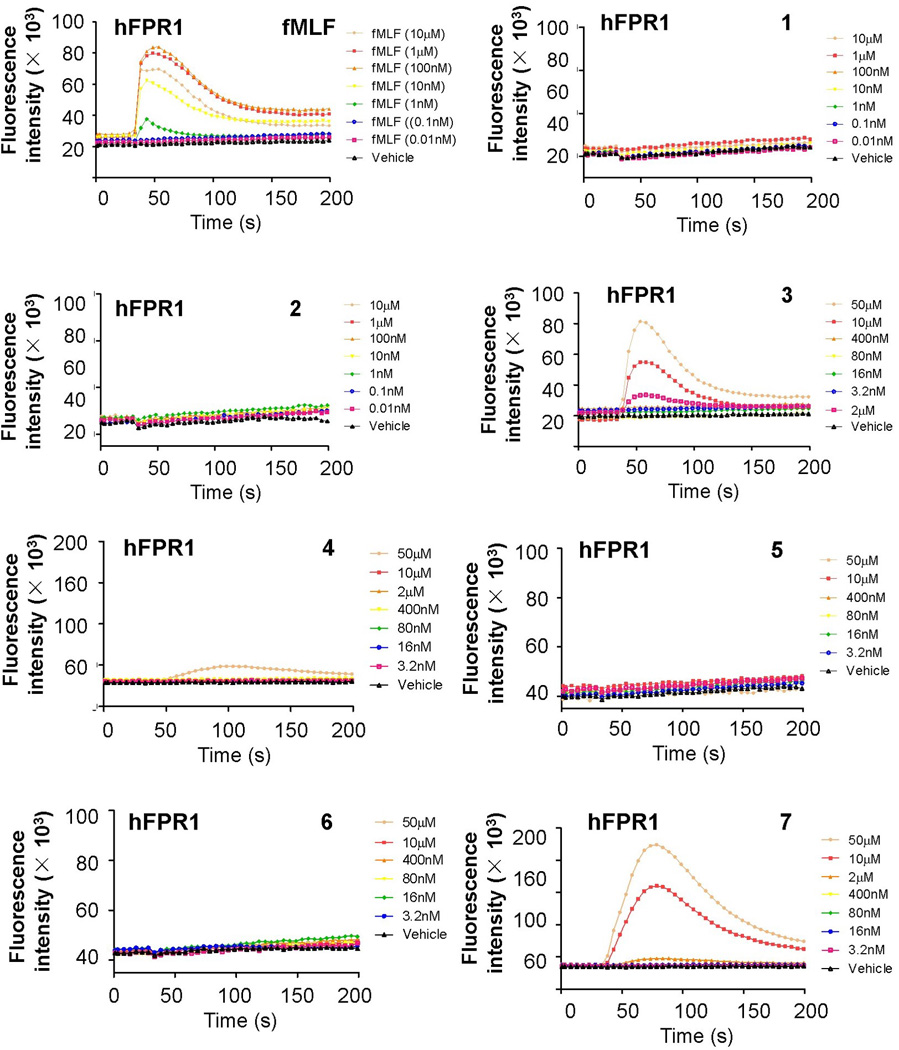
AApeptide-induced activation of calcium mobilization in human FPR1 transfected RBL cells.
The selectivity of these γ-AApeptides was further evaluated by testing their capability in the induction of calcium mobilization in human FPR2-transfected RBL cells. The FPR2-specific synthetic peptide W-peptide (WKYMVM) was used as a positive control.[3] It is known that fMLF is more selective towards FPR1 than FPR2, thus it is interesting to know if fMLF-mimicking γ-AApeptides have the similar selectivity. As shown in Figure 4, W-peptide exhibited potent activity in calcium mobilization assay. Sequences 1, 2, 5 and 6, which were inactive toward FP1, were also inactive toward FPR2 (data not shown). Interestingly, the other three γ-AApeptides 3, 4 and 7, which were active at FPR1, were also active at FPR2. However, they have different preference for the two receptors. While compound 4 showed the weakest activity to induce FPR1-mediated calcium mobilization, it displayed most potent activity towards FPR2. In contrast, both 3 and 7 were more selective for FPR1. At 10 µM, 7 was even more efficacious than fMLF at FPR1, however, it did not have any activity towards FPR2 in calcium mobilization assay at this concentration. This is consistent with previous findings that subtle changes in the short peptide/peptidomimetic sequences can affect the selectivity of compounds towards FPR1 and FPR2. [3]
Figure 4.
