

Abstract
4EGI-1, the prototypic inhibitor of eIF4E/eIF4G interaction, was identified in a high-throughput screening of small molecule libraries using a fluorescence polarization assay that measures inhibition of binding of an eIF4G-derived peptide to recombinant eIF4E. As such, the molecular probe 4EGI-1 holds a potential for studying molecular mechanisms involved in human disorders characterized by loss of physiologic restrains on translation initiation. A hit-to-lead optimization campaign was carried out to overcome the liability of the configurational instability in 4EGI-1, which stems from the (E)-to-(Z) isomerization of the hydrazone function. We identified compound 1a, in which the labile hydrazone was incorporated into a rigid indazole scaffold as a promising rigidified 4EGI-1 mimetic lead. In a structure-activity relationship study aimed at probing the structural latitude of this new chemotype as an inhibitor of eIF4E/eIF4G interaction and translation initiation we identified 1d, an indazole-based 4EGI-1 mimetic, as a new and improved lead inhibitor of eIF4E/eIF4G interaction and a promising molecular probe candidate for elucidating the role of cap-dependent translation initiation in a host of pathophysiological states.
Keywords: conformational rigidification, eIF4E/eIF4G interaction, eIF4F, indazoles, molecular probes, protein-protein interaction inhibitors, translation initiation
Introduction
Protein-protein interactions (PPIs) are ubiquitous and serve critical roles in many biological systems. By nature, the interacting interfaces of the two macromolecules are large (1940 ± 760 Å2)[1] and shallow with scattered binding hotspots that provide most of the binding energy. Compared to inhibition of enzyme-substrate or receptor-ligand interactions, disruption of PPIs by a small molecule presents an ambitious challenge.[2] Nevertheless, targeting PPIs by chemical biology approaches to develop effective small molecule probes and potential drug candidates will advance our understanding of PPIs involvement in normal- and patho-biology, and dramatically expand druggable genome allowing for novel therapeutic paradigms.[3] In recent years, this approach yielded some successful small molecule PPI inhibitors that were promoted into clinical trials.[4]
Translation, the process of protein biosynthesis, is comprised of three stages: initiation, elongation, and termination. Translation initiation (TI), a highly regulated process governed by numerous eukaryotic TI factors (eIFs), results in the assembly of elongation-competent 80S ribosome on the initiation codon.[5] The PPIs between the eIF4E and eIF4G, which is the focus of our studies, are essential for cap-dependent translation, by which most mammalian mRNAs are translated. In cap-dependent translation, an mRNA cap (m7GpppN, 7-methylguanosine-5′-triphospho nucleotide) structure is recognized by eIF4E, the cap-binding protein.[6] eIF4E interacts with eIF4G, a scaffolding protein, which also interacts with eIF4A, an RNA helicase, to form the eIF4F complex.[5, 7] Interaction of eIF4G with the dorsal side of eIF4E is centered on a conserved hydrophobic motif formed by the sequence Y(X)4LΦ where X can be any amino acid and Φ is any hydrophobic amino acid. The eIF4E/eIF4G interaction (and by extension formation of eIF4F complex) is regulated by eIF4E-binding proteins (4E-BPs), which includes the same hydrophobic motif as eIF4G and competes with eIF4G for binding to eIF4E. Hyper-phosphorylation of 4E-BPs by the mammalian Target of Rapamycin Complex 1 (mTORC1) reduces their affinity for eIF4E thus allowing for binding of eIF4E to eIF4G and formation of the eIF4F complex. The eIF4F complex binds mRNA and 40S ribosomal subunit in addition to other TI factors, unwinds the secondary structures in the 5′-untranslated (5′UTR) region of mRNA to scan the mRNA for the initiator AUG codon where the 40S ribosomal subunit is joined by the 60S subunit, thus forming the translation-competent 80S ribosome on the start codon.
In most normal cells, the low abundance of eIF4E relative to other eIFs[8] and its interaction with 4E-BPs turns it into a rate limiting factor for the cap-dependent translation.[9] However, this balance is functionally perturbed in many diseased states. Regulation of the eIF4F activity plays a critical role in normal biology, while its perturbation contributes to the patho-biology of many human disorders. eIF4F complex is critically required for cellular proliferation, differentiation and survival.[10] It also plays a critical role in long-term memory consolidation.[11] The eIF4F complex is implicated in the pathobiology of human disorders including autism,[12] fragile X syndrome,[13] tuberous sclerosis,[14] and some cancers. Autism is thought to be caused by increased translation of synaptic mRNA.[15] Dysregulation of synaptic protein synthesis may result from genetic alterations in the components of the eIF4F complex or its negative regulators, 4E-BPs, or activation of upstream signaling pathways that elevate eIF4F activity. The most prominent example of these pathways is signaling by mammalian target of rapamycin (mTOR), which by itself is regulated by the interplay of phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt), phosphatase, and tensin homolog (PTEN). Various genetic aberrations that lead to activation of mTOR are associated with autism spectrum disorders.[16] Importantly, many symptoms of autism spectrum disorders can be reversed by inhibiting mTOR activity[17] or by pharmacologically inhibiting the formation of the eIF4F complex.[12b] Another mental disorder, fragile-X-syndrome, is caused by defects in the functioning of the cytoplasmic fragile-X mental retardation protein (FMRP) interacting protein 1, which is an eIF4E binding protein that suppresses eIF4F driven cap-dependent translation.[18] Tuberous sclerosis complex (TSC) is another group of disorders caused by mutations in the tuberin and hamartin genes that lead to uncontrolled activation of mTOR and thereby eIF4F complex formation and unrestricted translation.[19] TSC disorders include benign tumors of lung, kidney, brain, and other organs and autism spectrum disorders. At the molecular level TSC is associated with activation of eIF4F as well as uncontrolled endoplasmic reticulum stress. The involvement of eIF4F complex in numerous pathological states highlights the urgent need for better pharmacological probes to study the role of this complex in normal- and patho-biology. Such probes may also provide a launching pad for the development of therapeutic agents for various inflammatory, proliferative, and neurodegenerative disorders.
We discovered 4EGI-1 (Scheme 1) in a high throughput screening campaign employing fluorescence polarization (FP) assay that searched chemical libraries for small molecules that bind to eIF4E and compete with a fluorescence-labeled eIF4G-derived peptide.[20] 4EGI-1 binds to eIF4E, disrupts eIF4E/eIF4G interaction, and inhibits cap-dependent TI in vitro and growth of human cancer xenografts in vivo.[20] Preliminary SAR studies highlighted the critical role of the hydrazone function in the minimal pharmacophore of 4EGI-1.[20a] However, while the hydrazone is essential for 4EGI-1’s inhibitory activity, it introduces inherent configurational instability, which is a major liability in controlling chemophysical properties and achieving defined and preselected biological activities. (E)-to-(Z) isomerization around the C=N bond is affected by solvents, solutes, pH, and temperature making the more stable and slightly more hydrophobic (Z)-isomer the predominant isomer. This isomerization phenomenon observed in a wide range of hydrazones of different nature is extensively documented in the literature.[21]
Scheme 1.

(E)-to-(Z) isomerization of 4EGI-1.
To overcome the configurational liability in 4EGI-1 we sought to eliminate the isomerization around its hydrazone function. Evidently, ring closure between the NH of the hydrazone (CH=N-NH-) and the ortho position of the benzyl moiety, which is recapitulated closely in an indazole system (Scheme 2). In this chemotype, the hydrazone becomes an integral part of an indazole scaffold, a condensed bicyclic system, and is locked in the (E)-configuration so that the potential isomerization around the C=N~N- bond is blocked. As such, 1-(4-(3,4-dichlorophenyl)thiazol-2-yl)-5-nitro-1H-indazole-3-carboxylic acid (1b) is structurally a rigidified (E)-4EGI-1-mimetic.
Scheme 2.
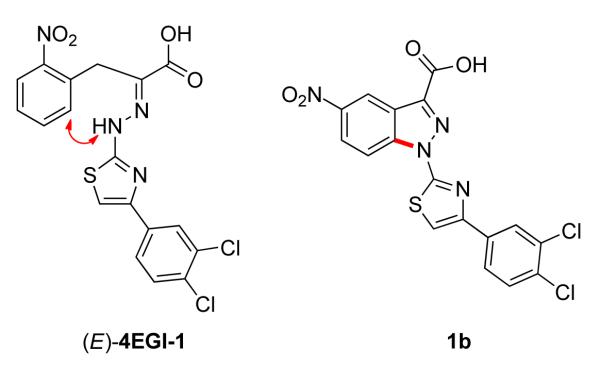
Recapitulation of the configuration of (E)-4EGI-1 around the C=N~N- bond in 3-substituted indazole scaffold, a true (E)-4EGI-1 mimetic.
Natural products containing an indazole core are quite rare, nevertheless, indazole appears as a privileged scaffold in several bioactive compounds of diverse therapeutic significance,[22] such as antidepressants,[23] contraceptives,[24] anticoagulants,[25] anti-HIV,[26] for treatment of metabolic syndromes,[27] analgesics,[28] anti-cancer drugs,[29] and chronic obstructive pulmonary disease therapeutics.[30]
In a recent study, an indazole core was introduced to configurationally lock, and thus mimic, an X-ray-derived target bound-conformation of a parent small molecule that is a potent inhibitor of reverse transcriptase.[26] This rigidification resulted in increased potency, specificity, and excellent pharmacokinetics. However, not all attempts to introduce an indazole ring into a bioactive compound result in improved activity. Incorporating the hydrazone function of a potent phosphodiesterase-4 inhibitor into the pyrazole ring of an indazole system resulted in significant loss of potency. This loss was attributed to the steric hindrance imposed by the indazole ring disrupting binding at the catalytic site.[30]
In here, we report the synthesis of a new class of configurationally constrained 4EGI-1 mimetic in which the hydrazone function is locked in an (E)-configuration as part of an indazole-based cyclic scaffold and evaluate them as inhibitors of eIF4E/eIF4G interaction and of cap-dependent TI.
Results and Discussion
Chemistry
The two synthetic approaches we employed in the preparation of the indazole-based constrained mimetics of (E)-4EGI-1 library are outlined in Scheme 3. The first, a convergent synthesis in which two fragments, 1H-indazole-3-carboxylic acid and 2-halo-4-phenylthiazole, were combined via a regioselective N-arylation to yield the final 4EGI-1 mimetic (Scheme 3, route i); and the second, a linear synthesis in which a 4EGI-1-like hydrazone was assembled in a stepwise manner followed by an intramolecular cyclization, involving the N-arylation of the NH of the hydrazone, generating the anticipated indazole-containing 4EGI-1 mimetic (Scheme 3, route ii).
Scheme 3.

Retrosynthetic analysis of the two strategies for the preparation of 3-substitued indazole as a configurationally constrained 4EGI-1 mimetic: i) convergent synthesis combines two independently synthesized building blocks, the 1H-indazole-3-carboxtlic acid and a 2-halo-4-phenylthiazole; ii) Linear synthesis that assembles in a stepwise manner a linear precursor that in the final step undergoes an intramolecular reaction to generate the anticipated constrained indazole-based 4EGI-1 mimetic.
In the convergent synthesis pathway (Scheme 3, i), the preparation of the 2-halo-4-phenyl thiazolyl fragment was carried out by a well-documented pathway. First, the bromoacetophenones 2a-h were transformed to the corresponding thiocyanateacetophenones by treatment of potassium thiocyanate[31] and subsequently underwent an intramolecular cyclization upon treatment with either gaseous HCl or HBr, to generate the corresponding 2-halo-4-phenylthiazole,[32] 4a-h, in moderate yields (Scheme 4). Nitration of 1H-indazole-3-carboxylic acid in fuming nitric acid yielded the other fragment, 5-nitro-1H-indazole-3-carboxylic acid.[33] Subsequent N-arylation of the indazole, or 5-nitroindazole, in the presence of powdered sodium hydroxide in DMSO, by 2-halo-4-phenylthiazole 4 at 110 °C, afforded the anticipated indazole-derived 4EGI-1 mimetic (1′a-c and 1a-g) in low to moderate yields (Scheme 4). Replacement of bromoacetophenones 2a-g with 2-bromo-1-phenylpropan-1-one 2h led to the introduction of a methyl group on C-5 of the thiazolyl ring (Scheme 4).
Scheme 4.

Convergent synthesis of constrained indazole-based 4EGI-1 mimetic via N-arylation of 1H-indazole-3-carboxylic acid by 2-halo-4-phenylthiazole. Reagents and conditions: i) KSCN (2 eq), acetonitrile, reflux, overnight; ii) HCl(g) or HBr (g), dry ether, 0 °C to rt, overnight; iii) HNO3/H2SO4 (conc.); iv) 1H-indazole-3-carboxylic acid or 5-nitro-1H-indazole-3-carboxylic acid, NaOH (2.2 eq) in DMSO, N2.
The second strategy for the synthesis was developed with the goal of obtaining higher yields of the focused 3-substituted indazole library of 1 (Scheme 3, ii). The synthetic pathway was designed to generate a derivative of 2-(2-bromo-4-phenyl)-2-[(4-arylthiazol-2-yl)hydrazono]acetic acid 7 that will form a fused pyrazole ring (i.e. indazole) through an intramolecular cyclization (Scheme 5). The transformation of arylhydrazones, usually formed by the condensation of a hydrazine derivative and 2-benzaldehydes or ketones, into indazoles is well established. This intramolecular aromatic nucleophilic substitution, with emphasis on the appropriate reaction conditions (dictated by the leaving group and the hydrazine moiety), is extensively documented in the literature.[22, 25, 34]
Scheme 5.

Linear synthesis of 3-substituted indazoles as configurationally constrained 4EGI-1 mimetic through stepwise assembly of a hydrazone followed by intramolecular cyclization of this linear precursor to form the anticipated product. Reagents and conditions: i) thiosemicarbazide (1eq), 1,4-dioxane, rt, overnight; ii) 6 (1eq), ethanol:water:acetic acid (10:4.5:0.5, v/v/v), reflux, overnight; iii) CuI (0.1 eq), DMEDA (0.3 eq), Na2CO3 (2.2 eq), ethanol:water (1:1, v/v); i’) NaNO3/H2SO4, 0 °C[39]; ii’) KOH in rt[40]; iii’) a) NaNO2/HBr at 0 °C, b) CuBr/HBr(aq) at rt.
In the first step of this pathway, we utilized a Hantzsch-type reaction[35] between thiosemicarbazide[36] and α-halo-acetophenones (2a-d, 2g and 2i-t) or 2-bromo-1-phenylpropan-1-one (2h and 2u). In most cases, this reaction led to the formation of two cyclic products, the desired 2-hydrazinyl-4-phenylthiazole (5a-d, 5g and 5i-t) (or 2-hydrazinyl-4-phenyl-5-methyl-thiazole (5h and 5u)) and the non-relevant 6-phenyl-6H-1,3,4-thiadiazin-2-amines 5*.[36] These side products 5*a-u were formed in variable quantities and in several cases in equal amount as the desired 5-membered ring intermediates 5a-u. For characterization purposes we have isolated few of the 2-aminothiadiazines 5* (e.g. 5*a, k, m, see experimental section) and confirmed their structural integrity. Practically, we could use the crude mixtures containing both 5 and 5* and subject them to the subsequent condensation with oxoacetic acid derivatives 6, obtaining the same yield of hydrazones 7 as in preparations where we used isolated hydrazines 5. This synthetic shortcut is possible because the 2-aminothiadiazines 5* are weaker nucleophiles than the 2-hydrazinyl-4-phenylthiazoles 5 and cannot compete in the condensation reaction. Moreover, the disparity in the polarity between 5* and 7 is much greater than between 5* and 5 making the isolation of 7 by chromatography a straightforward task.
Of note is the lack of understanding of the anticipated product distribution in the Hantzsch reactions of α-halocarbonyl-containing compounds with thiosemicarbazide. Many studies report the formation of mixtures of 2-hydrazinyl-4-phenylthiazole- and 2-aminothiadiazine-derivatives similar to what we have observed.[37] On the other hand, there are reports on reaction conditions that lead to the exclusive formation of 2-aminothiadiazine derivatives.[36, 38] In the absence of systematic studies that will provide mechanistic insight on the different reaction pathways we remain unable to control the reaction course to generate the 5-membered ring as the exclusive or predominant product.
A three step transformation of isatin into the critical 2-(2-bromo-5-nitro-phenyl)-2-oxoacetic acid 6 (Scheme 5) was accomplished by following two previously reported procedures that include nitration of isatin[39] and subsequent basic hydrolysis into the related 2-amino-5-nitro-2-oxoacetic acid.[40] For the last step, we adapted the Sandmeyer reaction to convert the aromatic amine into 6. Condensation of the 2-hydrazinyl-4-phenylthiazoles 5 with the 2-(2-bromophenyl)-2-oxoacetic acid 6 generated the targeted linear precursor 2-(2-bromo-4-phenyl)-2-[(4-arylthiazol-2-yl)hydrazono]-acetic acid 7. In general, we obtained a mixture of (E)- and (Z)-hydrazones in which (E) is the predominant one (>94%). The major isomer was isolated and purified for characterization, but this was not essential: Monitoring the intramolecular cyclization reaction by HPLC-MS indicated that both isomers in the mixture were fully consumed and transformed to the anticipated indazole 1. We assume that dynamic (Z)-to-(E) isomerization under the conditions of the cyclization reaction explains the disappearance of the (Z)-isomer. Our arylhydrazones 7 required a combination of base (Na2CO3/N,N’-dimethylethylenediamine) and CuI used as a catalyst, which proved to be successful in many N-arylation reactions.[41] This combination afforded synthesis of constrained indazole-based (E)-4EGI-1 mimetic library 1 (Scheme 5), in moderate to excellent yields.
