

Abstract
Bioorthogonal chemistry has enabled the selective labeling and detection of biomolecules in living systems. Bioorthogonal smart probes, which become fluorescent or deliver imaging or therapeutic agents upon reaction, allow for the visualization of biomolecules or targeted delivery even in the presence of excess unreacted probe. This review discusses the strategies used in the development of bioorthogonal smart probes and highlights the potential of these probes to further our understanding of biology.
Introduction
Bioorthogonal reactions proceed readily and selectively in biological systems. By tagging a biomolecule of interest with one of the two reaction partners (the chemical reporter group), the biomolecule can be selectively labelled by bioorthogonal reaction with a secondary reagent bearing a fluorescent probe or affinity handle (Fig. 1A). The small size of chemical reporter groups allows for their efficient incorporation into biomolecules by hijacking native biosynthetic pathways. This has facilitated the visualization and study of biomolecules not directly encoded by the genome. The topic of bioorthogonal reactions has been extensively reviewed, with recent reviews providing detailed discussions on the suitability of individual reactions for different applications.1–5
Fig. 1.

(A) Bioorthogonal reactions enable the selective modification of biomolecules. (B) Fluorescent smart probes undergo an enhancement in fluorescence upon bioorthogonal reaction. (C) Caged compounds deliver their cargo only after bioorthogonal reaction.
The ability of bioorthogonal chemistry to selectively label biomolecules has often been paired with fluorescence microscopy, where the spatiotemporal dynamics of these labeled biomolecules can be monitored. In bioorthogonal labeling reactions, an excess of fluorescent secondary reagent is typically required to maximize labeling efficiency of the reporter-tagged biomolecule. This excess reagent will have to be carefully removed through a series of washing steps to guarantee that only covalently attached probe remains. Otherwise, fluorescence from unreacted probe may obscure any signal from the labeled biomolecule of interest. As overall reaction rate is directly proportional to both rate constant and reagent concentration, a higher rate constant means less secondary reagent is needed. This minimizes the amount of unreacted probe present at the end of the reaction, reducing overall background signal. A major motivation in the field of bioorthogonal chemistry has therefore been to improve reaction kinetics.
A complementary approach to minimize background fluorescence from unreacted probe is through the development of bioorthogonal smart probes, which are activated by bioorthogonal reaction. Fluorescent probes (also known as fluorogenic or turn-on probes), for example, undergo an enhancement in fluorescence upon bioorthogonal reaction (Fig. 1B). By using these probes, fluorescence is only observed where the bioorthogonal reaction occurs. For imaging applications, bioorthogonal smart probes can prove crucial in situations where it is difficult to remove excess probe, such as for labeling reactions performed intracellularly or in vivo. In addition, there are instances where washing steps are undesirable, such as the real-time visualization of dynamic biological processes.
Bioorthogonal smart probes also include compounds that release drugs or imaging agents only upon bioorthogonal reaction (Fig. 1C). These caged compounds enable targeted delivery if a certain cell type preferentially incorporates a reporter group-tagged molecule. Selectivity is achieved if the caging process dramatically alters the activity or properties of the drug or imaging agent.
Here, we discuss design strategies used for the development of bioorthogonal smart probes, with particular emphasis on fluorescent probes. Considerations when evaluating these smart probes include the level of signal enhancement that occurs upon bioorthogonal reaction; probes with higher turn-on enable more sensitive detection. Additionally, for fluorescent probes, longer wavelength excitation and emission are beneficial as complications from phototoxicity, background autofluorescence, or poor tissue penetrance are minimized. Our review is organized by bioorthogonal reaction type, with each section beginning with a brief introduction of the reaction. The final section highlights selected applications of bioorthogonal smart probes, such as their use in monitoring bioorthogonal reaction progress and sequence-specific oligonucleotide detection.
Aldehyde/Ketone-Nucleophile Condensations
The condensation of aldehydes with α-effect amine nucleophiles, such as alkoxyamines or hydrazines to form oxime or hydrazone linkages, respectively, was one of the first commonly used chemoselective ligation reactions in biological systems (Fig. 2). An obvious limitation to the bioorthogonality of this reaction is the presence of aldehydes and ketones in intracellular metabolites. Yet, aldehydes and ketones are absent on the exterior of the cell, making this approach effectively bioorthogonal for reactions performed on the cell surface.
Fig. 2.