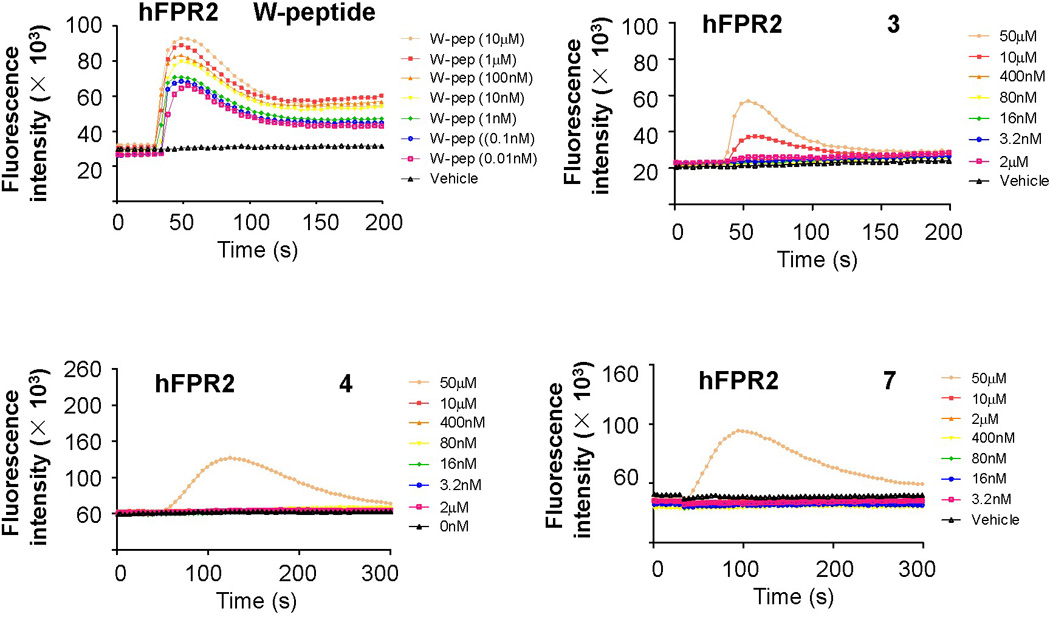
γ-AApeptide-induced activation of calcium mobilization in human FPR2 transfected RBL cells. Data shown are representative of three independent experiments.
In addition to calcium mobilization, it is known that binding of fMLF to FPR1 activates the MAP kinases ERK1 and ERK2 in human neutrophils.[3] To investigate whether the lead AApeptides has the ability to simulate this signaling pathway, RBL cells transfected to express human FPR1 were treated with either fMLF or γ-AApeptides 3, 4 and 7. ERK (including both ERK1 and ERK2) activation was determined using an antibody against the phosphorylated MAP kinases. Figure 5 shows that the ability of γ-AApeptides for the activation of the ERK signaling pathway is highly consistent to their capability to induce calcium mobilization shown in Figure 3.
Figure 5.