Comparison of the two synthetic pathways shows that, in spite of the larger number of synthetic steps, the linear synthesis is advantageous over the convergent one. In almost all cases we obtained better yields of the same 3-substituted indazoles (e.g. 1g - 47% vs. 12%; 1a – 30% vs. 21%; and 1b – 34% vs. 14%). The N-arylation step that combines the two fragments, 5-nitroindazole and 4a-h (route i in Scheme 3), is the main culprit for the lower overall yield of the convergent synthetic pathway. Taken together, our synthetic work generated a focused library of pure (>95% by RP-HPLC) constrained indazole-based (E)-4EGI-1 mimetic 1a-u, established their structural integrity by 1H-, 13C-NMR, and HR-MS, and laid the synthetic groundwork for further structural optimization.
Introduction of a nitro group on C-5 of the indazole ring compromised the solubility of these compounds: while 1′a-c had good solubility in methanol and DMSO at rt, their nitrated homologs 1a-c became poorly soluble. The nitro group is an integral part of the 4EGI-1 pharmacophore and contributes to the activity of its indazole-derived mimetic (vide infra). In order to increase the solubility of the 5-nitroindazole derivatives, we introduced polar groups on the phenylthiazolyl moiety. As expected, substituting the phenyl ring with polar groups such as hydroxy, carboxy, or amine groups resulted in improved solubility of up to 32-fold. For example, an improvement in DMSO solubility at room temperature was observed as we went from 1a (3.125 mm), the non-decorated constrained indazole-based 4EGI-1 mimetic, to 1c (50 mm), and to 1t (100 mm). Notably, introduction of a methyl on C-5 of the thiazole ring was also accompanied by improved solubility as demonstrated by comparing the non-methylated analog 1a (3.125 mm) with the methylated 1h (37.5 mm). The better solubility of 1h as compared to 1a, suggests that steric hindrance introduced by C-5 methylation may prevent facile aggregation and nucleation, which could accelerate precipitation.[42]
Biology
The newly synthesized constrained indazole-based 4EGI-1 mimetic library 1 was evaluated in a cell-free fluorescent polarization (FP) eIF4E/eIF4G interaction assay. This assay reports on the ability of a small molecule to disrupt the interaction of fluorescent-labeled eIF4G-derived peptide, which contains the eIF4E-binding consensus sequence, with eIF4E,[20a] a proxy for disruption of the eIF4F complex. We chose a set of compounds (1a, 1d, and 1l) to further evaluate their activity in cell-based secondary mechanistic assays: expression of oncogenic proteins encoded by weak mRNAs and disruption of endogenous eIF4E/eIF4G interaction in intact cells. Finally, we determined the cell proliferation inhibitory activity of these selected compounds to serve as a reporter of a global activity that may be comprised of their on-target as well as off-target activities.
Competitive inhibition of the eIF4E/eIF4G interaction by the constrained 4EGI-1 analogs
We have previously reported on using the same FP assay for the discovery and development of small molecules that disrupt eIF4E/eIF4G interactions.[20a] Briefly, we synthesized a fluorescein-tagged eIF4G-derived peptide that acts as a surrogate to the endogenous eIF4G. The synthetic fluorescent peptide contains the consensus eIF4E-binding motif (Y(X)4LΦ). We also generated a recombinant glutathione S-transferase-tagged fusion protein GST-DN26-eIF4E (in which eIF4E was truncated by the first N-terminal 26 amino acids). Herein, the (Z)-4EGI-1 was used in the FP assay as a positive control and the vehicle (DMSO) as a negative control. We report the potency of the new constrained indazole-based 4EGI-1 mimetic 1 as the ratio between the IC50 of the (Z)-4EGI-1 and the IC50 of compound 1 (Table 1).
Table 1.
Binding of the configurationally constrained indazole-based 4EGI-1 mimetics to eIF4E as measured by fluorescence polarization assay (FP).
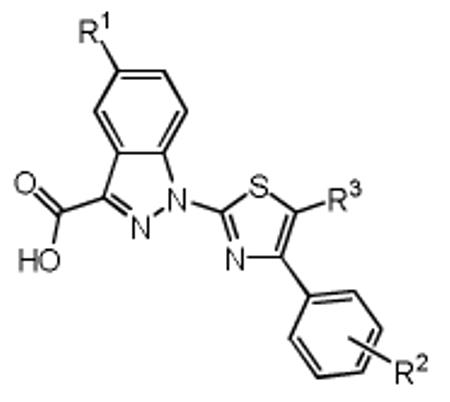
| Entry | Compound | R1 | R2 | R3 | IC50 (Z)-4EGI-1 /IC50 1a |
|---|---|---|---|---|---|
| 1 | (E)-4EGI-1 | NO2 | 3,4-dichloro | H | 0.84±0.07 (41.5)b |
| 2 | (Z)-4EGI-1 | NO2 | 3,4-dichloro | H | 1.00±0.00 (32.0)b |
| 3 | 1′a | H | H | H | NAc |
| 4 | 1′b | H | 3,4-dichloro | H | NAc |
| 5 | 1′c | H | 4-OCH3 | H | 0.40±0.05 |
| 6 | 1a | NO2 | H | H | 0.82±0.01 |
| 7 | 1b | NO2 | 3,4-dichloro | H | 1.40±0.11 |
| 8 | 1c | NO2 | 4-OCH3 | H | 1.70±0.18 |
| 9 | 1d | NO2 | 3-OCH3 | H | 4.04±0.52 |
| 10 | 1e | NO2 | 4-Cl | H | 0.91±0.08 |
| 11 | 1f | NO2 | 2,4-difluoro | H | 0.98±0.03 |
| 12 | 1g | NO2 | 4-F | H | 0.61±0.06 |
| 13 | 1h | NO2 | H | Me | 2.10±0.14 |
| 14 | 1i | NO2 | 2-OCH3 | H | 1.92±0.12 |
| 15 | 1j | NO2 | 2,5-dimethoxy | H | 1.05±0.01 |
| 16 | 1k | NO2 | 2,4-dichloro | H | 2.61±0.24 |
| 17 | 1l | NO2 | 4-pyrrolidine | H | 2.70±0.12 |
| 18 | 1m | NO2 | 4-morpholine | H | 2.24±0.05 |
| 19 | 1n | NO2 | 4-N(Me)2 | H | 1.81±0.03 |
| 20 | 1o | NO2 | 4-CO2H | H | 2.83±0.07 |
| 21 | 1p | NO2 | 2-CO2H | H | NAc |
| 22 | 1q | NO2 | 2-OH | H | 1.02±0.03 |
| 23 | 1r | NO2 | 3-OH | H | 0.75±0.01 |
| 24 | 1s | NO2 | 4-OH | H | 4.12±0.40 |
| 25 | 1t | NO2 | 3,4-dihydroxy | H | 4.09±0.12 |
| 26 | 1u | NO2 | 3,4-dihydroxy | Me | 2.26±0.35 |
The relative binding affinity of the library of compound 1 as measured by their activity in the fluorescence polarization assay. The relative activity of (Z)-4EGI-1 is given as 1.
The value in brackets is the IC50 in μm.
Not active.
Locking (E)-4EGI-1 in the corresponding indazole-based mimetic 1b resulted in a significant increase in relative binding affinity (cf. IC50 (Z)-4EGI-1/IC50 (E)-4EGI-1 = 0.84 to IC50 (Z)-4EGI-1/ IC50 1b = 1.40) (entries 1 and 7 in Table 1). Evidently, locking the hit compound 4EGI-1 in the (E)-mimicking configuration, as in the indazole system, is either neutral or enhances the affinity for eIF4E, compared to either the (E)- or the (Z)-isomers of 4EGI-1. This result was encouraging and suggested that the indazole scaffold is a constrained chemotype that mimics 4EGI-1 in inhibiting eIF4E/eIF4G interaction. Compounds 1a-u exhibit a range of competitive binding affinities that from no-activity to 4-fold higher affinity than (Z)-4EGI-1 (Table 1). Comparing the relative competitive-binding affinity of 1′a-c with 1a-c highlights the notable contribution of 5-NO2 group to the binding affinity of these nitrated derivatives to eIF4E. Previous SAR studies conducted in our laboratory revealed that the ortho-nitro group on of the phenylpyruvic moiety of 4EGI-1 itself was an indispensable feature of the minimal pharmacophore.
Substitution on the para position of the 4-phenylthiazolyl moiety plays an important role in the interaction of the indazole-derived ligands with eIF4E. While 4-chloro-, 4-fluoro-, and 2,4-difluoro-phenyl substituents, as in the respective 1e, 1g, and 1f, are less potent than, or equipotent to the hit 4EGI-1, the 2,4- and 3,4-dichlorophenyl substituents, as in derivatives 1k and 1b, respectively, are more potent than (Z)-4EGI-1. Diversifying the nature of substituents on the para position of the 4-phenylthiazolyl moiety, to include polar and potentially charged ones, contributed not only to improved solubility but also generated some of the most potent competitive binders to eIF4E. The change in apparent binding affinity of the 2-, 3-, and 4-hydroxy- (1q, 1r, and 1s) and 3,4-dihydroxyphenyl (1t) substituted analogs exemplifies the complex structure-activity relationship in this focused library. While 1r, the 3-hydroxy substituted analog, is less potent than 4EGI-1 and the 2-hydroxy and 4-hydroxy substituted analogs (1q and 1s, respectively) the 3,4-dihydroxy analog 1t and the 4-hydroxy analog 1s are the most potent analogs in this series (IC50 (Z)-4EGI-1/IC50 1 = 4.09 and 4.12, respectively). O-Methylation of the hydroxyl affects apparent binding affinity in a position-dependent manner: while O-methylation of the 2-OH and 3-OH enhances relative binding affinity (cf. 1.02 for 1q vs. 1.92 for 1i and 0.75 for 1r vs. 4.04 for 1d), O-methylation of 4-OH results in significant loss of apparent binding affinity (4.12 for 1s vs. 1.70 for 1c). Interestingly, the apparent binding affinity of the 2,4-dimethoxy analog is significantly lower than that of the 2-methoxy and 4-methoxy substituted analogs (cf. 1.05 for 1j vs. 1.92 and 1.70 for 1i and 1c, respectively). Introduction of potentially positive charged disubstituted amines in the para position of the 4-phenylthiazolyl group enhances apparent binding affinity gradually from 4-dimethylamino to 4-morpholino and 4-pyrrolidino (1.81 for 1n, 2.24 for 1m, and 2.70 for 1l). The positional dependency of binding affinity is also underscored in the marked differences between the inactive 2-and the potent 4-CO2H substituted derivatives, 1p and 1o, respectively. In summary, it is evident that apparent binding affinity of the constrained 4EGI-1 mimetic to eIF4E is greatly affected by the nature and position of substituents on the 4-phenylthiazolyl moiety. We are greatly encouraged by the tolerance of polar and potentially charge-bearing substituents that could be important modifiers of physicochemical and pharmacokinetic properties.
Inhibition of eIF4E/eIF4G interaction in cells
Encouraged by the results of the cell-free FP assay where the constrained indazole-based (E)-4EGI-1 mimetic displayed high activity, we selected representative analogs from this series for target validation in cell-based assays. Inhibition of eIF4E/eIF4G PPI in CRL-2813 cells by 1d was demonstrated in an eIF4E pull-down assay and compared with (E)-4EGI-1 (Figure 1). Western blot analysis of cap-affinity pulled-down material from lysates of melanoma cells treated with either 1d, (E)-4EGI-1, or vehicle (DMSO) revealed that 1d or (E)-4EGI-1 treatment depletes eIF4G from eIF4E complex without any effect on the amount of eIF4E. As previously reported for 4EGI-1,[20a] here too, we observed enhancement of 4E-BP1 binding in (E)-4EGI-1 or 1d treated cells relative to DMSO-treated cells (Figure 1).
Figure 1.
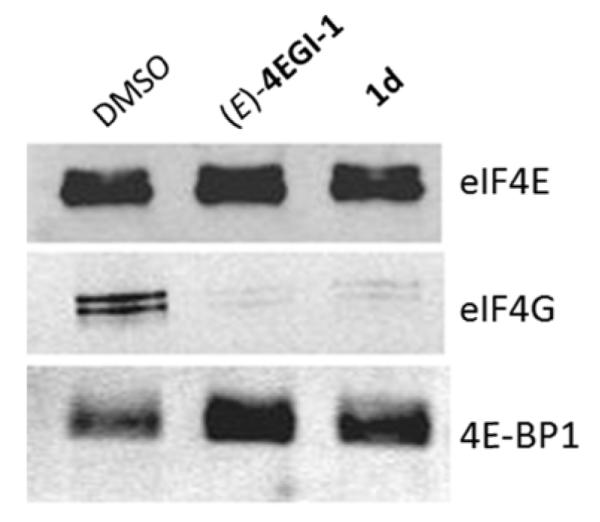
Compound 1d, an indazole-based constrained mimetic of 4EGI-1, disrupts eIF4E/eIF4G interaction similarly to 4EGI-1. CRL-2813 human melanoma cells were treated with DMSO, (E)-4EGI-1, or compound 1d at final concentration of 50 μm for a time period of 8 hours. Cell lysates were incubated with cap-conjugated agarose resin, and the retained material was eluted with m7-GTP. The eluted material was separated by SDS-PAGE and immunoblotted with antibodies specific to eIF4E, eIF4G, and 4E-BP1.
Inhibition of oncogenic but not housekeeping protein expression
We anticipate that inhibition of translation initiation by disrupting eIF4F assembly should preferentially inhibit expression of growth promoting, anti-apoptotic, and oncogenic proteins with minimal effect on the expression of housekeeping proteins. To validate this concept, we treated CRL-2813 cells with two constrained indazole-based 4EGI-1 mimetic: 1a, and 1d that represent two levels of apparent binding affinity to eIF4E, 4EGI-1 and DMSO as positive and negative controls, respectively (Table 1). Indeed, similar to 4EGI-1, selected indazole-based compounds inhibited significantly the expression of survivin, an oncogenic protein that inhibits apoptosis and expression of cyclin D1, a promoter of G1/S cell cycle progression (Figure 2). Importantly, none of the constrained indazole-based 4EGI-1 mimetic analogs had any impact on the expression of β-actin or glyceraldehyde 3-phosphate dehydrogenase (GAPDH), two bona fide housekeeping proteins. Interestingly, while active in the FP assay, 1l does not appear to be active in the cell-based assays (see also Figure 4). This may either be due to poor cell penetration, in vivo instability, or binding with higher affinity to other cellular proteins.
Figure 2.
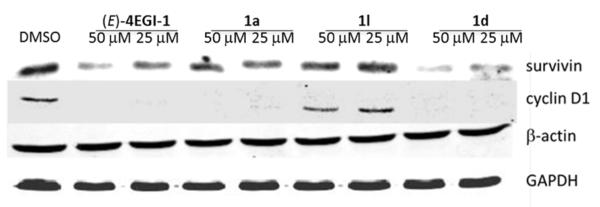
Inhibition of oncogenic proteins expression by constrained indazole-based 4EGI-1 mimetic. Inhibition of the expression of oncogenes cyclin D1 and survivin in human melanoma CRL-2813 cells that were treated with 4EGI-1 and constrained indazole-based 4EGI-1 mimetic at final concentration of 50 and 25 μm for a time period of 8 hrs. Immunoblots for β-actin and GAPDH indicate equal loading and, as anticipated, lack of inhibition of expression for housekeeping proteins.
Figure 4.
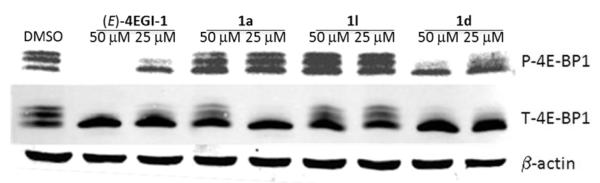
Effect of constrained indazole-based 4EGI-1 mimetic on the expression downstream effectors of mTOR. CRL-2813 were treated with DMSO, (E)-4EGI-1, 1a, 1l or 1d at final concentration of 50 and 25 μm for a time period of 8 hrs. Changes in the expression of p70S6K, phosphorylated- and total-4E-BP1 (P- and T-4E-BP1, respectively) are shown. Immunoblot for β-actin is shown as a control for total protein in the extract, as well as evidence for lack of effect of these compounds on housekeeping proteins.
Inhibition of cancer cells proliferation
Adherent human melanoma cells (CRL-2813), in addition to representing a prevalent human cancer, were found to be the most responsive to inhibition of proliferation by 4EGI-1.[20a] This sensitivity to anti-proliferative agents is most likely attributed to the presence of a BRAF mutation.[43] We have previously shown that Ras-Raf-MAPK driven cell proliferation is dependent on cyclin D1 expression.[44] The significant inhibition of cyclin D1 expression by our compounds may therefore explain, at least in part, the sensitivity of the melanoma cells to these agents. We therefore chose the CRL-2813 cells for evaluating the anti-proliferative activity of the three selected compounds, 1a, 1d, and 1l, from the configurationally constrained (E)-4EGI-1 mimetic library (Table 2).