The condensation of aldehydes with alkoxyamines or hydrazines/hydrazides to form oximes or hydrazones, respectively.
Aldehydes and ketones have been introduced onto cell surfaces via the biosynthetic incorporation of unnatural substrates, the periodate oxidation of cell surface glycans, and the enzymatic oxidation of alcohols or thiols in endogenous biomolecules.6–12 The condensation of α-effect amine nucleophiles occurs rapidly under acidic conditions, but the presence of additives can enable efficient labeling at neutral pH.10,13 Alkoxyamines and hydrazines condense with aldehydes with second-order rate constants of 1 to 10 M−1s−1 in pH 4.5 to 6.5 aqueous buffer, whereas hydrazides react orders of magnitude more slowly. The resulting adducts are prone to hydrolysis in aqueous environments, since the forward reaction involves a release of an equivalent of water. Oxime linkages, however, are significantly more stable than hydrazones,14 and the stability of these adducts can be further increased through the use of modified alkoxyamine or hydrazine reagents.15,16
Aryl hydrazines undergo significant changes in their electronic properties upon condensation with carbonyl functionalities. The color change observed when staining ketones and aldehydes during thin layer chromatography with 2,4-dinitrophenylhydrazine is one manifestation of this phenomenon. Thus, hydrazine-functionalized fluorophores may be expected to show fluorescence modulation upon reaction with ketones or aldehydes. As well, the lone pair of the distal hydrazine nitrogen may quench fluorescence through internal charge transfer (ICT) processes.17 Hydrazine-functionalized nitrobenzoxadiazoles, BODIPYs, and coumarins have been developed whose fluorescence is activated by reaction with ketones or aldehydes (1–3, Fig. 3).18–24 It should be noted that fluorescence enhancements of these compounds are highest in less polar solvents, and it has been reported that the hydrazone adducts of compound 1 are virtually non-fluorescent in neutral aqueous solutions. Additionally, aryl hydrazines and their adducts are prone to oxidation, which can complicate handling and storage.
Fig. 3.

Smart hydrazine probes. Hydrazine-functionalized nitrobenzoxadiazole (1), BODIPY (2), and coumarin (3). Spectroscopic properties correspond to the hydrazone products derived from these probes (benzaldehyde adduct for 1, propiolaldehyde adduct for 2, and salicylaldehyde adduct for 3).
Approaches that utilize alkoxyamines offer the benefits of ease of handling and the formation of a significantly more stable adduct in biological systems. Unfortunately, the fluorescence properties of alkoxyamine-functionalized fluorophores, in contrast to hydrazines, do not appear to be significantly perturbed by reaction with ketones or aldehydes.25 In another approach, Reymond and coworkers prepared weakly fluorescent alkoxyamine-substituted coumarin 4 (Fig. 4).26 After oxime formation (5), elimination yields nitrile and the highly fluorescent coumarin phenolate product 6. This elimination can be promoted through the use of higher pH or bovine serum albumin additive. The authors used these probes to detect biologically relevant aldehydes in vitro. While this strategy does not generate stable adducts, a reinvestigation of this approach in the context of bioorthogonal chemistry may be worth pursuing.
Fig. 4.
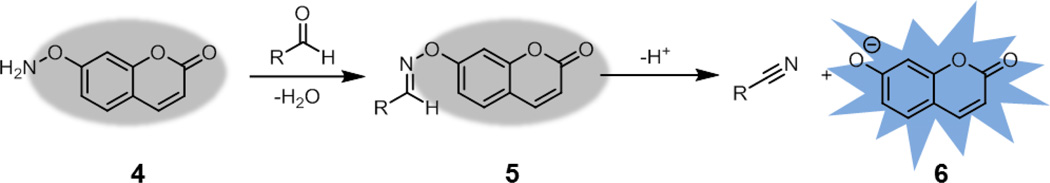
Aldehyde detection via oxime bond fragmentation. After formation of oxime 5, fragmentation produces a nitrile and the free phenolate 6.
Progress has been made in the development of smart probes suitable for the detection of aldehyde-functionalized biomolecules, yet the design of new probes, especially those based off alkoxyamines, is an important future direction. Likewise, the identification of alternative reagents that rapidly form stable adducts with ketones or aldehydes under aqueous conditions will provide new avenues for the visualization of these functional groups in biological systems.27,28
The Staudinger Ligation
The Staudinger ligation is a modification of the original Staudinger reduction of an azide by phosphines. Diverging from the Staudinger reduction, the intermediate aza-ylide is intercepted by a nearby ester, resulting in the formation a phosphorane, which collapses to form a phosphine oxide and a stable amide linkage (Fig. 5).29 As the two reaction partners are completely abiotic, selective ligation can occur in all biological compartments, in contrast to aldehyde or ketone condensation reactions. The second-order rate constant of the reaction is ~10−3 M−1s−1 in acetonitrile. Over the course of the reaction, the phosphine is oxidized to a phosphine oxide while the ester is cleaved to release methanol.30 These two processes have been exploited for the development of smart phosphine probes activated by the Staudinger ligation.
Fig. 5.