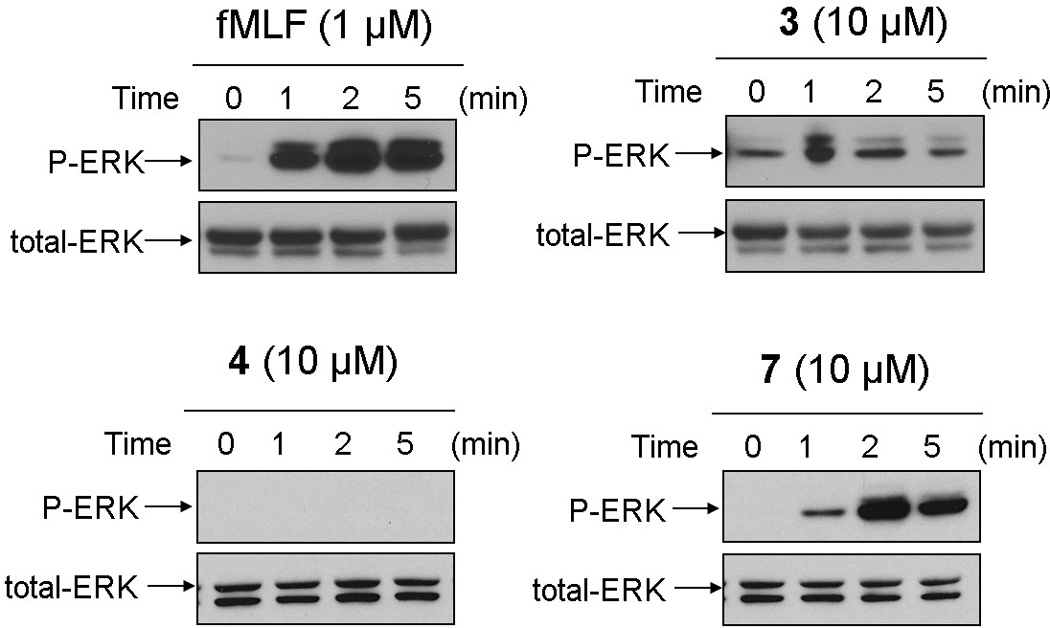
Activation of ERKs by fMLF and γ-AApeptides 3, 4 and 7 in human FPR1-transfected RBL cells. These cells were stimulated with fMLF peptide (1 µM, upper-left), γ-AApeptides 3 (10 µM, upper-right), 4 (10 µM, lower-left) and 7 (10 µM, lower-right), respectively. After various time intervals the cells were harvested and the phosphorylated ERKs were determined by Western blotting with anti-phospho-ERK antibodies. Data shown are representative of three independent experiments.
As shown in Figure 5, cells responded to fMLF with rapid tyrosine phosphorylation of ERK, which was consistently observed in previous studies. [3] At the tested concentration of 10 µM, 4 failed to show any capability to induce ERK phosphorylation. However, following stimulation with either 3 or 7, the rapid and potent response was observed at 1 min and peaked between 2 and 5 min. The ability of γ-AApeptides to activate the ERK signaling pathway further demonstrates their potential to mimic fMLF peptide.
In addition to activation of calcium mobilization and the ERK signaling pathway, fMLF triggers chemotaxis and ROS production, which are key features of host defense in response to bacterial infection. [2b] To assess whether γ-AApeptides can mimic these functions of fMLF, we investigated the ability of the lead γ-AApeptides 3, 4 and 7 for the activation of chemotaxis and ROS production. As shown in Figure 6, although less potent than fMLF, 3 and 7 can effectively trigger the production of ROS (Figure 6a) and induce chemotaxis (Figure 6b). Consistent to calcium mobilization and ERK activation, 7 is the most potent molecule in the ROS production and chemotaxis assays, while 4 does not exhibit noticeable activity at concentrations up to 10 µM.
Figure 6.
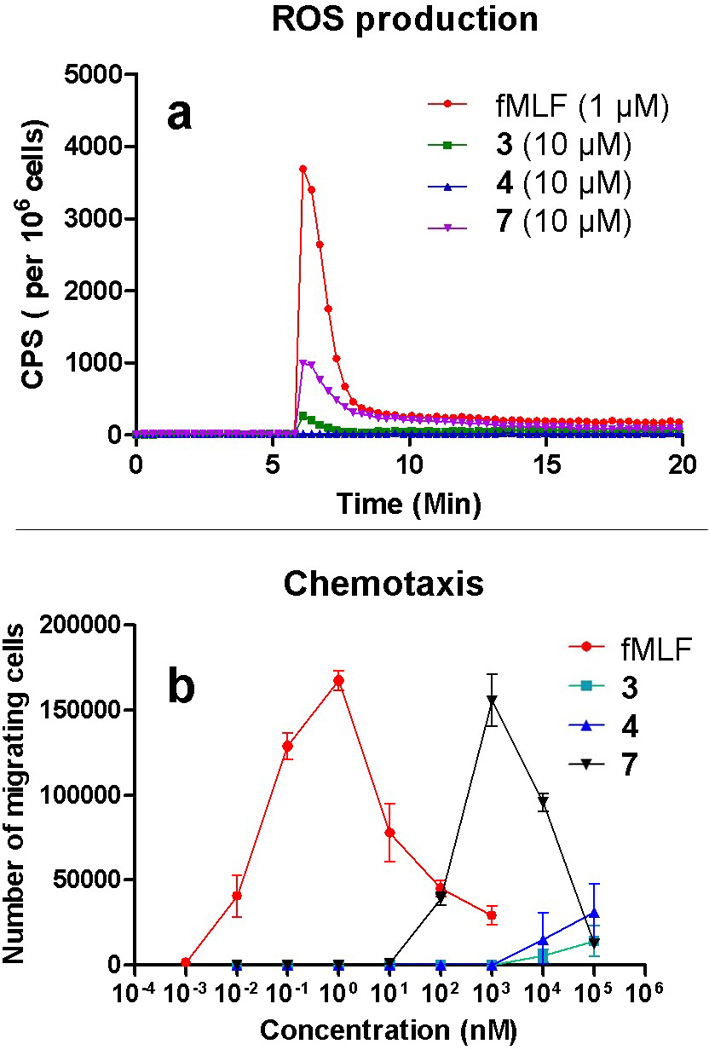
a, effects of the lead γ-AApeptides on ROS production in differentiated HL-60 cells. b, effects of the lead γ-AApeptides on chemotaxis of differentiated HL-60 cells. Data shown are representative of three independent experiments.