Table 2.
cLogP and IC50 values (μm) of selected compounds from the library of the constrained mimetics of 4EGI-1 in inhibition of cell proliferation assay by SRB in human melanoma CRL-2813.
| Compound | R1 | R2 | R3 | cLogP | SRB Assay IC50 (μm) in CRL-2813 |
|
|---|---|---|---|---|---|---|
| 1 | (E)-4EGI-1 | NO2 | 3,4-dichloro | H | 4.76 | 1.23±0.15 |
| 2 | (Z)-4EGI-1 | NO2 | 3,4-dichloro | H | 4.76 | 12.17±0.95 |
| 3 | 1a | NO2 | H | H | 3.99 | 5.65±1.77 |
| 4 | 1d | NO2 | 3-OCH3 | H | 4.00 | 4.40±0.57 |
Effect on regulators of the translation initiation machinery
We have previously shown that 4EGI-1 inhibits expression of mTOR and phosphorylation of 4E-BP1.[44] Similar inhibition of mTOR expression was observed with compound 1d as can be seen in Figure 3. Indazole derivative 1d also inhibited p70S6K, a substrate for mTOR. This finding prompted us to investigate the effects of selected constrained indazole-based 4EGI-1 mimetic on the expression and phosphorylation of 4E-BP1, downstream effector of mTOR. Western blot analysis of 4EGI-1-, 1a-, 1l-, or 1d-treated CRL-2813 cell lysates shows that similar to 4EGI-1, 1a and 1d reduced phosphorylation of 4E-BP1 (P-4E-BP1) (Figure 4). Quantitative analysis of the ratio of phosphorylated 4E-BP1 (P-4E-BP1) to total 4E-BP1 (T-4E-BP1) indicates significant effect for 4EGI-1 and 1d, one of the most potent constrained indazole-based 4EGI-1 mimetic (Table 1), but a lesser effect for 1a. Consistent with its modest effect on the expression of cyclin D1 or survivin (see Figure 2) compound 1l had a marginal effect on phoshoylation of 4E-BP1 (Figure 5). As anticipated, treatment with these agents did not affect expression of the housekeeping proteins β-actin and GAPDH. These data indicate that, interfering with eIF4E/eIF4G PPI (Figure 1) inhibits translation initiation not only by blocking eIF4F formation but also by modifying expression of upstream mediators of phosphorylation of 4E-BP1 and p70S6K (Figures 4 and 6) perhaps in a feed-forward loop that further contributes to inhibition of translation initiation.
Figure 3.
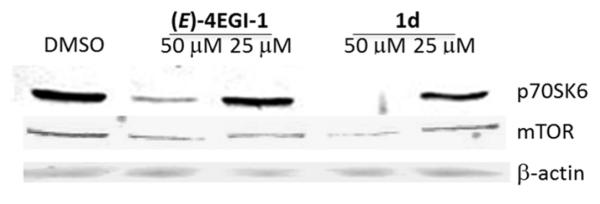
Effect of constrained indazole-based 4EGI-1 mimetic on the expression of mTOR. CRL-2813 were treated with DMSO, (E)-4EGI-1 or 1d at final concentration of 50 and 25 μm for a time period of 8 hrs and lysates were immunoblot with antibodies specific to mTOR and β-actin.
Figure 5.

Effect of (E)-4EGI-1 and compounds 1a, 1l, and 1d on the ratio of levels of phosphorylated 4E-BP1 (P-4E-BP1) to levels of total 4E-BP1 (T-4E-BP1) in CRL-2813, at final concentrations of 25 and 50 μm. Blots from two independent experiments were quantified using Odyssey software. The compounds reduced the levels of phosphorylated 4E-BP1 relative to the total amount.
Figure 6.

Effect of 4EGI-1 and its constrained indazole-based 4EGI-1 mimetic on mRNAs of cyclin D1, survivin, and p70S6K, whose expression was reduced in the presence of these analogs. Lysates of CRL-2813 cells that were incubated with the small molecules were analyzed by quantitative RT-PCR. The results from three independent experiments were calculated, and then were normalized to the levels of mRNA18S. The data are shown as the means ±S.D. and analyzed using one-way ANOVA with Dunnett post-test. The significance in difference was assigned at a level of less than a 5% probability (P<0.05).  mRNAcyclin
D1/mRNA18S,
mRNAcyclin
D1/mRNA18S,
 mRNAsurvivin/mRNA18S, and
mRNAsurvivin/mRNA18S, and  mRNAp70S6K/mRNA18S.
mRNAp70S6K/mRNA18S.
Effect of the constrained 4EGI-1 mimetic on protein expression is translational
To determine the mechanism of suppression of oncogenic protein expression by constrained indazole-based 4EGI-1 mimetic (Figures 4 and 6) we carried out real-time quantitative polymerase chain reaction (RT-PCR) analysis of the corresponding mRNAs using the 18S ribosomal RNA as an internal reference (Figure 6). Similar to 4EGI-1, the indazoles-derived 4EGI-1 mimetic 1a and 1d did not reduce the levels of cyclin D1, survivin, and p70S6K mRNAs. We therefore conclude that inhibition of oncogenic protein expression by constrained 4EGI-1 derivatives is translational.
Conclusions
In light of the recent progress made in our understanding of the role of eIF4F complex in normal and patho-biology, development of molecular probes that inhibit eIF4F complex formation is of great significance. Herein, we report a major advancement in the hit-to-lead optimization of 4EGI-1, the prototypic inhibitor of eIF4E/eIF4G interaction and cap-dependent translation initiation.[20a] We describe the rationale behind the design of the indazole-based constrained analogs of the hit compound 4EGI-1, development of an efficient synthetic pathway to obtain a focused library of such 3-substituted indazoles, and biological characterization of this library. In this new (E)-4EGI-1-mimetic chemotype, the original dynamically (E)-to-(Z) isomerization-susceptible hydrazone moiety becomes an integral part of the indazole scaffold and is locked in the (E)-mimicking configuration, preventing it from isomerizing into the (Z)-4EGI-1 mimicking form. Our findings strongly suggest that the (E)-configuration of 4EGI-1 contributes to the disruption of the interaction of eIF4E with eIF4G and to the inhibition of translation initiation. With this in mind, we demonstrate that structural rigidification can be combined with polarity- and solubility-enhancing substituents, resulting in molecular probes that avidly bind to eIF4E and effectively inhibit translation initiation. In summary, 1d emerges as a promising molecular probe for studying the role of eIF4F complex in normal- and patho-biology. We continue to explore a wide range of substituents on the phenyl rings and position 5 of the thiazolidine ring in order to further improve the potency and bioavailability of these indazole-based 4EGI-1 mimetics with the intention of testing them as therapeutic agents in a variety of proliferative and neuro-degenerative disorders. Furthermore, side by side comparison of these agents with chemical probes targeting other critical steps in the TI cascade[45] will shed light on the relative role of various TI factors in normal- and patho-biology.
Experimental Section
Chemistry
General
Intermediates and final compounds were purified using a reverse phase column (C18) on Isolera flash chromatography system from Biotage using a gradient of 0 to 100% methanol in water, unless otherwise noted. Low resolution HPLC-MS analysis was performed on Waters Alliance-Micromass ZQ with ESI source using an analytical Waters Symmetry C18 column (3.5μm, 2.1×100 mm) operating at 0.5 mL/min with a linear gradient of 0-100% of acetonitrile in water (both solvents contain 0.1% v/v formic acid) over 10 min. The purity of the compounds was determined by Waters Alliance analytical HPLC system operating at a flow rate of 1.0 mL/min, using a linear gradient of 0-100% acetonitrile in water (both solvents contain 0.1% v/v of trifluoroacetic acid) over 28 min, on a Waters XBridge BEH 130 C18 column (5μm, 4.6×100 mm). 1H, 13C, 19F, and 2D-NMR spectra were recorded at rt, unless otherwise noted, on a Varian Inova spectrophotometer (400 MHz for 1H, 100 MHz for 13C, and 376 MHz for 19F), or on Varian Inova spectrophotometer (600 MHz for 1H, 150 MHz for 13C), as noted. Chemical shifts were referenced to the residual solvent peaks in the deuterated solvent used. HR-MS spectra were taken on Agilent 6210 Time-of-Flight LC-MS with ESI source, the difference between the measured ion mass and the expected ion mass was less than 5 ppm. Melting points were measured on Electrothermal Mel-Temp manual melting point apparatus and are uncorrected.
General procedure A: Formation of thiocyanatoacetophenones (3a-h)
2-Bromoacetophenone (10 mmol) was dissolved in acetonitrile (50 ml). Potassium thiocyanate (20 mmol) was added and the mixture was refluxed for 2 hrs. The solvents were removed under vacuum and the residue extracted with a mixture of ethyl acetate and water. The organic phase was dried (MgSO4), filtered and the solvents removed under vacuum to yield the pure product.
1-Phenyl-2-thiocyanatoethanone (3a)
Off-white solid, mp 64 °C, 100% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.02 (d, J = 8.8 Hz, 2H), 7.72 (t, J = 7.2 Hz, 1H), 7.58 (t, J = 7.6 Hz, 2H), 5.11 (s, 2H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 192.8, 134.7, 129.4, 129.0, 113.3, 42.2; LC-MS (ESI+): Calcd. mass for C9H7NOS 177.02, found m/z 177.96 [M+H]+.
1-(3,4-Dichlorophenyl)-2-thiocyanatoethanone (3b)
Off-white solid, mp 103 °C, 100% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.25 (d, J = 2.0 Hz, 1H), 7.97 (dd, J = 8.4, 2.0 Hz, 1H), 7.52 (d, J = 8.4 Hz, 1H), 5.07 (s, 2H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 191.2, 137.5, 134.9, 132.5, 131.7, 131.0, 128.9, 113.0, 41.8; LC-MS (ESI+): Calcd. mass for C9H5Cl2NOS 244.95, found m/z 245.85, 247.83 [M+H]+.
1-(4-Methoxyphenyl)-2-thiocyanatoethanone (3c)
White solid, mp 121 °C, 100% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.00 (d, J = 8.8 Hz, 2H), 7.09 (d, J = 8.8 Hz, 2H), 5.06 (s, 2H), 3.87 (s, 3H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 191.0, 164.5, 131.5, 127.6, 114.6, 113.4, 56.1, 42.1; LC-MS (ESI+): Calcd. mass for C10H9NO2S 207.03, found m/z 207.98 [M+H]+.
1-(3-Methoxyphenyl)-2-thiocyanatoethanone (3d)
Recrystalized from 95% ethanol to give yellow needles, mp 71 °C, 100% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 7.54 (d, J = 8.0 Hz, 1H), 7.47 (m, 2H), 7.27 (d, J = 8.0, 2.4 Hz, 1H), 5.08 (s, 2H), 3.82 (s, 3H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 192.6, 159.9, 136.1, 130.6, 121.5, 120.8, 113.5, 113.3, 55.9, 42.2; LC-MS (ESI+): Calcd. mass for C10H9NO2S 207.03, found m/z 207.85 [M+H]+. AHPLC: 13.36 min, 100%.
1-(4-Chlorophenyl)-2-thiocyanatoethanone (3e)
White solid, mp 129 °C, 100% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.03 (d, J = 8.4 Hz, 2H), 7.66 (d, J = 8.8 Hz, 2H), 5.09 (s, 2H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 191.9, 139.7, 133.5, 131.0, 130.9, 129.6, 129.4, 113.2, 42.0; LC-MS (ESI+): Calcd. mass for C9H6ClNOS 210.99, found m/z 212.03 [M+H]+.
1-(2,4-Difluorophenyl)-2-thiocyanatoethanone (3f)
White solid, mp 64 °C, 100% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.04 (td, J = 8.8, 6.8 Hz, 1H), 7.50 (d, J = 11.6, 9.6, 2.4 Hz, 1H), 7.30 (dt, J = 8.8, 2.4 Hz, 1H), 4.96 (d, J = 2.8 Hz, 1H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 188.9 (d, J = 10.9, 3.9 Hz), 162.2 (dd, J = 269, 13.3 Hz), 162.8 (dd, J = 259, 13.2 Hz), 133.3, (dd, J = 10.9, 3.9 Hz), 120.3 (dd, J = 12, 4 Hz), 113.2 (d, J = 10.9 Hz), 113.1 (d, J = 21.8 Hz), 105.9 (t, J = 27 Hz), 45.0 (d, J = 7.7 Hz); LC-MS (ESI−): Calcd. mass for C9H5F2NOS 213.01, found m/z 212.09 [M−H]−.
1-(4-Fluorophenyl)-2-thiocyanatoethanone (3g)
White solid, mp 94 °C, 100% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.08 (dd, J = 8.6, 5.5 Hz, 2H), 7.39 (t, J = 8.8 Hz, 2H), 5.06 (s, 2H). 13C-NMR ([D6]-DMSO, 100 MHz) δ 191.4 (d, J = 2.0 Hz), 167.3 (t, J = 6.0 Hz), 164.8, 133.0 (dd, J = 9.7, 5.8 Hz), 132.5 (d, J = 10.1 Hz), 131.9, 131.6 (m), 131.3 (dd, J = 9.7, 2.7 Hz), 117.3 (d, J = 22.5 Hz), 115.7 (d, J = 22.4 Hz), 113.2 (t, J = 4.7 Hz), 42.0; LC-MS (ESI+): Calcd. mass for C9H6FNOS 195.02, found m/z 196.03 [M+H]+.
1-Phenyl-2-thiocyanatopropan-1-one (3h)
Brown oil, 100% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.05 (d, J = 7.2 Hz, 2H), 7.69 (d, J = 7.2 Hz, 1H), 7.56 (d, J = 7.6 Hz, 1H), 5.42 (q, J = 6.4, 1H), 1.63 (d, J = 6.4 Hz, 3H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 195.1, 134.6, 133.9, 129.4, 129.3, 111.5, 46.3, 18.4; LC-MS (ESI+): Calcd. mass for C10H9NOS 191.04, found m/z 191.94 [M+H]+.
General procedure B: Formation of 2-chloro-4-phenylthiazoles (4a-h)
Adopted from Merijanian et al.[32] HCl (g) was bubbled into a solution of 1-phenyl-2-thiocyanatoethanones (3) (10 mmol) in dry ether (100 ml) at 0 °C, until saturation. The suspension was left to warm spontaneously to rt, and stirred overnight. The volatiles were removed under vacuum, the residue dissolved in dichloromethane and washed with saturated solution of NaHCO3. Organic phase was dried (MgSO4), filtered and the solvents removed under vacuum. The residue was recrystalized from 95% ethanol.
2-Chloro-4-phenylthiazole (4a)
Off-white needles, mp 48 °C, 90% yield. 1H-NMR ([D6]-DMSO, 400 MHz): 8.12 (s, 1H), 7.91 (d, J = 7.2 Hz, 2H), 7.47 (t, J = 7.6 Hz, 1H), 7.39 (t, J = 7.2 Hz, 1H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 153.3, 151.1, 133.4, 129.4, 129.0, 126.2, 117.5; LC-MS (ESI+): Calcd. mass for C9H6ClNS 194.99, found m/z 195.91, 197.86 [M+H] +.
2-Chloro-4-(3,4-dichlorophenyl)thiazole (4b)
White needles, mp 96 °C, 72% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.29 (s, 1H), 8.10 (d, J = 1.6 Hz, 1H), 7.86 (dd, J = 8.4, 2.0 Hz, 1H), 7.67 (d, J = 8.4 Hz, 1H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 151.6, 150.6, 133.8, 132.2, 131.5, 131.4, 127.8, 126.3, 119.5; LC-MS (ESI+): Calcd. mass for C9H4Cl3NS 262.91, found m/z 263.83, 265.81, 267.79 [M+H]+.
2-Bromo-4-(4-methoxyphenyl)thiazole (4c)
White solid, mp 97 °C, 89% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 7.98 (s, 1H), 7.85 (d, J = 8.8 Hz, 2H), 7.01 (d, J = 8.8 Hz, 2H), 3.81 (s, 3H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 160.0, 154.8, 136.1, 127.7, 126.2, 116.9, 114.7, 55.7; LC-MS (ESI+): Calcd. mass for C10H8BrNOS 268.95, found m/z 269.78, 271.83 [M+H]+.
2-Chloro-4-(3-methoxyphenyl)thiazole (4d)
White needles, mp 53 °C, 78% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.11 (s, 1H), 7.46 (d, J = 7.6 Hz, 1H), 7.42 (d, J = 2.4 Hz, 1H), 7.33 (t, J = 8.0 Hz, 1H), 6.92 (dd, J = 8.0, 2.4 Hz, 1H), 3.79 (s, 3H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 160.1, 153.1, 150.9, 134.8, 130.5, 118.6, 117.8, 114.8, 111.5, 55.6; LC-MS (ESI+): Calcd. mass for C10H8ClNOS 225.00, found m/z 225.89, 227.87 [M+H]+. AHPLC: 16.97 min, 100%.
2-Chloro-4-(4-chlorophenyl)thiazole (4e)
White solid, mp 99 °C, 100% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.18 (s, 1H), 7.93 (d, J = 8. 0 Hz, 2H), 7.52 (d, J = 8.8 Hz, 2H). 13C-NMR ([D6]-DMSO, 100 MHz) δ 152.0, 151.4, 133.6, 132.3, 129.4, 128.0, 118.3; LC-MS (ESI+): Calcd. mass for C9H5Cl2NS 228.95, found m/z 229.92, 231.93 [M+H]+.