The archetypical Staudinger ligation. The phosphine reacts with an azide to generate a stable amide linkage and phosphine oxide with concomitant release of methanol.
In the phosphine oxidation approach, Bäuerle and coworkers have demonstrated that the fluorescence of coumarins is sensitive to the nature of the substituent at the 3-position.31 Thus, a coumarin functionalized with a phosphine at the 3-position was prepared by Bertozzi and coworkers (7, Fig. 6).32 The phosphorus lone pair may quench fluorescence due to ICT processes, and oxidation of the phosphine during the Staudinger ligation to form 8 will activate fluorescence. Compound 7 showed comparable kinetics to traditional Staudinger probes (k = 1.5 × 10−3 M−1s−1 in 1:1 acetonitrile/water). It underwent a 60-fold enhancement in fluorescence quantum yield upon Staudinger ligation, to a final quantum yield of 0.65, suggesting that quenching was fully relieved by phosphine oxidation. Unfortunately, phosphine oxidation can occur in the absence of azide due to ambient oxygen, and the attachment of phosphines onto fluorophores can accelerate this reaction, likely due to the generation of high-energy intermediates. This liability of non-specific phosphine oxidation may also limit other strategies beyond ICT for developing smart phosphine probes activated by the Staudinger ligation.
Fig. 6.

A smart phosphine probe activated by phosphine oxidation during the Staudinger ligation. Spectroscopic properties correspond to the product derived from reaction of 7 with an aliphatic azide.
To avoid this issue, a different strategy that relied on ester cleavage over the course of the reaction was pursued. In Förster resonance energy transfer (FRET)-based smart phosphine probes, a FRET quencher is attached to the alkoxide substituent of the ester while a fluorophore is conjugated directly to the triarylphosphine (9, Fig. 7A).33 After Staudinger ligation, the FRET quencher is released, revealing the fluorescence of the original fluorophore. This strategy was implemented by Bertozzi and coworkers using a fluorescein-derivative conjugated to the phosphine and Disperse Red 1 as the quencher. This compound had similar reaction kinetics to other Staudinger ligation probes [~4 × 10−3 M−1s−1 in 1:1 acetonitrile/phosphate buffered saline (PBS)], consistent with studies demonstrating that aza-ylide formation is rate determining.30
Fig. 7.