CONCLUSIONS
To summarize, we have designed a new class of peptidomimetics that mimic the structure and function of fMLF. This is also the first report of AApeptides as agonists that trigger GPCR signaling. From the focused library we synthesized on solid phase, a few AApeptides have shown to effectively induce calcium mobilization and Mitogen-Activated Protein Kinase (MAPK) signal transduction pathways in FPR-transfected RBL cells. More intriguingly, at high concentrations, certain γ-AApeptides are even more efficacious than fMLF in the induction of calcium mobilization. Similar to fMLF, these AApeptides are much more selective towards FPR1 rather than FPR2. Their agonistic activity is further supported by their ability to stimulate chemotaxis and production of superoxide in HL-60 cells. These results suggest that these fMLF-mimicking γ-AApeptides may emerge to be a new class of agents for the treatment of a variety of diseases. As fMLF peptides with C-terminus of COOH, CONH2 and COOMe can have different activity under different experimental conditions,[18] we envision that fMLF-mimicking γ-AApeptides may behave in a similar fashion. The synthesis and evaluation of these γ-AApeptides bearing different C-terminal functional groups are currently underway.
EXPERIMENTAL SECTION
General experimental methods
Knorr resin (0.66 mmol/g, 200–400 mesh) were provided by Chem-Impex International, Inc. All other reagents and solvents were purchased from either Sigma-Aldrich or Fisher Scientific. Formyl AApeptides were prepared on Knorr resin in peptide synthesis vessels on a Burrell Wrist-Action shaker. They were analyzed and purified on an analytical and a preparative Waters HPLC system, respectively, and then dried on a Labcono lyophilizer. NMR spectra of these AApeptides were obtained on a Varian Inova 400 instrument.
Materials for bioassays
The chemotactic peptides fMLF (N-formyl-Met-Leu-Phe) and W-peptide (WKYMVm) were purchased from Sigma (St. Louis, MO). FLIPR Calcium Assay Kit 5, used in the calcium mobilization assay, was purchased from Molecular Devices (Sunnyvale, Ca). The anti-ERK 1/2 and anti-phospho-ERK antibodies were from Cell Signaling Technologies (Beverly, MA). Other reagents were obtained from Sigma.
General synthetic procedure
Both α-AApeptides and γ-AApeptides were assembled on the solid phase following our previously reported protocol, and then the Fmoc groups on their N-terminus were removed by 20% piperidine/DMF. The formylation was achieved by the following procedure: Formic acid (3.0 eq, relative to resin loading) and isobutyl chloroformate (3.0 eq) were dissolved in 2 mL DCM, and cooled to 0 °C. N-Methylmorpholine (NMM) (3.0 eq) was added to the solution slowly. After 5 min, the solution mixture was added to the resin, and stirred for 2 h. Then the resin was washed with 3 mL DCM (3 ×) and 3 mL DMF (3 ×), respectively. The step of formylation was repeated twice. The AApeptides were cleaved from the resin in 95:5 TFA/H2O, and purified by HPLC. The final products were collected as white solid after lyophilization.