2-Chloro-4-(2,4-difluorophenyl)thiazole (4f)
White solid, mp 54 °C, 89% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.04 (td, J = 8.8, 6.4 Hz, 1H), 7.95 (d, J = 2.8 Hz, 1H), 7.41 (ddd, J = 12.0, 9.6, 2.8 Hz, 1H), 7.21 (tdd, J = 8.4, 2.8, 0.8 Hz, 1H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 162.4 (dd, J = 248.3, 12.4 Hz), 159.9 (dd, J = 252.3, 12.4 Hz), 151.2, 146.2, 131.3 (dd, J = 9.3, 3.8 Hz), 125.5 (d, J = 13.1 Hz), 118.0 (dd, J = 10.9, 3.1 Hz), 112.6 (dd, J = 21.1, 3.1 Hz), 105.3 (t, J = 10.3 Hz). 19F-NMR ([D6]-DMSO, 376 MHz) δ −109.4 (quintet, J = 8 Hz), −110.1 (qd, J = 9, 2.6 Hz), LC-MS (ESI+): Calcd. mass for C9H4ClF2NS 230.97, found m/z 231.93, 233.95 [M+H]+.
2-Chloro-4-(4-fluorophenyl)thiazole (4g)
White solid, mp 78 °C, 76% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.06 (s, 1H), 7.91 (dd, J = 8.4, 5.4 Hz, 1H), 7.25 (t, J = 8.8 Hz, 1H). 13C-NMR ([D6]-DMSO, 400 MHz) δ 162.9 (d, J = 245.9 Hz), 152.5, 151.5, 130.3 (d, J = 2.2 Hz), 128.7 (d, J = 8.0 Hz), 117.6, 116.5 (d, J = 21.9 Hz); 13F-NMR ([D6]-DMSO, 376 MHz) δ −113.5 (septet); LC-MS (ESI+): Calcd. mass for C9H5ClFNS 212.98, found m/z 213.98, 216.0 [M+H]+.
2-chloro-5-methyl-4-phenylthiazole (4h)
Colorless oil, 75% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 7.63 (d, J = 8.0 Hz, 2H), 7.47 (t, J = 8.4 Hz, 2H), 7.40 (t, J = 8.0 Hz, 1H), 2.52 (s, 3H); 13C-NMR ([D6]-DMSO, 400 MHz) δ 149.4, 146.5, 133.8, 131.6, 129.0, 128.8, 128.6, 128.4, 12.8; LC-MS (ESI+): Calcd. mass for C10H8ClNS 209.01, found m/z 210.01, 212.03 [M+H]+.
General procedure C: Formation of N-substituted indazoles (1′a-c) and N-substituted 5-nitroindazoles (1a-h)
Indazole (1 mmol) or 5-nitro-1H-indazole-3-carboxylic acid[33] (1 mmol) in DMSO (3 ml) was added dropwise to a suspension of NaOH (2.5 eq, fine powder) in DMSO (1 ml). After 5 min, a solution of the appropriate 2-halo-4-phenylthiazole (4a-g), or 2-chloro-5-methyl-4-phenylthiazole (4h) (1 mmol) in DMSO (2 ml) was added and the reaction mixture was heated to 120 °C for 3 days. The solution was cooled to rt, and the formed precipitate was filtered, then re-suspended in water, neutralized by 0.1 M HCl, and filtered. See work-up for each compound below. Note: Once the nitro group was introduced on C-5 of the indazole ring, the solubility of these derivatives was significantly reduced to the extent that recording 13C-NMR was not possible as the compounds precipitated from the hot solution during the acquisition, sometimes even when the probe’s temperature was kept at 65 °C. Running a 13C-NMR scan with less concentrated samples would take longer than 13 hrs.
1-(4-Phenylthiazol-2-yl)-1H-indazole-3-carboxylic acid (1′a)
White solid, mp 370 °C, 31% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.63 (d, J = 8.0 Hz, 1H), 8.38 (d, J = 7.6 Hz, 1H), 8.06 (d, J = 7.2 Hz, 1H), 7.87 (s, 1H), 7.64 (t, J = 7.2 Hz, 1H), 7.49 (t, J = 7.6 Hz, 2H), 7.37 (m, 2H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 164.7, 162.2, 151.9, 149.2, 139.0, 134.3, 129.3, 129.0, 128.7, 126.2, 126.0, 124.7, 124.0, 113.5, 109.8; LC-MS (ESI+): Calcd. mass for C17H11N3O2S 321.06, found m/z 322.19 [M+H]+; HR-MS (ESI+): Calcd. for C17H12N3O2S: 322.06447, measured: 322.06490 [M+H]+, 344.04683 [M+Na]+; AHPLC: 17.74 min, 96.3%.
5-Nitro-1-(4-phenylthiazol-2-yl)-1H-indazole-3-carboxylic acid (1a)
Recrystalized from acetic acid. White needles, mp 298 °C, 40% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.98 (d, J=1.6 Hz, 1H), 8.91 (d, J = 9.6 Hz, 1H), 8.52 (dd, J = 9.2, 2.4 Hz, 1H), 8.08 (s+d, 3H), 7.52 (t, J = 7.6 Hz, 2H), 7.43 (t, J = 7.6 Hz, 1H); 13C-NMR ([D6]-DMSO, 150 MHz, recorded at 65 °C) δ 162.0, 160.7, 152.5, 145.3, 141.9, 141.0, 133.9, 129.3, 128.9, 126.4, 124.8, 124.3, 119.4, 115.6, 112.2; LC-MS (ESI+): Calcd. mass for C17H10N4O4S 366.04, found m/z 366.95 [M+H]+; HR-MS (ESI+): Calcd. for C17H11N4O4S: 367.04955, measured: 367.04880 [M+H]+, 389.03101 [M+Na]+; AHPLC: 17.83 min, 100%.
1-(4-(3,4-Dichlorophenyl)thiazol-2-yl)-1H-indazole-3-carboxylic acid (1′b)
White solid, mp 289 °C, 21% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 13.92 (bs, 1H), 8.66 (d, J = 8.4 Hz, 1H), 8.23 (d, J = 2.0 Hz, 1H), 8.20 (d, J = 8.4 Hz, 1H), 8.16 (s, 1H), 8.02 (dd, J = 8.4, 2.0 Hz, 1H), 7.76 (t, J = 7.2 Hz, 1H), 7.72 (d, J = 7.2 Hz, 1H), 7.52 (t, J = 7.6 Hz, 1H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 162.8, 161.7, 149.6, 140.4, 139.2, 134.5, 132.2, 131.5, 131.2, 130.3, 127.8, 126.5, 125.8, 124.7, 122.8, 114.3, 113.4; LC-MS (ESI+): Calcd. mass for C17H9Cl2N3O2S: 388.98, found m/z 390.02, 392.00 [M+H]+; HR-MS (ESI+): Calcd. for C17H10Cl2N3O2S: 389.98653, measured: 389.98689 [M+H]+, 411.96699 [M+Na]+; AHPLC: 22.99 min, 99.22%.
1-(4-(3,4-Dichlorophenyl)thiazol-2-yl)-5-nitro-1H-indazole-3-carboxylic acid (1b)
Recrystalized from acetic acid. Yellow solid, mp 339 °C, decmp, 33.8% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 9.23 (d, J=2.0 Hz, 1H), 8.73 (d, J = 9.2 Hz, 1H), 8.46 (dd, J = 9.2, 2.4 Hz, 1H), 8.29 (d, J = 2.4 Hz, 1H), 8.15 (s, 1H), 8.08 (dd, J = 8.4, 2.4 Hz, 1H), 7.74 (d, J = 8.4 Hz, 1H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 162.4, 161.9, 151.5, 149.5, 144.0, 140.6, 134.6, 132.2, 131.6, 131.1, 127.9, 126.6, 125.9, 123.9, 121.8, 114.4, 113.1; LC-MS (ESI−): Calcd. mass for C17H8Cl2N4O4S 433.96, found m/z 435.04, 433.13 [M−H]−, 391.08 389.10 [M-NO2]−; HR-MS (ESI−): Calcd. for C17H7Cl2N4O4S: 432.9571, measured: 432.9558 [M−H]−; AHPLC: 20.62 min, 98.93%.
1-(4-(3,4-Dichlorophenyl)thiazol-2-yl)-5-nitro-1H-indazole-3-carboxylic acid (1′c)
Recrystalized from acetic acid. White solid, mp 229 °C, 25% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.73 (d, J = 8.4 Hz, 1H), 8.23 (d, J = 8.0 Hz, 1H), 7.99 (d, J = 8.8 Hz, 2H), 7.80 (s, 1H), 7.76 (t, J = 7.2 Hz, 1H), 7.52 (t, J = 7.6 Hz, 1H), 7.04 (d, J = 8.8 Hz, 2H), 3.81 (s, 3H); 13C-NMR ([D6]-DMSO, 150 MHz) δ 161.4, 159.7, 158.3, 150.5, 137.7, 128.5, 126.1, 125.3, 124.0, 123.1, 121.2, 113.1, 112.8, 107.4, 54.1; LC-MS (ESI+): Calcd. mass for C18H13N3O3S 351.07, found m/z 352.03 [M+H]+; HR-MS (ESI+): Calcd. for C18H14N3O3S: 352.07504, measured: 352.07541 [M+H]+, 374.05702 [M+Na]+; AHPLC: 17.17 min, 96.41%.
1-(4-(4-Methoxyphenyl)thiazol-2-yl)-5-nitro-1H-indazole-3-carboxylic acid (1c)
Yellow solid, mp 281 °C, 60% yield, mp 281 °C. 1H-NMR ([D6]-DMSO, 400 MHz) δ 9.22 (d, J = 2.0Hz, 1H), 8.76 (d, J = 9.6 Hz, 1H), 8.46 (dd, J = 9.2, 2.4 Hz, 1H), 8.00 (d, J = 8.8 Hz, 2H), 7.80 (s, 1H), 7.05 (d, J = 8.8 Hz, 2H), 3.82(s, 3H); 13C-NMR ([D6]-DMSO, 400 MHz) δ 162.5, 161.3, 152.0, 144.0, 140.6, 127.8, 126.9, 125.7, 123.8, 121.7, 114.7, 114.4, 108.8, 55.7; LC-MS (ESI+): Calcd. mass for C18H13N4O5S 396.05, found m/z 397.03 [M+H]+; HR-MS (ESI+): Calcd. for C18H13N4O5S: 397.06012, measured: 397.06071 [M+H]+, 419.04280 [M+Na]+ ; AHPLC: 17.53 min, 98.23%.
1-(4-(3-Methoxyphenyl)thiazol-2-yl)-5-nitro-1H-indazole-3-carboxylic acid (1d)
Off-white solid, mp 286 °C, 39% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.98 (s, 1H), 8.87 (d, J = 9.6 Hz, 1H), 8.59 (d, J = 10.0 Hz, 1H), 8.09 (s, 1H), 7.64 (d, J = 7.2 Hz, 1H), 7.56 (s, 1H), 7.41 (t, J = 8.4 Hz, 1H), 6.98 (dd, J = 8.0, 2.0 Hz, 1H), 3.85 (s, 3H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 161.3, 160.2, 151.9, 144.0, 140.6, 135.4, 130.5, 125.7, 124.0, 121.6, 118.8, 114.4, 114.3, 118.9, 111.4, 55.7. MS (ESI+): Calcd. mass for C18H12N4O5S 396.05, found m/z 397.08 [M+H]+; HR-MS (ESI+): Calcd. for C18H13N4O5S: 397.06012, measured: 397.05951 [M+H]+; AHPLC: 17.59 min, 97.64%.
1-(4-(4-Chlorophenyl)thiazol-2-yl)-5-nitro-1H-indazole-3-carboxylic acid (1e)
Recrystalized from DMSO. Yellow solid, mp 348 °C, decomp, 33% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.97 (s, 1H), 8.87 (d, J = 8.8 Hz, 1H), 8.54 (dd, J = 9.2, 2.0 Hz, 1H), 8.11 (s, 1H), 8.08 (d, J = 8.8 Hz, 1H), 7.54 (d, J = 8.4 Hz, 2H); LC-MS (ESI+): Calcd. mass for C17H9ClN4O4S 400.00, found m/z 401.07, 403.09 [M+H]+; HR-MS (ESI−): Calcd. for C17H8ClN4O4S: 398.9960, measured: 398.9962 [M−H]−; AHPLC: 19.39 min, 98.63%.
1-(4-(2,4-Difluorophenyl)thiazol-2-yl)-5-nitro-1H-indazole-3-carboxylic acid (1f)
Recrystalized from acetic acid. Yellow solid, mp 299 °C, 28% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.98 (s, 1H), 8.79 (d, J = 8.8 Hz, 1H), 8.50 (d, J = 8.4 Hz, 1H), 8.23 (m, 1H), 7.84 (s, 1H), 7.35 (t, J = 9.6 Hz, 1H), 7.22 (m, 1H); LC-MS (ESI+): Calcd. mass for C17H8F2N4O4S 402.02, found m/z 402.96 [M+H]+; HR-MS (ESI−): Calcd. for C17H7F2N4O4S: 401.0162, measured: 401.0151 [M−H]−; AHPLC: 18.55 min, 99.27%.
1-(4-(4-Fluorophenyl)thiazol-2-yl)-5-nitro-1H-indazole-3-carboxylic acid (1g)
The reaction mixture was neutralized with 1N HCl, the precipitate filtered, washed with water and dried. Off-white solid, mp 309 °C, 100% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 9.27 (d, J = 2.4 Hz, 1H), 8.78 (d, J = 9.6 Hz, 1H), 8.46 (dd, J = 9.2, 2.0 Hz, 1H), 8.15 (dd, J = 8.4, 5.2 Hz, 2H), 7.96 (s, 1H), 7.35 (t, J =8.8 Hz, 2H); 13C-NMR ([D6]-DMSO, 100 MHz, recorded at 70 °C) δ 164.4 (d, J = 244.5 Hz), 160.1, 150.6, 144.4, 140.3, 129.8, 127.8 (d, J = 8.5 Hz), 123.8, 118.9, 115.4, (d, J = 21.0 Hz), 114.7, 111.1; 19F-NMR ([D6]-DMSO, 376 MHz) δ −113.29 (m); LC-MS (ESI+): Calcd. mass for C17H9FN4O4S 384.03, found m/z 384.88 [M+H]+, 366.90 [M-NO2]+; HR-MS (ESI−): Calcd. for C17H8FN4O4S: 383.0256, measured: 383.0251 [M−H]−; AHPLC: 17.94 min, 100%.
1-(5-Methyl-4-phenylthiazol-2-yl)-5-nitro-1H-indazole-3-carboxylic acid (1h)
White solid, mp 304 °C, decmp, 98% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 9.26 (d, J = 1.6 Hz, 1H), 8.64 (d, J = 9.2 Hz, 1H), 8.43 (dd, J = 9.2, 2.0 Hz, 1H), 7.82 (d, J = 7.6 Hz, 2H), 7.54 (t, J = 7.6 Hz, 2H), 7.44 (m, 1H), 2.61 (s, 3H); LC-MS (ESI+): Calcd. mass for C18H12N4O4S 380.06, found m/z 381.10 [M+H]+; HR-MS (ESI+): Calcd. for C18H13N4O4S: 381.06520, measured: 381.06635 [M+H]+; AHPLC: 18.84 min, 100%.
General procedure D: Formation of 2-hydrazinyl-4-phenylthiazoles (5 and 5*)
Thiosemicarbazide (1 mmol) was added to a solution of bromoacetophenone (1 mmol) in dioxane (15 ml) and stirred at rt overnight. For some of the derivatives, in addition to the desired 2-hydrazinyl-4-phenylthiazole (5), a 6-membered isomer (5*) was also formed. In these cases, the precipitate was filtered, dissolved in DMSO and loaded on a RP-Biotage column using a linear gradient of 0-100% methanol in water to allow the separation of the 2 isomers. Otherwise, the mixture of the isomers was used in the next step, and the 6-membered ring isomer was left unreacted. Additional work-up, NMR data, LC-MS spectra, yields (for 5* yield is based on LCMS chromatogram), and melting points are given below for the 2-hydrazinyl-4-phenylthiazole derivative (5) and in some cases for (5*) as well.
2-Hydrazinyl-4-phenylthiazole (5a)
Brown solid, mp 129 °C, 27% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.55 (s, 1H), 7.80 (d, J = 8.2 Hz, 2H), 7.36 (t, J = 7.8 Hz, 2H), 7.25 (t, J = 7.2 Hz, 1H), 7.09(s, 1H), 4.85 (bs, 2H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 176.4, 151.1, 135.7, 128.9, 127.6, 125.8, 102.3; LC-MS (ESI+): Calcd. mass for C9H9N3S 191.05, found m/z 192.12 [M+H]+, 175.07 [M-NH2]+; AHPLC: 7.88 min, 98.01%.
5-Phenyl-6H-1,3,4-thiadiazin-2-amine (5*a)
Brown solid, mp 125 °C, 40% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 10.08 (bs, 1H), 9.27 (bs, 1H), 7.85 (d, J = 6.2 Hz, 2H), 7.50 (m, 3H), 4.27 (s, 2H). 13C-NMR ([D6]-DMSO, 100 MHz) δ 164.7, 151.8, 133.4, 131.9, 129.5, 127.4, 22.6. LC-MS (ESI+): Calcd. for C9H9N3S: 191.25, found: 192.12 [M+H]+; AHPLC: 7.38 min, 98.92%.
4-(3,4-Dichlorophenyl)-2-hydrazinylthiazole (5b)
[20a] Yellow solid, 80% yield, mp 107 °C.
2-Hydrazinyl-4-(4-methoxyphenyl)thiazole (5c)
Brown solid, mp 188 °C, 87% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.48 (s, 1H), 7.70 (d, J = 6.4 Hz, 2H), 6.90 (d+s, 3H), 4.81 (s, 2H), 3.74 (s, 3H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 176.3, 158.9, 150.9, 128.6, 127.2, 114.2, 100.3, 55.5; LC-MS (ESI+): Calcd. mass for C10H11N3OS 221.06, found m/z 222.26 [M+H]+, 205.20 [M-NH2]+. AHPLC: 8.38 min, 97.46%.