Phosphine probes activated by ester cleavage in the Staudinger ligation. (A) A FRET-based smart phosphine probe (9) where a quencher is released upon Staudinger ligation. Spectroscopic properties correspond to the product derived from reaction of 9 with benzyl azide. (B) A caged luciferin-phosphine conjugate (11) that releases free luciferin upon Staudinger ligation.
Unlike coumarin-phosphine 7, the phosphine-oxide product derived from non-specific oxidation of 9 remained quenched. In contrast, compound 9 underwent a 170-fold enhancement in fluorescence quantum yield upon Staudinger ligation to form 10, reaching a final quantum yield of 0.64 in pH 7.0 PBS. This FRET-based smart phosphine probe was suitable for the detection of azide-functionalized glycoproteins on living cells. Importantly, direct visualization of these glycoproteins on live cells using traditional fluorophore phosphine conjugates resulted in high non-specific background staining unless highly charged fluorophores were used, demonstrating the advantages of smart probes.34 The limited cell permeability of 9 has precluded its use for the visualization of intracellular targets. In principle, this strategy is generalizable to longer wavelength fluorophores by changing both the fluorophore and the quencher. Finally, as FRET efficiency varies as a function of 1/r6, (r = distance from donor to acceptor), versions of this probe with more tightly-linked fluorophore and quencher should show even higher enhancement upon ligation.
This strategy was also used by Bertozzi and coworkers to develop a smart probe for bioluminescence imaging through the use of a caged luciferin (11, Fig. 7B).35 Acylation of luciferin blocks processing by luciferase to generate light.36 Only upon release of free luciferin after Staudinger ligation is a suitable substrate for luciferase available and bioluminescence observed. The selectivity of this probe was demonstrated by bioluminescence signal in cultured cells only in the presence of azide-functionalized glycoproteins. The inherent sensitivity of bioluminescence imaging allowed for azide detection using nanomolar concentrations of probe, orders of magnitude lower than required for fluorescence-based methods. In principle, this caging strategy can be used with any fluorophore in which acylation or alkylation results in a significant reduction in fluorescence. A limitation, however, of this strategy is that it would not yield a covalently-attached fluorescent adduct, reducing spatial resolution.
Finally, phosphines containing other electrophilic traps have also been developed, most notably in the context of the “traceless” Staudinger ligation for native amide bond formation in peptide synthesis.37,38 For bioorthogonal labeling applications, this may be advantageous when the bulky triarylphosphine oxide adduct is not desirable. These phosphines have been used as a platform for bioorthogonal smart probes in the context of drug delivery, where active drug is released upon reaction.39 While the Staudinger ligation has been supplanted by newer bioorthogonal reactions with more rapid kinetics, its exquisite selectivity, especially in the context of in vivo applications, provide motivation for further advances in smart probe design.40
Azide-Alkyne Click Cycloadditions
The azide-alkyne “click” cycloaddition is one of the most widely used bioorthogonal reactions. The reaction occurs in the presence of Cu(I) catalyst or in the absence of copper through the use of strained cyclooctynes to form stable triazole linkages (Fig. 8).41–43 The second-order rate constant of the copper-free cycloaddition with the parent cyclooctyne is ~10−3 M−1s−1 in acetonitrile. Modifying the electronics of the cyclooctyne or increasing ring strain via aryl/cyclopropane ring fusions and heteroatom incorporation have increased the rate constants to over 1 M−1s−1 in acetonitrile.43–49 The most efficient ligands for copper-catalyzed click reactions produce reaction rates greater than 104 M−1s−1 with respect to concentration of copper catalyst.50–53 Earlier contributions to this area of fluorogenic click probes, through 2010, have been reviewed by Wang and coworkers.54 Unlike the Staudinger ligation, where ester cleavage provided a general approach for the design of activatable probes, such strategies are not readily apparent in azide-alkyne click cycloadditions. Triazoles are components of many fluorophores, but these examples are highly dependent on the substitution pattern of both the alkyne and azide reaction partner. As a result, regenerating triazole-containing fluorophores by click reaction will not be a broadly applicable strategy for the visualization and detection of tagged biomolecules.55–59
Fig. 8.