Compound 1. 1H-NMR (CD3OD, 400 MHz) δ = 8.08 (1H, d), 7.21 (5H, m), 4.43 (1H, m), 4.22 (1H, m), 3.41-3.04 (5H, m), 2.86-1.84 (10H, m), 1.56-1.42 (3H, m), 0.89 (6H). 13C-NMR (CD3OD, 100 MHz) δ = 174.7, 172.6, 172.1, 162.4, 138.2, 128.9, 128.1, 126.3, 62.6, 51.0, 48.7, 37.3, 34.1, 33.9, 31.1, 30.9, 29.6, 27.5, 21.3, 13.8. HR-ESI: [M]+ cacl: 464.2457, found: 464.2397.
Compound 2. 1H-NMR (CD3OD, 400 MHz) δ = 8.06(1H, m), 7.28-7.15 (5H, m), 4.22 (2H, m), 3.44-3.06 (4H, m), 2.62-2.22 (2H, m), 1.93-1.30 (9H, m), 0.90-0.78 (11H, m). 13C-NMR (CD3OD, 100 MHz) δ = 176.2, 174.1, 163.8, 139.7, 130.5, 129.6, 127.8, 64.0, 53.5, 50.3, 38.7, 35.6, 35.5, 32.9, 32.5, 28.9, 23.3, 22.8, 22.7, 14.3. HR-ESI: [M]+ cacl: 447.2893, found: 447.2814.
Compound 3. 1H-NMR (CD3OD, 400 MHz) δ = 8.08 (1H, d), 7.25-7.13 (5H, m), 4.46-3.29 (5H, m), 2.91-2.41 (6H, m), 2.13-1.81 (5H, m), 1.63 (1H, m), 1.43 (1H, m), 1.23 (1H, m), 0. 85 (7H, m). 13C-NMR (CD3OD, 100 MHz) δ = 174.6, 174.1, 172.4, 171.9, 162.4, 141.1, 128.1, 128.0, 125.6, 52.8, 51.4, 50.7, 45.9, 40.8, 34.6, 31.4, 30.7, 29.7, 24.5, 22.3, 20.6, 13.8. HR-ESI: [M]+ cacl: 464.2457, found: 464.2438.
Compound 4. 1H-NMR (CD3OD, 400 MHz) δ = 8.07 (1H, d), 7.25-7.14 (5H, m), 4.30-3.88 (4H, m), 3.45-3.29 (2H, m), 2.87-2.48 (4H, m), 1.74-1.20 (9H, m), 0.92-0.82 (9H, m). 13C-NMR (CD3OD, 100 MHz) δ = 174.5, 174.0, 172.6, 171.8, 162.2, 141.0, 128.0, 125.6, 52.8, 52.3, 50.7, 50.2, 45.9, 40.8, 34.7, 31.5, 30.7, 27.7, 24.4, 21.9, 20.6, 12.8. HR-ESI: [M]+ cacl: 446.2893, found: 464.2879.
Compound 5. 1H-NMR (CD3OD, 400 MHz) δ = 8.06(1H, d), 7.47-7.45 (2H, m), 7.34-7.33 (3H, m), 4.47(3H, m), 4.07-3.93 (3H, m), 3.08 (2H, d), 2.48-1.17 (9H, m), 0.86 (6H, m). 13C-NMR (CD3OD, 100 MHz) δ =172.1, 171.9, 162.3, 130.8, 129.5, 128.1, 128.0, 57.5, 52.7, 51.4, 49.5, 45.4, 40.6, 31.3, 29.5, 24.4, 22.2, 20.7, 13.7. HR-ESI: [M]+ cacl: 486.1971, found: 486.1944.
Compound 6.1H-NMR (CD3OD, 400 MHz) δ = 8.08 (1H, d), 7.29-7.21 (5H, m), 4.50-3.29 (8H, m), 2.47(2H, m), 2.05-1.21 (8H, m), 0.86 (6H, m). 13C-NMR (CD3OD, 100 MHz) δ = 173.6, 172.9, 171.6, 162.3, 134.8, 129.0, 128.1, 126.4, 53.2, 51.3, 50.5, 46.1, 41.0, 39.8, 31.2, 29.7, 24.5, 22.3, 20.7, 13.8. HR-ESI: [M]+ cacl: 450.2301, found: 450.2264.
Compound 7. 1H-NMR (CD3OD, 400 MHz) δ = 8.07 (1H, d), 7.29-7.20 (5H, m), 4.35-3.34 (8H, m), 1.76-1.17 (9H, m), 0.86 (9H, m). 13C-NMR (CD3OD, 100 MHz) δ =173.6, 172.6, 171.6, 162.2, 134.7, 128.9, 128.1, 126.4, 53.2, 52.3, 50.9, 40.9, 39.5, 31.4, 27.7, 24.4, 22.4, 21.9, 20.6, 12.8. HR-ESI: [M]+ cacl: 432.2737, found: 432.2695.