2-Hydrazinyl-4-(3-methoxyphenyl)thiazole (5d)
Off white solid, mp 140 °C, 44% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.57 (bs, 1H), 7.36 (m, 2H), 7.26 (t, J = 8.0 Hz, 1H), 7.11 (s, 1H), 6.82 (d, J = 8.0 Hz, 1H), 4.85 (bs, 2H), 3.77 (s, 3H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 181.0, 164.6, 155.7, 141.8, 134.7, 123.0, 118.0, 115.9, 107.6, 107.5, 60.2; LC-MS (ESI+): Calcd. mass for C10H11N3OS 221.06, found m/z 222.32[M+H]+, 206.27 [M-NH]+; AHPLC: 8.20 min, 100%.
5-(3-Methoxyphenyl)-6H-1,3,4-thiadiazin-2-amine (5*d)
Off white solid, 16% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 7.44 (m, 3H), 7.13 (m, 1H), 4.29 (s, 2H), 3.80 (s, 3H). 13C-NMR ([D6]-DMSO, 100 MHz) δ 164.7, 160.0, 151.6, 134.8, 130.7, 119.9, 117.8, 112.4, 55.9, 22.7. LC-MS (ESI+): Calcd. for C10H11N3OS: 221.28, found: 222.19 [M+H]+; AHPLC: 7.99 min, 98.03%.
4-(4-Fluorophenyl)-2-hydrazinylthiazole (5g)
Gray solid, mp 108 °C, 60% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.57 (s, 1H), 7.82 (d, J = 5.6 Hz, 2H), 7.15 (m, 2H), 7.03 (s, 1H), 4.86 (s, 2H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 176.7, 162.0 (d, J = 244.6 Hz), 150.3, 132.5, 128.0 (d, J = 8.0 Hz), 115.9 (d, J = 21.2 Hz), 102.3; 19F-NMR ([D6]-DMSO, 376 MHz) δ −113.8; LC-MS (ESI−): Calcd. mass for C9H8FN3S 209.04, found m/z 193.04 [M-NH2]−; AHPLC: 8.30 min, 96.79%.
2-Hydrazinyl-5-methyl-4-phenylthiazole (5h)
White solid, mp 155 °C, 66% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.22 (bs, 1H), 7.54 (d, J = 8.8 Hz, 2H), 7.36 (t, J = 7.2 Hz, 2H), 7.25 (m, 1H), 4.72 (bs, 2H), 2.33 (s, 3H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 172.3, 146.0, 136.3, 128.5, 128.2, 127.1, 115.6, 12.7; LC-MS (ESI+): Calcd. mass for C10H11N3S 205.07, found m/z 206.06 [M+H]+, 189.01 [M-NH2]+; AHPLC: 8.78 min, 100%.
2-Hydrazinyl-4-(2-methoxyphenyl)thiazole (5i)
Off-white solid, mp 122 °C, 89% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.43 (s, 1H), 8.01 (dd, J = 7.6, 1.6 Hz, 1H), 7.23 (m, 1H), 7.21 (s, 1H), 7.05 (d, J = 8.0 Hz, 1H), 6.96 (t, J = 7.6 Hz, 1H), 4.80 (s, 1H), 3.88 (s, 3H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 174.5, 157.0, 146.9, 129.4, 128.4, 123.8, 120.7, 111.8, 106.7, 55.8. LC-MS (ESI+): Calcd. mass for C10H11N3OS 221.06, found m/z 222.19[M+H]+, 205.14 [M-NH2]+; AHPLC: 8.39 min, 100%.
4-(2,5-dimethoxyphenyl)-2-hydrazinylthiazole (5j)
Brown solid, mp 123 °C, 88% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.45 (s, 1H), 7.58 (d, J = 3.2 Hz, 1H), 7.24 (s, 1H), 6.96 (d, J = 9.2 Hz, 1H), 6.78 (dd, J = 8.8, 3.2 Hz, 1H), 4.79 (s, 2H), 3.81 (s, 3H), 3.70 (s, 3H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 174.8, 153.6, 151.6, 147.0, 124.8, 114.9, 113.5, 113.2, 107.4, 56.5, 56.0; LC-MS (ESI+): Calcd. mass for C11H13N3O2S 251.07, found m/z 252.06 [M+H]+, 235.08 [M-NH2]+; AHPLC: 8.80 min, 98.68%.
4-(2,4-Dichlorophenyl)-2-hydrazinylthiazole (5k)
Peach-colored solid, mp 145 °C, 88% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.59 (s, 1H), 7.86 (d, J = 8.4 Hz, 1H), 7.60 (d, J = 2.0 Hz, 1H), 7.43 (dd, J = 8.4, 2.0 Hz, 1H), 7.18 (s, 1H), 4.87 (s, 2H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 175.5, 146.4, 132.9, 132.7, 132.4, 131.7, 130.1, 127.8, 107.9; LC-MS (ESI+): Calcd. mass for C9H7Cl2N3S 258.97, found m/z 261.91, 259.93 [M+H]+, 244.89, 242.88 [M-NH2]+; AHPLC: 10.51 min, 100%.
5-(2,4-Dichlorophenyl)-6H-1,3,4-thiadiazin-2-amine (5*k)
Off white solid, mp 161 °C, 8% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 7.72 (d, J = 1.6 Hz, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.50 (dd, J = 8.4, 1.6 Hz, 1H), 6.94 (bs, 2H), 3.54 (s, 2H). 13C-NMR ([D6]-DMSO, 100 MHz) δ 149.7, 146.7, 136.6, 134.7, 132.7, 129.6, 127.9, 24.8. LCMS (ESI+): Calcd. for C9H7Cl2N3S: 260.14, found: 262.04, 260.06 [M+H]+; AHPLC: 9.97 min, 100%.
2-Hydrazinyl-4-(4-(pyrrolidin-1-yl)phenyl)thiazole (5l)
Yellow solid, mp 106 °C, 44% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 9.44 (s, 2H), 7.47 (d, J = 8.4 Hz, 2H), 6.74 (s, 1H), 6.61 (d, J = 8.4 Hz, 2H), 3.25 (m, 4H), 1.94 (m, 4H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 168.6, 148.5, 142.5, 130.6, 115.4, 112.1, 99.0, 48.1, 25.4; LC-MS (ESI+): Calcd. mass for C13H16N4S 260.11, found m/z 261.18 [M+H]+, 244.20 [M-NH]+; AHPLC: 10.61 min, 95.91%.
5-(4-(Pyrrolidin-1-yl)phenyl)-6H-1,3,4-thiadiazin-2-amine (5*l)
Yellow solid, 21% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 7.76 (d, J = 8.4 Hz, 2H), 6.63 (d, J = 8.4 Hz, 2H), 4.24 (s, 2H), 3.31 (m, 4H), 1.97 (m, 4H). 13C-NMR ([D6]-DMSO, 100 MHz) δ 163.9, 152.1, 150.2, 129.0, 112.0, 47.7, 25.4, 22.3. LC-MS (ESI+): Calcd. for C13H16N4S: 260.36, found: 261.12 [M+H]+; AHPLC: 9.33 min, 98.95%.
4-(4-(2-Hydrazinylthiazol-4-yl)phenyl)morpholine (5m)
Compound 5m was synthesized from 2m[46] according to general procedure D. Orange solid, mp 181 °C, 48% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 7.66 (d, J = 8.8 Hz, 2H), 6.91 (d, J = 9.2 Hz, 2H), 6.86 (s, 1H), 3.73 (m, 4H), 3.11 (m, 4H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 176.1, 151.2, 150.5, 127.0, 126.7, 115.2, 99.6, 66.5, 48.7; LC-MS (ESI+): Calcd. mass for C13H16N4OS 276.10, found m/z 277.09 [M+H]+, 260.02 [M-NH2]+; AHPLC: 7.51 min, 89.85%.
5-(4-morpholinophenyl)-6H-1,3,4-thiadiazin-2-amine (5*m)
Brown powder, mp 147 °C, 19% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 7.77 (d, J = 8.8 Hz, 2H), 6.98 (d, J = 9.2 Hz, 2H), 6.61(bs, 2H), 3.74 (m, 4H), 3.59 (s, 2H), 3.19 (m, 4H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 152.4, 148.1, 128.0, 125.5, 114.6, 66.4, 48.0, 21.8; LC-MS (ESI+): Calcd. mass for C13H16N4OS 276.10, found m/z 277.03 [M+H]+.
4-(2-Hydrazinylthiazol-4-yl)-N,N-dimethylaniline (5n)
Compound 5n was synthesized from 2n (2n was formed by dimethylation[47] of 1-(4-aminophenyl)ethanone followed by bromination,[46] according to general procedure D. Brown solid, mp 165 °C, 74% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.43 (s, 1H), 7.61 (d, J = 8.8 Hz, 2H), 6.76 (s, 1H), 8.21 (d, J = 8.8 Hz, 2H), 4.80 (bs, 2H), 2.90 (s, 6H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 176.1, 151.7, 150.0, 126.8, 124.3, 112.5, 98.4. LC-MS (ESI+): Calcd. mass for C11H14N4S 234.09, found m/z 235.08 [M+H]+, 218.03 [M-NH2]+.
4-(2-hydrazinylthiazol-4-yl)benzoic acid (5o)
Compound 5o was synthesized from 2o[46] according to general procedure D. Brown powder, mp 226 °C, 28% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.65 (s, 1H), 7.92 (m, 4H), 7.30 (s, 1H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 176.5, 167.6, 150.1, 149.6, 139.6, 130.1, 129.5, 125.8, 104.9; LC-MS (ESI+): Calcd. mass 235.04, found m/z 236.00 [M+H]+, 218.95 [M-NH2]+; AHPLC: 6.37 min, 95.88%
4-(2-Amino-6H-1,3,4-thiadiazin-4-yl)benzoic acid (5*o)
Brown powder, 50% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 10.18 (s, 1H), 9.37 (s, 1H), 8.00 (m, 4H), 4.32 (s, 2H). 13C-NMR ([D6]-DMSO, 100 MHz) δ 167.1, 164.8, 150.8, 137.3, 133.4, 130.2, 127.6, 22.6. LC-MS (ESI+): Calcd. for C10H9N3O2S: 235.26, found: 236.13 [M+H]+; AHPLC: 5.29 min, 100%.
2-(2-Hydrazinylthiazol-4-yl)benzoic acid (5p)
Compound 5p was synthesized from 2p,[48] according to general procedure D. Due to fast degradation upon purification 5p was used without purification. 21% yield. LC-MS (ESI+): Calcd. mass for C9H7BrO3 235.04, found m/z 236.00 [M+H]+, 217.96 [M-NH2]+.
2-(2-Hydrazinylthiazol-4-yl)phenol (5q)
Compound 5q was synthesized from 2q[49] according to general procedure D. Off-white solid, mp 190 °C, 32% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 11.84 (s, 1H), 8.91 (s, 1H), 7.62 (d, J = 8.0 Hz, 1H), 7.06 (m, 2H), 6.74 (m, 2H), 4.94 (s, 2H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 175.9, 155.8, 148.7, 148.5, 129.3, 126.7, 119.3, 118.9, 117.3, 101.3; LC-MS (ESI+): Calcd. mass for C9H9N3OS 207.05, found m/z 208.11 [M+H]+, 191.06 [M-NH2]+; AHPLC: 7.52 min, 95.58%.
2-(2-Amino-6H-1,3,4-thiadiazin-5-yl)phenol (5*q)
Yellow solid, 62% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 7.67 (d, J = 7.6 Hz, 1H), 7.30 (t, J = 7.6 Hz, 1H), 7.17 (bs, 2H), 6.91 (m, 2H), 3.89 (s, 2H). 13C-NMR ([D6]-DMSO, 100 MHz) δ 160.5, 152.5, 151.9, 131.6, 128.1, 118.8, 117.6, 117.4, 20.5. LC-MS (ESI+): Calcd. for C9H9N3OS: 207.25, found: 208.11 [M+H] +; AHPLC: 7.11 min, 100%.
3-(2-Hydrazinylthiazol-4-yl)phenol (5r)
Compound 5r was synthesized from 2r[49] according to general procedure D. Gray solid, mp 177 °C, 78% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 9.35 (bs, 1H), 8.51 (s, 1H), 7.21 (m, 2H), 7.13 (t, J = 8.0 Hz, 1H), 6.99 (s, 1H), 6.65 (m, 1H), 4.83 (bs, 2H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 176.2, 157.9, 151.2, 137.0, 129.8, 116.7, 114.6, 112.9, 102.2; LC-MS (ESI+): Calcd. mass for C9H9N3OS 207.05, found m/z 208.05 [M+H]+, 191.06 [M-NH2]+; AHPLC: 6.59 min, 93.38%.
4-(2-Hydrazinylthiazol-4-yl)phenol (5s)
Compound 5s was synthesized from 2s[49] according to general procedure D. Yellow solid, 168 °C, 14% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 9.36 (s, 1H), 8.37 (s, 1H), 7.53 (d, J = 8.8 Hz, 2H), 6.74 (s, 1H), 6.67 (d, J = 8.8 Hz, 2H), 4.73 (s, 2H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 176.1, 157.2, 151.3, 127.2, 127.1, 115.6, 99.4; LC-MS (ESI+): Calcd. mass for C9H9N3OS 207.05, found m/z 208.18 [M+H]+, 191.19 [M-NH2]+; AHPLC: 6.31 min, 92.30%.
4-(2-Amino-6H-1,3,4-thiadiazin-5-yl)phenol (5*s)
Brown powder, 57% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 7.66 (d, J = 8.8 Hz, 2H), 6.75 (d, J = 8.4 Hz, 2H), 3.52 (s, 2H). 13C-NMR ([D6]-DMSO, 100 MHz) δ 159.1, 149.1, 147.1, 128.4, 127.3, 115.8, 21.9. LC-MS (ESI−): Calcd. for C9H9N3OS: 207.25, found: 190.20 [M-OH]−; AHPLC: 9.66 min, 96.60%.
4-(2-Hydrazinylthiazol-4-yl)benzene-1,2-diol (5t)
Brown solid, mp 115 °C, 38% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.89 (bs, 2H), 8.44 (s, 1H), 7.20 (d, J = 2.4 Hz, 1H), 7.06 (dd, J = 8.0, 2.0 Hz, 1H), 6.74 (s, 1H), 6.70 (d, J = 8.0 Hz, 1H), 2.54 (s, 1H); 13C-NMR ([D6]-DMSO, 400 MHz) δ 175.9, 151.5, 145.4, 145.3, 127.6, 117.3, 115.9, 113.7, 99.4; LC-MS (ESI−): Calcd. mass for C9H9N3O2S 223.04, found m/z 222.32 [M−H]−, 206.26 [M-NH2]−; AHPLC: 5.63 min, 100%.
4-(2-Hydrazinyl-5-methylthiazol-4-yl)benzene-1,2-diol (5u)
Compound 5u was synthesized from 2u (The synthesis of compound 2u was adopted from Koch et al.[50] using 2-bromopropanoic acid and resorcinol: Off white solid, mp 151 °C, 82% yield. 1H-NMR ([D6]-acetone, 400 MHz) δ 8.85 (bs, 1H), 8.42 (s, 1H), 7.55 (m, 2H), 6.95 (d, J = 8.8 Hz, 1H), 5.56 (q, J = 6.6 Hz, 1H), 1.80 (d, J = 6.4 Hz, 1H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 182.4, 141.6, 135.9, 117.4, 113.5, 106.5, 105.8, 32.8, 10.7; LC-MS (ESI+): Calcd. mass for C9H9BrO3 243.97, found m/z 247.04, 244.99 [M+H]+) according to general procedure D. Off-white solid, mp 154 °C, 74% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 9.02 (bs, 2H), 7.03 (d, J = 2.0 Hz, 1H), 6.84 (dd, J = 8.4, 2.0 Hz, 1H), 6.77 (d, J = 8.0 Hz, 1H), 2.31 (s, 3H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 175.7, 170.0, 145.3, 145.1, 126.6, 119.7, 116.1, 115.8, 113.9, 12.7; LC-MS (ESI+): Calcd. mass for C10H11N3O2S 237.06, found m/z 238.05, [M+H]+, 221.00 [M-NH2]+.
5-Nitroisatine
Prepared according to Magiatis et al.[39] Orange solid, 100% Yield. 1HNMR ([D6]-DMSO, 400 MHz) δ 11.64 (s, 1H), 8.41 (dd, J = 8.8, 2.4 Hz, 1H), 8.17 (d, J = 2.4 Hz, 1H), 7.06 (d, J = 8.8 Hz, 1H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 182.8, 160.3, 155.6, 143.0, 133.5, 120.0, 118.5, 112.9; LC-MS (ESI−): Calcd. mass for C8H4N2O4 192.02, found m/z 190.86 [M−H]−, 162.91 [M-CO]−.
2-(2-Amino-5-nitrophenyl)-2-oxoacetic acid
Prepared according to Halváč et al.[39] 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.39 (d, J = 2.8 Hz, 1H+2H of NH2), 8.11 (dd, J = 9.2, 2.4 Hz, 1H), 6.95 (d, J = 9.2 Hz, 1H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 189.7, 166.0, 156.9, 135.6, 130.9, 130.4, 118.2, 110.3; LC-MS (ESI−): Calcd. mass for C8H6N2O5 210.03, found m/z 209.01 [M−H]−.