The copper-catalyzed and copper-free click reactions between alkynes and azides to form triazoles.
Significant progress has been made in the development of smart azide probes activated by cycloaddition with alkynes. These probes commonly take advantage of the fact that the lone pair of the proximal azide nitrogen is delocalized into the triazole after click reaction, and thus cannot participate in fluorescence quenching. A significant advantage of preparing azide probes is their ease of synthesis, as the azide can be readily prepared from amines and is tolerant of a variety of synthetic operations, making the preparation of large fluorophore libraries fairly straightforward.
The first smart azide probes were the azidocoumarins reported by Wang and coworkers.60 Similar to the previously described smart coumarin-phosphine probe 7, introduction of an azide into the 3-position of coumarins also significantly quenches fluorescence via ICT. Upon reaction with alkynes, the lone pair of the azide is delocalized into the triazole and fluorescence is enhanced. Wang and coworkers prepared a panel of azidocoumarins via an efficient and modular route. Of these compounds, 3-azido-7-hydroxycoumarin (12, Fig. 9) showed the most impressive fluorescence enhancement upon click reaction, around several hundred-fold in pH 7.4 PBS, to yield a highly fluorescent triazole product. The robust enhancement in fluorescence of this probe and its decent water solubility and cell permeability have made this probe a popular choice for the detection of alkynes on intracellular and extracellular targets in biological systems.61,62 By replacing the hydroxyl group with other electron donating groups such as amines, excitation and emission maxima of these azidocoumarins is shifted to longer wavelengths, but fluorescence enhancement is also diminished.
Fig. 9.

Various smart azide probes quenched by ICT: 3-azido-7-hydroxycoumarin (12) and azide-functionalized 1,8-naphthalimide (13), BODIPY (14), benzothiazole (15), and carbazole (16). Spectroscopic properties correspond to representative triazole products derived from these azide probes.
The 1,8-naphthalimide system has also been used for developing smart azide probes.63 Again, the presence of an azide in 4-position of naphthalimide (13) quenches fluorescence via ICT. After triazole formation, quenching is relieved and fluorescence is enhanced. Quenching via ICT may also be operant in other scaffolds, such as in azide-functionalized BODIPY, benzothiazole, and carbazoles (14–16), although other factors may be at play.64–67
The lone pair of azides can also quench fluorescence via photoinduced electron transfer (PeT), which differs from ICT in that the azide is electronically decoupled from the fluorophore in the ground state.68 Wang and coworkers showed that azide-functionalized anthracene compounds undergo an up to 75-fold increase in fluorescence upon triazole formation (17, Fig. 10).69 The absorption of these compounds remained constant during the click reaction, consistent with a PeT mechanism.
Fig. 10.