Cell Culture
The human FPR1 and FPR2 stable transfectants of rat basophilic leukemia cell RBL-2H3 were generated previously in our lab, and maintained in DMEM supplemented with 20% FBS. HL-60 cells were maintained in RPMI1640 supplemented with 10% FBS. The neutrophil-like differentiation of HL-60 cells was obtained by addition of 1.3% DMSO for 8 days.
Calcium Mobilization Assay
Human FPR1 or human FPR2 stable transfected RBL cells were grown to 70–80% confluence in black-wall/clear-bottom 96-well assay plates. Before assay, the cells were washed with HBSS (with Ca2+ and Mg2+) containing 20 mM HEPES and incubated in the same buffer. All Ca2+ mobilization assays were conducted with the use of a FLIPR Calcium Assay Kit 5. Cells were loaded for 1 h at 37 °C with FLIPR calcium sensitive dye, according to the manufacturer’s protocols. The addition of agonists was robotically controlled, and samples were read on a FlexStation II (Molecular Devices). Cells were excited at 485 nm, and Ca2+ fluorescence was detected with an emission wavelength of 525 nm.
Chemotaxis Assay
Differentiated HL-60 cells were resuspended in RPMI1640 containing 0.01%BSA at 5 X 106 cells/ml. A 24-well flat bottom Ultra low attachment surface plate (Costar) was precoated with RPMI1640 containing 0.01% BSA for 1 hour at 37°C. The buffer was removed from the plate, and agonists of different concentrations were added to each well (600 µl/well). After each agonist was added to the wells, a Millicell sterilized culture plate insert with 3 µm pore size was added to each well, followed by immediate addition of 100 µL of HL-60 cells. Plates were incubated at 37°C for 30 min, and chemotactic movement was halted by removal of the insert. Cells that migrated to the bottom chamber were detached by addition of 60 µL EDTA (20mM) and counted under microscope. To distinguish between chemotactic movement and chemokinetic movement, the number of cells that migrated in the presence of ligand in both top and bottom chambers was subtracted from samples with ligand present only in the bottom chambers.
Western-blot Analysis of Mitogen-Activated Protein Kinases activation
Activation of the p44/p42 MAP kinases (ERK1/2) was determined based on activation-associated phosphorylation. hFPR1-RBL cells were cultured in 6-well plates and stimulated with either fMLF (1 µM) or any one of the compounds (10 µM) as indicated. The cells were stimulated for 1, 2, and 5 minutes at 37°C and terminated by adding 150 µL of ice-cold SDS-PAGE loading buffer (15% (v/v) glycerol, 125 mM Tris-Cl, pH 6.8, 5 mM EDTA, 2% (w/v) SDS, 0.1% bromphenol blue, and 2.5% mercaptoethanol). Samples were transferred to microcentrifuge tubes and sonicated three times for 3 s each to disperse DNA contents. After boiling, samples were analyzed by SDS-polyacrylamide gel electrophoresis and blotted using anti-ERK1/2 and anti-phospho-ERK1/2 antibodies. The resulting immunocomplex was visualized by SuperSignal West Pico Chemiluminescence kit (Pierce, Rockford, IL) according to manufacturer’s instruction.
Measurement of Superoxide Production
Superoxide (O2−) generation was measured using an isoluminol enhanced chemiluminescent method. Differentiated HL-60HL-60 cells were assayed in HBSS containing 0.5% BSA with a cell density of 1 X106 cells/ml. Cells were incubated at 37°C for 5 min in the presence of 100 µM isoluminol and 40 U/ml HRP. After preincubation, 200 µl of the above mixture was added to each well of a white 96-well flat-bottom plate. Superoxide production was measured in a Wallac Victor2 1420 Multilabel Counter. Basal chemiluminescence was measured for 5 min, after which HL-60HL-60 cells were stimulated with agonists at indicated concentrations and chemiluminescence was measured for 20 min for each agonists.
Supplementary Material
ACKNOWLEDGEMENTS
This work is supported by NSF CAREER award 1351265 (JC), and NIH RO1AI033503 (RDY).