2-(2-bromo-5-nitrophenyl)-2-oxoacetic acid (6)
The procedure was adopted from Pospíšil et al.[51]: A solution of NaNO2 (3.92 gr, 56.8 mmol) in water (14 gr) was added dropwise at 0 °C to 2-(2-amino-5-nitrophenyl)-2-oxoacetic (5.0 gr, 23.8 mmol) in aqueous HBr (14 mL of 48 wt. % in H2O into 14 mL water) maintaining the temperature at 0 °C. After the addition was complete, the solution was allowed to stir at this temperature for another 30 minutes, and then transferred slowly into a suspension of CuBr (6.2 gr, 43.2 mmol) in aqueous HBr (14 mL of 48 wt. % in H2O into 14 mL water). The resulting mixture was stirred at rt for an hour, and then extracted with ethyl acetate. The organic phase was washed with saturated NaHCO3 and brine, filtered through Celite, dried (MgSO4) and evaporated to yield a yellow solid (82% yield), mp 109 °C. 1HNMR ([D6]-DMSO, 400 MHz) δ 8.49 (d, J = 2.8 Hz, 1H), 8.27 (dd, J = 8.4, 2.8 Hz, 1H), 8.03 (d, J = 8.8 Hz, 1H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 187.5, 162.1, 147.2, 138.4, 135.3, 127.9, 127.3, 125.9; LC-MS (ESI−): Calcd. mass for C8H4BrNO5 272.93, found m/z 273.94, 271.96 [M−H]−, 201.97, 199.92 [M-NO2]; AHPLC: 7.66 min, 100%.
General procedure E: Formation of the hydrazones (7)
Compound 6 (2 mmol) and the appropriate 2-hydrazinyl-4-phenylthiazole (2 mmol) were refluxed for 3 hrs in a mixture of ethanol (10 ml), acetic acid (0.5 ml) and water (4.5 ml). The precipitate that formed was filtered and was usually pure enough to be used in the next step. Otherwise, further purification was carried out by RP Biotage using a gradient of 0-100% methanol in water for 12 column volumes.
2-(2-Bromo-5-nitrophenyl)-2-(2-(4-phenylthiazol-2-yl)hydrazono)acetic acid (7a)
Yellow solid, mp 216 °C, 91% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.28 (s, 1H), 8.20 (dd, J = 8.8, 2.8 Hz, 1H), 8.02 (d, J = 8.8 Hz, 1H), 7.79 (d, J = 7.6 Hz, 2H), 7.46 (bs, 1H), 7.37 (m, 2H), 7.28 (t, J = 7.2 Hz, 1H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 164.2, 147.7, 134.5, 130.9, 129.1, 126.8, 126.0, 125.9, 106.4; LC-MS (ESI+): Calcd. mass for C17H11BrN4O4S 445.97, found m/z 446.82, 448.88 [M+H]+; AHPLC: 15.33 min, 96.27%.
2-(2-Bromo-5-nitrophenyl)-2-(2-(4-(3,4-dichlorophenyl)thiazol-2-yl)hydrazono) acetic acid (7b)
Yellow solid, mp 209 °C, 52% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.29 (d, J = 2.8 Hz, 1H), 8.21 (dd, J = 8.8, 2.4 Hz, 1H), 8.03 (m, 2H), 7.78 (dd, J = 8.8, 2.0 Hz, 1H), 7.69 (bs, 1H), 7.63 (d, J = 8.8 Hz, 1H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 164.0, 147.7, 134.6, 131.9, 131.4, 130.9, 130.4, 127.7, 126.1, 126.9, 126.0; LC-MS (ESI+): Calcd. mass for C17H9BrCl2N4O4S 513.89, found m/z 518.75, 516.70, 514.72 [M+H]+; AHPLC: 18.11 min, 100%.
2-(2-Bromo-5-nitrophenyl)-2-(2-(4-(4-methoxyphenyl)thiazol-2-yl)hydrazono)acetic acid (7c)
Yellow solid, mp 204 °C, 66% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.28 (d, J = 2.8Hz, 1H), 8.21 (dd, J = 9.2, 2.8 Hz, 1H), 8.04 (d, J = 8.8 Hz, 1H), 7.73 (d, J = 8.8 Hz, 2H), 7.28 (s, 1H), 6.94 (d, J = 8.8 Hz, 2H), 3.77 (s, 3H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 164.2, 159.4, 147.7, 134.5, 131.0, 127.3, 126.8, 126.0, 114.5, 55.6; LC-MS (ESI+): Calcd. mass for C18H13BrN4O5S 475.98, found m/z 477.09, 479.14 [M+H]+, 431.14, 433.13 [M-NO2]+; AHPLC: 15.11 min, 95.81%.
2-(2-Bromo-5-nitrophenyl)-2-(2-(4-(3-methoxyphenyl)thiazol-2-yl)hydrazono) acetic acid (7d)
Light yellow solid, mp 197 °C, 84% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.30 (d, J = 2.8 Hz, 1H), 8.22 (dd, J = 8.8, 2.8 Hz, 1H), 8.04 (d, J = 8.8 Hz, 1H), 8.02 (s, 1H), 7.50 (s, 1H), 7.34 (m, 2H), 7.29 (t, J = 7.6 Hz, 1H), 6.86 (dd, J = 8.4, 2.0 Hz, 1H), 3.76 (s, 3H). 13C-NMR ([D6]-DMSO, 100 MHz, 65 °C) δ 164.0, 160.2, 147.9, 134.5, 130.8, 130.1, 126.9, 126.7, 125.8, 118.5, 114.3, 111.5, 55.7. LC-MS (ESI+): Calcd. mass for C18H13BrN4O5S 475.98, found m/z 476.96, 478.91 [M+H]+; AHPLC: 15.30 min, 94.32%.
2-(2-Bromo-5-nitrophenyl)-2-(2-(4-(4-fluorophenyl)thiazol-2-yl)hydrazono)acetic acid (7g)
Yellow solid, mp 220 °C, 96% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.30 (d, J = 2.4 Hz, 1H), 8.22 (dd, J = 8.4, 2.8 Hz, 1H), 8.04 (d, J = 8.4 Hz, 1H), 7.84 (dd, J = 8.4, 5.6 Hz, 2H), 7.46 (s, 1H), 7.22 (m, 2H); 13C-NMR ([D6]-DMSO, 400 MHz) δ 164.1, 162.2 (d, J = 245.1 Hz), 147.7, 134.5, 130.9, 128.0 (d, J = 7.7Hz), 126.8, 126.0, 115.9 (d, J = 21.8 Hz); LC-MS (ESI−): Calcd. mass for C17H10BrFN4O4S 463.96, found m/z 462.88, 464.90 [M−H]−; AHPLC: 15.66 min, 100%.
2-(2-Bromo-5-nitrophenyl)-2-(2-(5-methyl-4-phenylthiazol-2-yl)hydrazono)acetic acid (7h)
Yellow solid, mp 213 °C, 91% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.21 (s, 1H), 8.16 (d, J = 7.6 Hz, 1H), 8.00 (d, J = 8.4 Hz, 1H), 7.52 (m, 2H), 7.39 (m, 2H), 7.31 (m, 1H), 2.39 (s, 3H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 164.4, 147.6, 134.4, 131.0, 128.8, 128.4, 128.0, 126.7, 125.7, 12.7. LC-MS (ESI+): Calcd. mass for C18H13BrN4O4S 459.98, found m/z 463.01, 461.03 [M+H]+, 417.00, 415.01 [M-NO2]+; AHPLC: 15.43 min, 100%.
2-(2-Bromo-5-nitrophenyl)-2-(2-(4-(2-methoxyphenyl)thiazol-2-yl)hydrazono)acetic acid (7i)
Yellow solid, mp 168 °C, 94% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.26 (s, 1H), 8.19 (d, J = 8.8 Hz, 1H), 8.01 (d, J = 89.2 Hz, 1H), 7.89 (d, J = 7.2 Hz, 1H), 7.46, (s, 1H), 7.26 (t, J = 7.6 Hz, 1H), 7.08 (d, J = 8.4 Hz, 1H), 6.94 (t, J = 7.6 Hz, 1H), 3.88 (s, 3H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 164.3, 157.0, 147.7, 134.5, 131.0, 129.3, 129.2, 126.8, 125.9, 120.8, 112.0, 55.9. LC-MS (ESI+): Calcd. mass for C18H13BrN4O5S 475.99, found m/z 477.09, 479.08 [M+H]+; AHPLC: 15.41 min, 100%.
2-(2-Bromo-5-nitrophenyl)-2-(2-(4-(2,5-dimethoxyphenyl)thiazol-2-yl)hydrazono)acetic acid (7j)
Yellow solid, mp 165 °C, 78% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.25 (s, 1H), 8.19 (d, J = 8.4 Hz, 1H), 8.02 (d, J = 8.4 Hz, 1H), 7.51 (s, 1H), 7.49 (s, 1H), 7.00 (d, J = 9.2 Hz, 1H), 6.82 (d, J = 9.2, 2.4 Hz, 1H), 3.83 (s, 3H), 3.65 (s, 3H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 164.2, 153.4, 151.3, 147.7, 134.5, 131.0, 126.8, 125.9, 114.4, 114.1, 113.3, 56.3, 55.9; LC-MS (ESI+): Calcd. mass for C19H15BrN4O6S 505.99, found m/z 507.24, 509.16[M+H]+, 461.16, 463.21 [M-NO2]+; AHPLC: 15.49 min, 100%.
2-(2-Bromo-5-nitrophenyl)-2-(2-(4-(2,4-dichlorophenyl)thiazol-2-yl)hydrazono)acetic acid (7k)
Light pink solid, mp 206 °C, 98% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.31 (d, J = 2.8 Hz, 1H), 8.22 (dd, J = 9.2, 2.8 Hz, 1H), 8.04 (d, J = 8.8 Hz, 1H), 7.80 (d, J = 8.8 Hz, 1H), 7.68 (d, J = 2.0 Hz, 1H), 7.53(s, 1H), 7.47 (dd, J = 8.8, 2.4 Hz, 1H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 164.1, 147.7, 134.5, 133.3, 132.7, 132.2, 130.9, 130.2, 127.9, 126.8, 126.1. LC-MS (ESI+): Calcd. mass for C17H9BrCl2N4O4S 513.89, found m/z 514.93, 516.95, 518.90 [M+H]+; AHPLC: 18.06 min, 93.65%.
2-(2-Bromo-5-nitrophenyl)-2-(2-(4-(4-(pyrrolidin-1-yl)phenyl)thiazol-2-yl)hydrazono)acetic acid (7l)
Dark brown solid, mp 201 °C, 50% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.26 (d, J = 2.4 Hz, 1H), 8.20 (d, J = 8.8, 2.4 Hz, 1H), 8.03 (d, J = 8.8 Hz, 1H), 7.59 (d, J = 8.8 Hz, 2H), 7.03 (s, 1H), 6.51 (d, J = 8.8 Hz, 2H), 3.23 (m, 4H), 1.94 (m, 4H); 13C-NMR ([D6]-DMSO, 400 MHz) δ 164.3, 147.8, 147.7, 134.4, 131.0, 127.0, 126.8, 125.8, 111.9, 47.7, 25.4; LC-MS (ESI+): Calcd. mass for C21H18BrN5O4S 515.03, found m/z 516.04, 518.02 [M+H]+; AHPLC: 14.50 min, 93.49%.
2-(2-Bromo-5-nitrophenyl)-2-(2-(4-(4-morpholinophenyl)thiazol-2-yl)hydrazono)acetic acid (7m)
Yellow solid, mp 191 °C, 73% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.20 (d, J = 2.4 Hz, 1H), 8.19 (dd, J = 8.4, 2.8 Hz, 1H), 7.96 (d, J = 8.8 Hz, 1H), 7.59 (d, J = 9.2 Hz, 2H), 7.13 (s, 1H), 6.86 (d, J = 8.8 Hz, 2H), 3.65 (m, 4H), 3.06 (m, 4H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 164.5, 151.2, 147.9, 134.7, 131.2, 127.1, 115.4, 66.7, 48.6. LC-MS (ESI+): Calcd. mass for C21H18BrN5O5S 531.02, found m/z 532.04, 534.09 [M+H]+; AHPLC: 12.60 min, 95.94%.
2-(2-Bromo-5-nitrophenyl)-2-(2-(4-(4-(dimethylamino)phenyl)thiazol-2-yl)hydrazono)acetic acid (7n)
Yellow solid, mp 191 °C, 65% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.26 (d, J = 2.8 Hz, 1H), 8.20 (dd, J = 8.8, 2.8 Hz, 1H), 8.03 (d, J = 8.8 Hz, 1H), 7.61 (d, J = 8.8 Hz, 2H), 7.08 (s, 1H), 6.70 (d, J = 8.8 Hz, 2H), 2.91 (s, 6H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 164.3, 150.4, 147.7, 134.4, 131.0, 126.9, 126.8, 125.8, 112.4, 40.4. LC-MS (ESI+): Calcd. mass for C19H16BrN5O4S 489.01, found m/z 490.19, 492.10 [M+H]+; AHPLC: 10.44 min, 100%.
4-(2-(2-((2-Bromo-5-nitrophenyl)(carboxy)methylene)hydrazinyl)thiazol-4-yl) benzoic acid (7o)
Yellow powder, mp 216 °C, 83% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.30 (d, J = 2.4 Hz, 1H), 8.22 (dd, J = 9.2, 2.8 Hz, 1H), 8.05 (d, J = 8.8 Hz, 1H), 7.94 (m, 4H), 7.68 (s, 1H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 167.4, 164.1, 147.7, 138.4, 134.5, 130.9, 130.2, 130.1, 126.8, 126.0, 125.9, 109.0; LC-MS (ESI+): Calcd. mass for C18H11BrN4O6S 489.96, found m/z Calcd. for 491.04, 493.03 [M+H]+, 445.03, 443.01 [M-NO2]+; AHPLC: 12.49 min, 95.47%.
2-(2-(2-((2-Bromo-5-nitrophenyl)(carboxy)methylene)hydrazinyl)thiazol-4-yl) benzoic acid (7p)
Yellow solid, mp 183 °C, 68% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.26 (d, J = 2.4 Hz, 1H), 8.19 (dd, J = 8.4, 2.8 Hz, 1H), 8.01 (d, J = 8.4 Hz, 1H), 7.65 (d, J = 7.6 Hz, 1H), 7.54 (m, 2H), 7.43 (m, 1H), 7.01 (s, 1H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 169.7, 164.3, 147.6, 134.4, 132.9, 131.1, 131.0, 130.3, 129.3, 128.6, 126.7, 125.9, 125.4; LC-MS (ESI+): Calcd. mass for C18H11BrN4O6S 489.96, found m/z 493.03, 491.04 [M+H]+, 475.04, 473.06 [M-NO2]+; AHPLC: 12.43 min, 91.84%.
2-(2-Bromo-5-nitrophenyl)-2-(2-(4-(2-hydroxyphenyl)thiazol-2-yl)hydrazono)acetic acid (7q)
Yellow solid, mp 198 °C, 78% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 10.6 (s, 1H), 8.27 (d, J = 2.8 Hz, 1H), 8.17 (dd, J = 9.2, 2.8 Hz, 1H), 7.99 (d, J = 8.8 Hz, 1H), 7.73 (d, J = 7.2 Hz, 1H), 7.49 (s, 1H), 7.06 (t, J = 7.8 Hz, 1H), 6.81 (d, J = 8.0 Hz, 1H), 6.75 (d, J = 7.4 Hz, 1H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 164.1, 155.4, 147.7, 134.6, 130.9, 129.4, 127.9, 126.8, 126.2, 119.5, 117.1, 107.7; LC-MS (ESI+): Calcd. mass for C17H11BrN4O5S 461.96, found m/z 465.13, 463.08 [M+H]+, 419.11, 417.13 [M-NO2]+; AHPLC: 15.38 min, 96.30%.
2-(2-Bromo-5-nitrophenyl)-2-(2-(4-(3-hydroxyphenyl)thiazol-2-yl)hydrazono)acetic acid (7r)
Orange solid, mp 192 °C, 38% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 9.37, (s, 1H), 8.22 (d, J = 2.8 Hz, 1H), 8.14 (dd, J = 8.8, 2.8 Hz, 1H), 7.96 (d, J = 8.8 Hz, 1H), 7.29 (s, 1H), 7.11 (m, 3H), 6.63 (dd, J = 8.0, 1.6 Hz, 1H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 164.2, 158.0, 147.7, 134.5, 131.0, 130.0, 126.8, 116.9, 115.4, 113.0; LC-MS (ESI+): Calcd. mass for C17H11BrN4O5S 461.96, found m/z 463.01, 464.99[M+H]+; AHPLC: 12.86 min, 89.97%.
2-(2-Bromo-5-nitrophenyl)-2-(2-(4-(4-hydroxyphenyl)thiazol-2-yl)hydrazono)acetic acid (7s)
Yellow solid, 158 °C, 51% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 9.60 (s, 1H), 8.27 (d, J = 2.4 Hz, 1H), 8.22 (d, J = 2.8 Hz, 1H), 8.19 (d, J = 2.4 Hz, 1H), 8.03 (d, J = 8.8 Hz, 2H), 7.61 (d, J = 8.8 Hz, 2H), 7.16 (s, 1H), 6.76 (d, J = 8.4, 2H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 164.2, 157.8, 147.7, 135.3, 134.5, 131.0, 127.4, 126.8, 126.2, 125.9, 115.8; LC-MS (ESI+): Calcd. mass for C17H11BrN4O5S 461.96, found m/z 462.95, 464.99 [M+H]+, 416.93, 418.98 [M-NO2]+; AHPLC: 12.44 min, 91.32%.
2-(2-Bromo-5-nitrophenyl)-2-(2-(4-(3,4-dihydroxyphenyl)thiazol-2-yl)hydrazono) acetic acid (7t)
Yellow solid, mp 185 °C, 89% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.27 (d, J = 2.4 Hz, 1H), 8.20 (s, 1H), 8.04 (s, 1H), 7.12 (m, 2H), 6.73 (s, 1H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 164.3, 147.7, 146.0, 145.7, 134.5, 131.0, 126.8, 125.9, 117.5, 116.0, 113.8; LC-MS (ESI+): Calcd. mass for C17H11BrN4O6S 477.96, found m/z 479.14, 481.13 [M+H]+, 433.13, 435.11[M-NO2]+; AHPLC: 11.31 min, 100%.
2-(2-Bromo-5-nitrophenyl)-2-(2-(4-(3,4-dihydroxyphenyl)-5-methylthiazol-2-yl)hydrazono)acetic acid (7u)
Yellow solid, mp 177 °C, 78% yield. 1H-NMR ([D6]-acetone, 400 MHz) δ 8.25 (d, J = 2.0 Hz, 1H), 8.16 (dd, J = 11.7, 2.7 Hz, 1H), 7.96 (d, J = 8.6 Hz, 1H), 6.98 (s, 1H), 6.84 (dd, J = 8.2, 1.6 Hz 1H), 6.77 (d, J = 8.2 Hz, 1H), 2.33 (s, 3H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 165.1, 146.6, 144.5, 144.0, 138.1, 135.4, 133.0, 129.1, 124.9, 123.9, 123.8, 119.1, 114.4, 114.0, 10.2; LC-MS (ESI+): Calcd. mass for C18H13BrN4O6S 491.97, found m/z 492.83, 494.81 [M+H]+; AHPLC: 11.45 min, 95.10%.
General procedure F: Formation of the N-substituted nitro-indazoles (1a-u) from hydrazone (7)
Hydrazone 7 (0.5 mmol), CuI (0.1 eq), DMEDA (0.3 eq) and Na2CO3 (2.2 eq) were refluxed in a mixture of ethanol in water (1:1, 2 ml) for 2-12 hrs. The full conversion of compound 7 to the desired product was monitored by HPLC-MS under the conditions described in the general section. The reaction mixture was cooled to rt, neutralized with 1N HCl, and the precipitate was filtered, dissolved in DMSO and loaded on RP-Biotage column for further purification, as described in the general section above. For compounds 1a-h see experimental data under general procedure C.
1-(4-(2-Methoxyphenyl)thiazol-2-yl)-5-nitro-1H-indazole-3-carboxylic acid (1i)
Yellow solid, mp 298 °C, 89% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 9.09 (s, 1H), 8.81 (d, J = 8.8 Hz, 1H), 8.51 (d, J = 9.6 Hz, 1H), 8.24 (d, J = 7.6 Hz, 1H), 7.98 (s, 1H), 7.39 (t, J = 7.8 Hz, 1H), 7.18 (d, J = 8.4 Hz, 1H), 7.13 (t, J = 7.4 Hz, 1H), 3.98 (s, 3H); 13C-NMR ([D6]-DMSO, 100 MHz, recorded at 70 °C) δ 159.6, 157.3, 148.4, 144.8, 140.9, 129.9, 129.7, 124.9, 124.3, 122.5, 121.2, 120.2, 115.3, 115.0, 112.5, 56.2. LC-MS (ESI+): Calcd. mass for C18H12N4O5S 396.05, found m/z 396.95 [M+H]+; HR-MS (ESI−): Calcd. for C18H11N4O5S: 395.0460, measured: 395.0456 [M−H]−; AHPLC: 18.03 min, 96.17%.
1-(4-(2,5-Dimethoxyphenyl)thiazol-2-yl)-5-nitro-1H-indazole-3-carboxylic acid (1j)
White solid, mp 298 °C, 89% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.90 (s, 1H), 8.73 (d, J = 9.2 Hz, 1H), 8.60 (d, J = 9.2 Hz, 1H), 7.99 (s, 1H), 7.61 (s, 1H), 7.07 (d, J = 8.8 Hz, 1H), 6.92 (d, J = 8.8 Hz, 1H), 3.88 (s, 3H), 3.79 (s, 3H). LC-MS (ESI+): Calcd. mass for C19H14N4O6S 426.06, found m/z 426.91 [M+H]+; HR-MS (ESI+): Calcd. for C19H15N4O6S: 427.07068, measured: 427.07188 [M+H]+, 449.05356 [M+Na]+; AHPLC: 17.84 min, 99.43%.
1-(4-(2,4-Dichlorophenyl)thiazol-2-yl)-5-nitro-1H-indazole-3-carboxylic acid (1k)
Light pink solid, mp 332 °C, 87% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 9.27 (s, 1H), 8.65 (d, J = 9.2 Hz, 1H), 8.44 (d, J = 9.6 Hz, 1H), 8.09 (d, J = 8.4 Hz, 1H), 8.02 (s, 1H), 7.80 (d, J = 2.0 Hz, 1H), 7.61 (dd, J = 8.4, 2.4 Hz, 1H); MS (ESI+): Calcd. mass for C17H8Cl2N4O4S 433.96, found m/z 433.96, 435.98 [M+H]+; HR-MS (ESI−): Calcd. for C17H7Cl2N4O4S: 432.9571, measured: 434.970 [M−H]−; AHPLC: 20.89 min, 97.49%.
5-Nitro-1-(4-(4-(pyrrolidin-1-yl)phenyl)thiazol-2-yl)-1H-indazole-3-carboxylic acid (1l)
Brown solid, mp 290 °C, 61% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 9.00 (s, 1H), 8.87 (d, J = 9.2 Hz, 1H), 8.57 (dd, J = 9.6, 2.0 Hz, 1H), 7.83 (d, J = 8.8 Hz, 2H), 7.65 (s, 1H), 6.59 (d, J = 8.8 Hz, 2H), 3.31 (m, 4H), 2.02 (m, 4H); 13C-NMR ([D6]-DMSO, 100 MHz, recorded at 65 °C) δ 162.0, 160.1, 153.5, 148.4, 145.2, 141.7, 140.9, 127.5, 124.6, 124.3, 121.3, 119.3, 115.6, 112.2, 107.2, 47.8, 25.3; LC-MS (ESI+): Calcd. mass for C21H17N5O4S 435.10, found m/z 436.10 [M+H]+; HR-MS (ESI+): Calcd. for C21H18N5O4S: 435.10740, measured: 436.10594 [M+H]+, 458.08842 [M+Na]+; AHPLC: 18.52 min, 96.59%.
1-(4-(4-Morpholinophenyl)thiazol-2-yl)-5-nitro-1H-indazole-3-carboxylic acid (1m)
Brown solid, mp 270 °C, 62% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 8.90 (s, 1H), 8.79 (d, J = 9.6 Hz, 1H), 8.51 (d, J = 7.6, 1.6 Hz, 1H), 7.89 (d, J = 8.4 Hz, 1H), 7.85 (s, 1H), 7.18 (d, J = 7.2 Hz, 1H); LC-MS (ESI+): Calcd. mass for C21H17N5O5S 451.09, found m/z 451.97 [M+H]+; HR-MS (ESI+): Calcd. for C21H18N5O5S: 452.10232, measured: 452.10334 [M+H]+; AHPLC: 15.21 min, 95.95%.
1-(4-(4-(Dimethylamino)phenyl)thiazol-2-yl)-5-nitro-1H-indazole-3-carboxylic acid (1n)
Brown solid, mp 257 °C, 61% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 9.18 (d, J = 2.0 Hz, 1H), 8.78 (d, J = 8.8 Hz, 1H), 8.47 (dd, J = 9.2, 2.4 Hz, 1H), 8.27 (s, 1H), 7.87 (d, J = 9.2 Hz, 2H), 7.61 (s, 1H), 6.80 (d, J = 9.2 Hz, 2H), 2.96 (s, 6H); LC-MS (ESI+): Calcd. mass for C19H15N5O4S 409.08, found m/z 410.32 [M+H]+; HR-MS (ESI−): Calcd. for C19H16N5O4S: 410.09175, measured: 410.09373 [M+H]+; AHPLC: 12.60 min, 96.12%.
1-(4-(4-Carboxyphenyl)thiazol-2-yl)-5-nitro-1H-indazole-3-carboxylic acid (1o)
Brown solid, mp 316 °C, 42% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 9.01 (s, 1H), 8.93 (d, J = 8.0 Hz, 1H), 8.60 (d, J = 8.8 Hz, 1H), 8.27 (s, 1H), 8.22 (d, J = 8.8 Hz, 2H), 7.05 (d, J = 7.6 Hz, 2H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 167.4, 160.9, 151.1, 145.0, 140.8, 137.5, 130.8, 130.3, 130.0, 129.1, 126.3, 124.8, 119.4, 115.7, 114.4, 110.0; LC-MS (ESI+): Calcd. mass 410.03, found m/z 411.04 [M+H]+; HR-MS (ESI+): Calcd. for C18H11N4O6S: 411.03938, measured: 411.03859 [M+H]+; 13.71 min, 95.83%.
1-(4-(2-Carboxyphenyl)thiazol-2-yl)-5-nitro-1H-indazole-3-carboxylic acid (1p)
Yellow solid, mp 275 °C, 49% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 9.01 (d, J = 2.0 Hz, 1H), 8.76 (d, J = 9.6 Hz, 1H), 8.52 (dd, J = 9.6, 2.4 Hz, 1H), 7.91 (s, 1H), 7.82 (d, J = 7.6 Hz, 1H), 7.70 (d, J = 7.6 Hz, 1H), 7.63 (dt, J = 7.6, 1.2 Hz, 1H), 7.54 (t, J = 7.6 Hz, 1H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 170.6, 162.2, 159.7, 151.9, 148.2, 145.1, 140.9, 133.7, 132.6, 131.0, 129.7, 129.0, 128.9, 124.7, 124.2, 119.4, 115.4, 114.6. LC-MS (ESI+): Calcd. mass for C18H10N4O6S 410.03, found m/z 410.98 [M+H]+; HR-MS (ESI+): Calcd. for C18H11N4O6S: 411.03938, measured: 411.03998 [M+H]+, 433.02222 [M+Na]+, 448.99526 [M+K]+; AHPLC: 13.83 min, 96.71%.
1-(4-(2-Hydroxyphenyl)thiazol-2-yl)-5-nitro-1H-indazole-3-carboxylic acid (1q)
Yellow solid, mp 299 °C, 96% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 10.55 (s, 1H), 9.18 (s, 1H), 8.72 (d, J = 8.8 Hz, 1H), 8.45 (dd, J = 9.2, 1.2 Hz, 1H), 8.16 (dd, J = 8.0, 1.6 Hz, 1H), 7.98 (s, 1H), 7.16 (t, J = 15.5 Hz, 1H), 4.00 (d, J = 8.4 Hz, 1H), 6.92 (t, J = 7.2 Hz, 1H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 160.0, 155.7, 150.1, 148.9, 144.1, 140.6, 129.5, 129.3, 123.9, 121.5, 120.6, 119.7, 116.8, 114.5, 114.1; LC-MS (ESI+): Calcd. mass for C17H10N4O5S 382.04, found m/z 382.78 [M+H]+; HR-MS (ESI−): Calcd. for C17H19N4O5S: 381.0299, measured: 381.0286 [M−H]−; AHPLC: 15.85 min, 98.79%.
1-(4-(3-Hydroxyphenyl)thiazol-2-yl)-5-nitro-1H-indazole-3-carboxylic acid (1r)
Yellow solid, mp>400 °C, 68% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 9.23 (s, 1H), 8.66 (d, J = 8.8 Hz, 1H), 8.39 (d, J = 8.8 Hz, 1H), 7.84 (s, 1H), 7.52 (s, 1H), 7.45 (d, J = 7.2 Hz, 1H), 7.27 (t, J = 8.0 Hz, 1H), 6.80 (d, J = 7.2 Hz, 1H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 161.2, 159.9, 158.5, 158.4, 152.2, 143.9, 140.57, 135.3, 130.3, 124.6, 123.7, 121.8, 117.0, 115.9, 114.2, 113.4, 110.8. LC-MS (ESI+): Calcd. mass for C17H10N4O5S 382.04, found m/z 383.08 [M+H]+. HR-MS (ESI+): Calcd. for C17H11N4O5S: 383.04447, measured: 383.04599 [M+H]+, 405.02794 [M+Na]+; AHPLC: 14.40min, 98.09%.
1-(4-(4-Hydroxyphenyl)thiazol-2-yl)-5-nitro-1H-indazole-3-carboxylic acid (1s)
Brown solid, 231 °C, 87% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 9.73 (s, 1H), 8.99 (d, J = 2.0 Hz, 1H), 8.91 (d, J = 9.2 Hz, 1H), 8.56 (dd, J = 9.6, 2.0 Hz, 1H), 7.90 (d, J = 8.8 Hz, 2H), 7.79 (s, 1H), 6.88 (d, J = 8.4 Hz, 2H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 160.4, 158.4, 152.7, 148.7, 148.1, 145.1, 140.9, 127.9, 125.1, 124.8, 124.3, 119.5, 116.1, 115.7, 109.1; MS (ESI+): Calcd. mass for C17H10N4O5S 382.04, found m/z 382.97 [M+H]+; HR-MS (ESI+): Calcd. for C17H11N4O5S: 383.04447, measured: 383.04629 [M+H]+, 405.02794 [M+Na]+; AHPLC: 14.09 min, 95.07%.
1-(4-(3,4-Dihydroxyphenyl)thiazol-2-yl)-5-nitro-1H-indazole-3-carboxylic acid (1t)
Brown solid, mp 258 °C, 72% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 9.27 (bs, 1H), 9.09 (bs, 1H), 9.00 (s, 1H), 8.87 (d, J = 9.6 Hz, 1H), 8.56 (d, J = 9.2 Hz, 1H), 7.72 (s, 1H), 7.45 (s, 1H), 7.37 (d, J = 8.4 Hz, 1H), 6.85 (d, J = 8.4 Hz, 1H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 160.2, 152.8, 146.6, 145.9, 145.0, 125.5, 124.7, 119.5, 118.0, 116.3, 115.4, 113.9, 109.1; LC-MS (ESI+): Calcd. mass for C17H10N4O6S 398.03, found m/z 399.14 [M+H]+, 381.16 [M-OH]+; HR-MS (ESI+): Calcd. for C17H11N4O6S: 399.0394, measured: 399.0404 [M+H]+; AHPLC: 12.71 min, 100%.
1-(4-(3,4-Dihydroxyphenyl)-5-methylthiazol-2-yl)-5-nitro-1H-indazole-3-carboxylic acid (1u)
Yellow solid, mp 244 °C, 67% yield. 1H-NMR ([D6]-DMSO, 400 MHz) δ 9.09 (s, 1H), 8.70 (d, J = 9.2 Hz, 1H), 8.37 (dd, J = 9.6, 2.4 Hz, 1H), 7.17 (d, J = 1.6 Hz, 1H), 7.01 (dd, J = 1.6 Hz, 1H), 7.79 (d, J = 8.4 Hz, 1H), 2.49 (s, 3H); 13C-NMR ([D6]-DMSO, 100 MHz) δ 156.4, 148.1, 145.9, 145.8, 144.0, 140.3, 125.9, 123.7, 122.7, 120.8, 119.8, 116.3, 115.9, 114.4, 12.7; LC-MS (ESI+): Calcd. mass for C18H12N4O6S 412.05, found m/z 413.09 [M+H]+; HR-MS (ESI+): Calcd. for C18H13N4O6S: 413.05503, measured: 413.05543 [M+H]+; AHPLC: 13.23 min, 97.63%.
Biology
Cell growth assay
CRL-2813 human melanoma cells and CRL-2351 human breast cancer cells were grown per ATCC instructions (http://www.atcc.org). Cells were plated in standard 96-well tissue culture plates and treated with test compounds (dissolved in DMSO) at final concentrations of 17.14, 5.14, 1.54 and 0.46 μM. On the fifth day after addition of the compound, the plates were fixed with 10% cold trichloroacetic acid at 4 °C for 1hr, washed 5 times with water and air dried overnight at 4 °C. Sulforhodamine B (SRB) in 1% acetic acid was added to each well and incubated at rt for 30 min. Unbound dye was removed by washing the wells extensively with water containing 1% acetic acid, and the plates were air dried overnight at 4 °C. TRIS-base 10 mm was added to each well and the plates were read at 520 nm using Thermo Scientific MultiScan FC plate reader.[45a]
Western blotting
The WB analysis was performed as described.[45a] Briefly, CRL-2813 cells were seeded in 15×100 cm plates and treated with DMSO or the tested compounds at final concentration of 50, 25 μm for 8 hours at >75% confluency. Cells were washed with ice-cold PBS once, and lysed. Equal protein samples were separated by SDS–PAGE and western blot analysis was performed as described. The following antibodies were utilized: cyclin D1 (Santa Cruz Biotechnology, sc-8396), survivin (Cell Signaling, #2808), mTOR (Cell Signaling, # 2983), p70S6K (Santa Cruz Biotechnology, sc-8418), eIF4E (Cell Signaling, #9742), P-eIF4E (Cell Signaling, #9741), T-4E-BP1 (Cell Signaling, #9452), P-4E-BP1 (Cell Signaling, #9459), β-actin (Santa Cruz Biotechnology, sc-1615).