REFERENCES
- 1.Tosi MF. J. Allergy Clin. Immunol. 2005;116:241–249. doi: 10.1016/j.jaci.2005.05.036. quiz 250. [DOI] [PubMed] [Google Scholar]
- 2.a) Kirpotina LN, Khlebnikov AI, Schepetkin IA, Ye RD, Rabiet MJ, Jutila MA, Quinn MT. Mol. Pharmacol. 2010;77:159–170. doi: 10.1124/mol.109.060673. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, Serhan CN, Murphy PM. Pharmacol. Rev. 2009;61:119–161. doi: 10.1124/pr.109.001578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He R, Tan L, Browning DD, Wang JM, Ye RD. J. Immunol. 2000;165:4598–4605. doi: 10.4049/jimmunol.165.8.4598. [DOI] [PubMed] [Google Scholar]
- 4.He HQ, Liao D, Wang ZG, Wang ZL, Zhou HC, Wang MW, Ye RD. Mol. Pharmacol. 2013;83:389–398. doi: 10.1124/mol.112.081315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Mollica A, Paradisi MP, Varani K, Spisani S, Lucente G. Bioorg. Med. Chem. 2006;14:2253–2265. doi: 10.1016/j.bmc.2005.11.001. [DOI] [PubMed] [Google Scholar]; b) Wan HX, Zhou C, Zhang Y, Sun M, Wang X, Yu H, Yang X, Ye RD, Shen JK, Wang MW. Biochem. Pharmacol. 2007;74:317–326. doi: 10.1016/j.bcp.2007.04.016. [DOI] [PubMed] [Google Scholar]; c) Karlsson J, Fu H, Boulay F, Bylund J, Dahlgren C. Biochem. Pharmacol. 2006;71:1488–1496. doi: 10.1016/j.bcp.2006.02.010. [DOI] [PubMed] [Google Scholar]; d) Fu H, Karlsson J, Bylund J, Movitz C, Karlsson A, Dahlgren C. J. Leukoc. Biol. 2006;79:247–256. doi: 10.1189/jlb.0905498. [DOI] [PubMed] [Google Scholar]
- 6.Giordano C, Lucente G, Masi A, Paradisi MP, Sansone A, Spisani S. Bioorg. Med. Chem. 2006;14:2642–2652. doi: 10.1016/j.bmc.2005.11.043. [DOI] [PubMed] [Google Scholar]
- 7.Yang KH, Fang H, Ye JS, Gong JZ, Wang JT, Xu WF. Die Pharmazie. 2008;63:779–783. [PubMed] [Google Scholar]
- 8.a) Torino D, Mollica A, Pinnen F, Feliciani F, Spisani S, Lucente G. Bioorg. Med. Chem. 2009;17:251–259. doi: 10.1016/j.bmc.2008.11.010. [DOI] [PubMed] [Google Scholar]; b) Lucente G, Giordano C, Sansone A, Torino D, Spisani S. Amino Acids. 2009;37:285–295. doi: 10.1007/s00726-008-0145-3. [DOI] [PubMed] [Google Scholar]; c) Mollica A, Feliciani F, Stefanucci A, Costante R, Lucente G, Pinnen F, Notaristefano D, Spisani S. J. Pept. Sci. 2012;18:418–426. doi: 10.1002/psc.2414. [DOI] [PubMed] [Google Scholar]
- 9.a) Hu Y, Li X, Sebti SM, Chen J, Cai J. Bioorg. Med. Chem. Lett. 2011;21:1469–1471. doi: 10.1016/j.bmcl.2011.01.005. [DOI] [PubMed] [Google Scholar]; b) Niu Y, Hu Y, Li X, Chen J, Cai J. New J. Chem. 2011;35:542–545. [Google Scholar]; c) Niu Y, Hu Y, Wu H, Cai J. Methods Mol. Biol. 2013;1081:35–46. doi: 10.1007/978-1-62703-652-8_3. [DOI] [PubMed] [Google Scholar]
- 10.Wu H, Amin MN, Niu Y, Qiao Q, Harfouch N, Nimer A, Cai J. Org. Lett. 2012;14:3446–3449. doi: 10.1021/ol301406a. [DOI] [PubMed] [Google Scholar]
- 11.Wu H, Li Y, Ge B, Niu Y, Qiao Q, Tipton J, Cao C, Cai J. Chem. Commun. 2014;50:5206–5208. doi: 10.1039/c3cc46685j. [DOI] [PubMed] [Google Scholar]
- 12.Yang Y, Niu Y, Hong H, Wu H, Zhang Y, Engle J, Barnhart T, Cai J, Cai W. Chem. Commun. 2012;48:7850–7852. doi: 10.1039/c2cc33620k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Niu Y, Wu H, Li Y, Hu Y, Padhee S, Li Q, Cao C, Cai J. Org. Biomol. Chem. 2013;11:4283–4290. doi: 10.1039/c3ob40444g. [DOI] [PubMed] [Google Scholar]; b) Wu H, Niu Y, Padhee S, Wang RE, Li Y, Qiao Q, Ge B, Cao C, Cai J. Chem. Sci. 2012;3:2570–2575. [Google Scholar]; c) Niu Y, Padhee S, Wu H, Bai G, Harrington L, Burda WN, Shaw LN, Cao C, Cai J. Chem. Commun. 2011;47:12197–12199. doi: 10.1039/c1cc14476f. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Li Y, Smith C, Wu H, Padhee S, Manoj N, Cardiello J, Qiao Q, Cao C, Yin H, Cai J. ACS Chem. Biol. 2014;9:211–217. doi: 10.1021/cb4006613. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Niu Y, Padhee S, Wu H, Bai G, Qiao Q, Hu Y, Harrington L, Burda WN, Shaw LN, Cao C, Cai J. J. Med. chemistry. 2012;55:4003–4009. doi: 10.1021/jm300274p. [DOI] [PubMed] [Google Scholar]; f) Padhee S, Hu Y, Niu Y, Bai G, Wu H, Costanza F, West L, Harrington L, Shaw L, Cao C, Cai J. Chem. Commun. 2011;47:9729–9731. doi: 10.1039/c1cc13684d. [DOI] [PubMed] [Google Scholar]; g) Niu Y, Wang RE, Wu H, Cai J. Future Med. Chem. 2012;4:1853–1862. doi: 10.4155/fmc.12.111. [DOI] [PubMed] [Google Scholar]
- 14.a) Niu Y, Bai G, Wu H, Wang RE, Qiao Q, Padhee S, Buzzeo R, Cao C, Cai J. Mol. Pharm. 2012;9:1529–1534. doi: 10.1021/mp300070w. [DOI] [PubMed] [Google Scholar]; b) Niu Y, Jones AJ, Wu H, Varani G, Cai J. Org. Biomol. Chem. 2011;9:6604–6609. doi: 10.1039/c1ob05738c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Mollica A, Paglialunga Paradisi M, Torino D, Spisani S, Lucente G. Amino Acids. 2006;30:453–459. doi: 10.1007/s00726-006-0260-y. [DOI] [PubMed] [Google Scholar]; b) Jiarpinitnun C, Kiessling LL. J. Am. Chem. Soc. 2010;132:8844–8845. doi: 10.1021/ja102640c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Wu H, Li Y, Bai G, Niu Y, Qiao Q, Tipton JD, Cao C, Cai J. Chem. Commun. 2014;50:5206–5208. doi: 10.1039/c3cc46685j. [DOI] [PubMed] [Google Scholar]; b) Wu H, Teng P, Cai J. Eur. J. Org. Chem. 2014:1760–1765. [Google Scholar]
- 17.a) Gordon EJ, Gestwicki JE, Strong LE, Kiessling LL. Chem. Biol. 2000;7:9–16. doi: 10.1016/s1074-5521(00)00060-0. [DOI] [PubMed] [Google Scholar]; b) Gestwicki JE, Strong LE, Borchardt SL, Cairo CW, Schnoes AM, Kiessling LL. Bioorg. Med. Chem. 2001;9:2387–2393. doi: 10.1016/s0968-0896(01)00162-6. [DOI] [PubMed] [Google Scholar]; c) Kiessling LL, Gestwicki JE, Strong LE. Angew. Chem. Int. Ed. 2006;45:2348–2368. doi: 10.1002/anie.200502794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Mollica A, Stefanucci A, Costante R, Pinnen F. Antiinflamm. Antiallergy Agents Med. Chem. 2012;11:20–36. doi: 10.2174/187152312803476246. [DOI] [PubMed] [Google Scholar]; b) Dentino AR, Raj PA, DeNardin E. Arch. Biochem. Biophys. 1997;337:267–274. doi: 10.1006/abbi.1996.9791. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.