RT-PCR
RT-PCR analysis was carried out essentially as described by Chen 2011.[45a] Briefly, in a 96-well plate CRL-2813 cells were incubated for 6 hrs in the presence of the tested compounds at final concentrations of 25 and 50 μm. Total RNA was extracted with FastLane Cell One-Step RT-PCR Kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol. Contaminating DNA was removed by DNase I treatment. 1-Step Real-time PCR was performed on a Bio-Rad iCycler IQ5 system by using B-R 1-Step SYBR Green qRT-PCR Kit (Quanta BioSciences, Gaithersburg, MD) according to the manufacturer’s specifications. The thermal cycler conditions were as follows: 10 minutes at 50 °C, hold for 5 minutes at 95 °C, followed by 2-step PCR for 45 cycles of 95 °C for 15 seconds followed by 60 °C for 30 seconds. All PCR reactions were performed in triplicate in at least 2 independent PCR runs. Mean values of these repeated measurements were used for calculation. To calibrate the results, all the transcript quantities were normalized to 18S rRNA (was 18S ribosomal RNA-like mRNA in mouse). The following primers were used in real-time PCR reactions: for cyclin D1: Hs_CCND1_1_SG QuantiTect Primer Assay, QT00495285, for survivin: Hs_BIRC5_2_SG QuantiTect Primer Assay, QT01679664, for p70S6K: Hs_RPS6KB2_1_SG QuantiTect Primer Assay, QT00086632, and Human 18s rRNA (5′ CGGCGACGACCCATTCGAAC 3′ and 5′ GAATCGAACCCTGATTCCCCGTC 3′).
Supplementary Material
Acknolegements
This work was supported by NCI grant #RO1CA121357 to M.C. and a sponsored research agreement from Egenix Inc.
References
- [1] a).Lo Conte L, Chothia C, Janin J. J. Mol. Biol. 1999;285:2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]; b) Fuller JC, Burgoyne NJ, Jackson RM. Drug Discov. Today. 2009;14:155–161. doi: 10.1016/j.drudis.2008.10.009. [DOI] [PubMed] [Google Scholar]
- [2] a).Scott DE, Ehebauer MT, Pukala T, Marsh M, Blundell TL, Venkitaraman AR, Abell C, Hyvönen M. ChemBioChem. 2013;14:332–342. doi: 10.1002/cbic.201200521. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Surade S, Blundell TL. Chem. Biol. 2012;19:42–50. doi: 10.1016/j.chembiol.2011.12.013. [DOI] [PubMed] [Google Scholar]; c) Gonzalez-Ruiz D, Gohlke H. Curr. Med. Chem. 2006;13:2607–2625. doi: 10.2174/092986706778201530. [DOI] [PubMed] [Google Scholar]; d) Azzarito V, Long K, Murphy NS, Wilson AJ. Nat. Chem. 2013;5:161–173. doi: 10.1038/nchem.1568. [DOI] [PubMed] [Google Scholar]; e) Thompson AD, Dugan A, Gestwicki JE, Mapp AK. ACS Chem. Biol. 2012;7:1311–1320. doi: 10.1021/cb300255p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3] a).Wilson AJ. Chem. Soc. Rev. 2009;38:3289–3300. doi: 10.1039/b807197g. [DOI] [PubMed] [Google Scholar]; b) Zinzalla G, Thurston DE. Future Med.Chem. 2009;1:65–93. doi: 10.4155/fmc.09.12. [DOI] [PubMed] [Google Scholar]
- [4] a).Buchwald P. IUBMB life. 2010;62:724–731. doi: 10.1002/iub.383. [DOI] [PubMed] [Google Scholar]; b) Whitty A, Kumaravel G. Nat. Chem. Biol. 2006;2:112–118. doi: 10.1038/nchembio0306-112. [DOI] [PubMed] [Google Scholar]; c) Berg T. Angew. Chem. Int. Ed. 2003;42:2462–2481. doi: 10.1002/anie.200200558. [DOI] [PubMed] [Google Scholar]
- [5].Sonenberg N, Hinnebusch AG. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gingras AC, Raught B, Sonenberg N. Annu. Rev. Biochem. 1999;68:913–963. doi: 10.1146/annurev.biochem.68.1.913. [DOI] [PubMed] [Google Scholar]
- [7].Jackson RJ, Hellen CU, Pestova TV. Nature Rev. Mol. Cell Biol. 2010;11:113–127. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hiremath LS, Webb NR, Rhoads RE. J. Biol. Chem. 1985;260:7843–7849. [PubMed] [Google Scholar]
- [9].Richter JD, Sonenberg N. Nature. 2005;433:477–480. doi: 10.1038/nature03205. [DOI] [PubMed] [Google Scholar]
- [10].Sonenberg N. Curr. Opin. Cell. Biol. 1993;5:955–960. doi: 10.1016/0955-0674(93)90076-3. [DOI] [PubMed] [Google Scholar]
- [11].Hoeffer CA, Cowansage KK, Arnold EC, Banko JL, Moerke NJ, Rodriguez R, Schmidt EK, Klosi E, Chorev M, Lloyd RE, Pierre P, Wagner G, LeDoux JE, Klann E. Proc. Nat. Ac. Sci. USA. 2011;108:3383–3388. doi: 10.1073/pnas.1013063108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12] a).O’Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, Carvill G, Kumar A, Lee C, Ankenman K, Munson J, Hiatt JB, Turner EH, Levy R, O’Day DR, Krumm N, Coe BP, Martin BK, Borenstein E, Nickerson DA, Mefford HC, Doherty D, Akey JM, Bernier R, Eichler EE, Shendure J. Science. 2012;338:1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gkogkas CG, Khoutorsky A, Ran I, Rampakakis E, Nevarko T, Weatherill DB, Vasuta C, Yee S, Truitt M, Dallaire P, Major F, Lasko P, Ruggero D, Nader K, Lacaille JC, Sonenberg N. Nature. 2013;493:371–377. doi: 10.1038/nature11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13] a).Connor SA, Hoeffer CA, Klann E, Nguyen PV. Learn. Mem. 2011;18:207–220. doi: 10.1101/lm.2043811. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Napoli I, Mercaldo V, Boyl PP, Eleuteri B, Zalfa F, De Rubeis S, Di Marino D, Mohr E, Massimi M, Falconi M, Witke W, Costa-Mattioli M, Sonenberg N, Achsel T, Bagni C. Cell. 2008;134:1042–1054. doi: 10.1016/j.cell.2008.07.031. [DOI] [PubMed] [Google Scholar]
- [14] a).Yang Q, Inoki K, Kim E, Guan KL. Proc. Nat. Ac. Sci. USA. 2006;103:6811–6816. doi: 10.1073/pnas.0602282103. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tsai PT, Hull C, Chu Y, Greene-Colozzi E, Sadowski AR, Leech JM, Steinberg J, Crawley JN, Regehr WG, Sahin M. Nature. 2012;488:647–651. doi: 10.1038/nature11310. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kobayashi T, Minowa O, Sugitani Y, Takai S, Mitani H, Kobayashi E, Noda T, Hino O. Proc. Nat. Ac. Sci. USA. 2001;98:8762–8767. doi: 10.1073/pnas.151033798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kelleher RJ, 3rd, Bear MF. Cell. 2008;135:401–406. doi: 10.1016/j.cell.2008.10.017. [DOI] [PubMed] [Google Scholar]
- [16] a).Amiri A, Cho W, Zhou J, Birnbaum SG, Sinton CM, McKay RM, Parada LF. J. Neurosci. 2012;32:5880–5890. doi: 10.1523/JNEUROSCI.5462-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhou J, Parada LF. Curr. Opin. Neurobiol. 2012;22:873–879. doi: 10.1016/j.conb.2012.05.004. [DOI] [PubMed] [Google Scholar]; c) Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF. Neuron. 2006;50:377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Auerbach BD, Osterweil EK, Bear MF. Nature. 2011;480:63–68. doi: 10.1038/nature10658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, Ramesh V, Silva AJ. Nat. Med. 2008;14:843–848. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sharma A, Hoeffer CA, Takayasu Y, Miyawaki T, McBride SM, Klann E, Zukin RS. J. Neurosci. 2010;30:694–702. doi: 10.1523/JNEUROSCI.3696-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19] a).Tee AR, Fingar DC, Manning BD, Kwiatkowski DJ, Cantley LC, Blenis J. Proc. Nat. Ac. Sci. USA. 2002;99:13571–13576. doi: 10.1073/pnas.202476899. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kang YJ, Lu MK, Guan KL. Cell Death Differ. 2011;18:133–144. doi: 10.1038/cdd.2010.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20] a).Moerke NJ, Aktas H, Chen H, Cantel S, Reibarkh MY, Fahmy A, Gross JD, Degterev A, Yuan J, Chorev M, Halperin JA, Wagner G. Cell. 2007;128:257–267. doi: 10.1016/j.cell.2006.11.046. [DOI] [PubMed] [Google Scholar]; b) Chen L, Aktas BH, Wang Y, He X, Sahoo R, Zhang N, Denoyelle S, Kabha E, Yang H, Freedman RY, Supko JG, Chorev M, Wagner G, Halperin JA. Oncotarget. 2012;3:869–881. doi: 10.18632/oncotarget.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21] a).Chaur MN, Collado D, Lehn JM. Chemistry. 2011;17:248–258. doi: 10.1002/chem.201002308. [DOI] [PubMed] [Google Scholar]; b) Brandt A, Cerquetti M, Corsi GB, Pascucci G, Simeoni A, Martelli P, Valcavi U. J. Med. Chem. 1987;30:764–767. doi: 10.1021/jm00388a003. [DOI] [PubMed] [Google Scholar]
- [22].Cerecetto H, Gerpe A, Gonzalez M, Aran VJ, de Ocariz CO. Mini-Rev. Med. Chem. 2005;5:869–878. doi: 10.2174/138955705774329564. [DOI] [PubMed] [Google Scholar]
- [23].Gschwend HW. Vol. US 3681382 A. Ciba Geigy Co.; USA: 1972. [Google Scholar]
- [24].Corsi G, Palazzo G. J. Med. Chem. 1976;19:778–783. doi: 10.1021/jm00228a008. [DOI] [PubMed] [Google Scholar]
- [25].Lee YK, Parks DJ, Lu T, Thieu TV, Markotan T, Pan W, McComsey DF, Milkiewicz KL, Crysler CS, Ninan N, Abad MC, Giardino EC, Maryanoff BE, Damiano BP, Player MR. J. Med. Chem. 2008;51:282–297. doi: 10.1021/jm701217r. [DOI] [PubMed] [Google Scholar]
- [26].Gomez R, Jolly SJ, Williams T, Vacca JP, Torrent M, McGaughey G, Lai MT, Felock P, Munshi V, Distefano D, Flynn J, Miller M, Yan Y, Reid J, Sanchez R, Liang Y, Paton B, Wan BL, Anthony N. J. Med. Chem. 2011;54:7920–7933. doi: 10.1021/jm2010173. [DOI] [PubMed] [Google Scholar]
- [27].Zhang X, Song F, Kuo GH, Xiang A, Gibbs AC, Abad MC, Sun W, Kuo LC, Sui Z. Bioorg. Med. Chem. Lett. 2011;21:4762–4767. doi: 10.1016/j.bmcl.2011.06.067. [DOI] [PubMed] [Google Scholar]
- [28] a).Brown BS, Keddy R, Perner RJ, DiDomenico S, Koenig JR, Jinkerson TK, Hannick SM, McDonald HA, Bianchi BR, Honore P, Puttfarcken PS, Moreland RB, Marsh KC, Faltynek CR, Lee CH. Bioorg. Med. Chem. Lett. 2010;20:3291–3294. doi: 10.1016/j.bmcl.2010.04.047. [DOI] [PubMed] [Google Scholar]; b) Bodmer-Narkevitch V, Anthony NJ, Cofre V, Jolly SM, Murphy KL, Ransom RW, Reiss DR, Tang C, Prueksaritanont T, Pettibone DJ, Bock MG, Kuduk SD. Bioorg. Med. Chem. Lett. 2010;20:7011–7014. doi: 10.1016/j.bmcl.2010.09.121. [DOI] [PubMed] [Google Scholar]
- [29].Okuzumi T, Ducker GS, Zhang C, Aizenstein B, Hoffman R, Shokat KM. Mol. Biosyst. 2010;6:1389–1402. doi: 10.1039/c003917a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kummerle AE, Schmitt M, Cardozo SV, Lugnier C, Villa P, Lopes AB, Romeiro NC, Justiniano H, Martins MA, Fraga CA, Bourguignon JJ, Barreiro EJ. J. Med. Chem. 2012;55:7525–7545. doi: 10.1021/jm300514y. [DOI] [PubMed] [Google Scholar]
- [31].Delbecq F, Cordonnier G, Pommery N, Barbry D, Henichart JP. Bioorg. Med. Chem. Lett. 2004;14:1119–1121. doi: 10.1016/j.bmcl.2003.12.076. [DOI] [PubMed] [Google Scholar]
- [32].Merijanian A, Sharma GM, Moushati J, Gunderson K. J. Org. Chem. 1986;51:543–545. [Google Scholar]
- [33].Schmidt A, Beutler A, Habeck T, Mordhorst T, Snovydovych B. Synthesis. 2006;2006:1882–1894. [Google Scholar]
- [34] a).Lukin K, Hsu MC, Fernando D, Leanna MR. J. Org. Chem. 2006;71:8166–8172. doi: 10.1021/jo0613784. [DOI] [PubMed] [Google Scholar]; b) Schmidt A, Beutler A, Snovydovych B. Eur. J.Org. Chem. 2008;2008:4073–4095. [Google Scholar]
- [35].Kempson J. Hantzsch Thiazole Synthesis. John Wiley & Sons, Inc.; Hoboken, N. J: 2011. [Google Scholar]
- [36].Schröder J, Henke A, Wenzel H, Brandstetter H, Stammler HG, Stammler A, Pfeiffer WD, Tschesche H. J. Med. Chem. 2001;44:3231–3243. doi: 10.1021/jm010887p. [DOI] [PubMed] [Google Scholar]
- [37] a).Avetissyan AA, Khchatryan LA, Papoyan RF. Hayastani Kimiakan Handes. 2010;63:121. [Google Scholar]; b) Novikova AP, Perova NM, Egorova LG, Bragina EI. Khimiya Geterotsiklicheskiki Soedinenii. 1991;6:843–846. [Google Scholar]
- [38] a).Kidwai M, Venkataramanan R, Dave B. J. Heterocyclic Chem. 2002;39:1045–1047. [Google Scholar]; b) Gothwal P, Srivastava YK. Der. Chemica Sinica. 2012;3:318–322. [Google Scholar]
- [39].Magiatis P, Polychronopoulos P, Skaltsounis AL, Lozach O, Meijer L, Miller DB, O’Callaghan JP. Neurotox. Terat. 2010;32:212–219. doi: 10.1016/j.ntt.2009.12.005. [DOI] [PubMed] [Google Scholar]
- [40].Hlaváč J, Soural M, Hradil P, Fryšová I, Slouka J. J. Heterocyclic Chem. 2004;41:633–636. [Google Scholar]
- [41] a).Antilla JC, Baskin JM, Barder TE, Buchwald SL. J. Org. Chem. 2004;69:5578–5587. doi: 10.1021/jo049658b. [DOI] [PubMed] [Google Scholar]; b) Antilla JC, Klapars A, Buchwald SL. J. Am. Chem. Soc. 2002;124:11684–11688. doi: 10.1021/ja027433h. [DOI] [PubMed] [Google Scholar]; c) Kylmala T, Udd S, Tois J, Franzen R. Tet. Lett. 2010;51:3613–3615. [Google Scholar]
- [42].Ishikawa M, Hashimoto Y. J. Med. Chem. 2011;54:1539–1554. doi: 10.1021/jm101356p. [DOI] [PubMed] [Google Scholar]
- [43].Paraiso KHT, Xiang Y, Rebecca VW, Abel EV, Chen YA, Munko AC, Wood E, Fedorenko IV, Sondak VK, Anderson ARA, Ribas A, Dalla Palma M, Nathanson KL, Koomen JM, Messina JL, Smalley KSM. Cancer Res. 2011;71:2750–2760. doi: 10.1158/0008-5472.CAN-10-2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Aktas H, Cai H, Cooper GM. Mol. Cell. Biol. 1997;17:3850–3857. doi: 10.1128/mcb.17.7.3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45] a).Chen T, Ozel D, Qiao Y, Harbinski F, Chen L, Denoyelle S, He X, Zvereva N, Supko JG, Chorev M, Halperin JA, Aktas BH. Nat. Chem. Biol. 2011;7:610–616. doi: 10.1038/nchembio.613. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bai H, Chen T, Ming J, Sun H, Cao P, Fusco DN, Chung RT, Chorev M, Jin Q, Aktas BH. ChemBioChem. 2013;14:1255–1262. doi: 10.1002/cbic.201300177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Diwu Z, Beachdel C, Klaubert DH. Tet. Lett. 1998;39:4987–4990. [Google Scholar]
- [47].Herbivo C, Comel A, Kirsch G, Raposo MMM. Tetrahedron. 2009;65:2079–2086. [Google Scholar]
- [48].Harris RLN, Huppatz JL. Aust. J. Chem. 1977;30:2225–2240. [Google Scholar]
- [49].King LC, Ostrum GK. J.Org. Chem. 1964;29:3459–3461. [Google Scholar]
- [50].Koch K, Biggers MS. J. Org. Chem. 1994;59:1216–1218. [Google Scholar]
- [51].Pospisil J, Muller C, Furstner A. Chemistry. 2009;15:5956–5968. doi: 10.1002/chem.200802681. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.