Smart azide probes utilizing PeT: azide-functionalized anthracene (17), fluorescein (18), and Si-rhodamine (19, R = oligoethylene glycol groups). Spectroscopic properties correspond to representative triazole products derived from these azide probes.
Bertozzi and coworkers took advantage of PeT from the pendant aryl ring of xanthene fluorophores to develop smart azide probes.70 Xanthene fluorophores exhibit visible light excitation and emission, minimizing complications from background autofluorescence in biological samples. Nagano and coworkers have elegantly demonstrated that PeT efficiency in fluoresceins can be predicted using computational chemistry.71 Bertozzi and coworkers reasoned that the change in electron density of the pendant aryl azide to an aryl triazole would result in significant fluorescence enhancement if the proper substituents on the pendant aryl ring were present. After screening for these proper substituents using computational methods, they identified an azidonaphthyl-substituted fluorescein, 18, which underwent a 34-fold enhancement in fluorescence quantum yield upon triazole formation in pH 7.4 PBS and was suitable for the visualization of terminal alkyne-functionalized glycoproteins in fixed mammalian cells.
Given the generality of PeT, smart azide probes with near-infrared emission were next developed by Bertozzi and coworkers. At these wavelengths, background fluorescence from endogenous biomolecules is minimal and tissue penetrance highest. These probes were designed around the Si-rhodamine scaffold developed by Nagano and coworkers, and were identified using the same strategy as for the fluorogenic azidofluoresceins.72,73 One azido-Si-rhodamine, 19, underwent a 48-fold enhancement in fluorescence quantum yield upon triazole formation in pH 7.4 PBS. Water-soluble versions of 19 were well-suited for the visualization of linear alkyne-functionalized glycoproteins on the surface of live mammalian cells and cyclooctyne-functionalized peptidoglycan in the Gram-positive bacteria. The fluorescence quantum yields of the triazole products, derived from 18 and 19, 0.8 and 0.2, respectively, suggest that PeT quenching is fully relieved following click reaction. Finally, this PeT strategy should be generalizable to other fluorophore scaffolds beyond xanthenes.74
An interesting property of these two probes is that the reduction of aryl azides, a potential degradation pathway in biological systems by endogenous thiols, results in a further reduction in fluorescence.75 This may be advantageous for the labeling of intracellular targets, as fluorescence from unreacted probe is minimized over time. In contrast, this degradation pathway can be problematic if azide reduction results in a dramatic enhancement in fluorescence.63
A limitation of azide-functionalized probes is that they require either the incorporation of relatively bulky cyclooctynes or the use of a copper catalyst. Recent progress in increasing the biocompatibility of copper-click reactions have utilized copper-chelating groups on azide probes, which minimizes the amount of copper necessary for the reaction and enables labeling of intracellular linear alkynes.76,77 Further advances in this area may further expand the scope of biomolecules that can be visualized by azide-functionalized smart probes.
Unlike azides, alkynes do not have a lone pair to inherently quench fluorescence via charge transfer pathways. Smart alkyne probes are therefore more challenging to design and the mechanisms by which these probes operate may be difficult to rationalize. In seminal work by Fahrni and coworkers, they discovered that coumarins containing an alkyne at the 7-position undergo an enhancement in fluorescence upon triazole formation (20, Fig. 11).78,79 The more electron-donating character of the triazole relative to the alkyne lowers the energy of the (π,π*) state well below the (n,π*) state. This prevents fluorescence quenching via (π,π*)1−(n,π*)3 intersystem crossing, and results in a 18-fold enhancement in fluorescence quantum yield upon click reaction.
Fig. 11.

Smart alkyne and cyclooctyne probes activated by reactions with azides: linear alkynes include 7-alkynyl coumarin (20, R = methyl or alkoxymethyl) and alkyne-functionalized xanthene (21), 1,8-naphthalimide (22), benzothiazole (23), and benzoxadiazole (24), while cyclooctynes include coumBARAC (25) and Fl-DIBO (26). Spectroscopic properties correspond to representative triazole products derived from these alkyne probes.
Other smart alkyne probes have also been identified by rapid access to libraries of probes via Sonogashira coupling. For example, a panel of six alkynyl xanthene compounds were prepared and reacted with a set of 17 azide probes – this combinatorial approach led to the identification of a combination of alkyne probe, azide, and solvent which resulted in a 9-fold enhancement in brightness upon triazole formation (21).80 In addition, smart alkynyl naphthalimides, benzothiazoles, and benzoxadiazoles are known (22–24).63,81–83 Of these probes, alkynyl coumarins, naphthalimides, and benzothiazoles have been shown to be suitable for the detection of azide-functionalized biomolecules in biological systems.63,82,84
Smart cyclooctynes combine the advantages of the azide as a reporter group, such as small size and good stability, with the ability to react in the absence of potentially cytotoxic copper catalyst. The difficulty in synthesizing cyclooctynes, however, has hampered the preparation of fluorescent cyclooctynes for evaluation. Still, several pioneering examples exist of such smart cyclooctyne probes.
The alkynyl coumarin identified by Fahrni and coworkers has been adapted for copper-free click chemistry by incorporating the alkyne as part of a cyclooctyne (25, coumBARAC). Upon reaction with azides, the compound showed a 10-fold enhancement in fluorescence quantum yield.85 More recently, Boons and coworkers reported a cyclooctyne (26, Fl-DIBO) which undergoes an impressive 1300-fold enhancement in brightness upon reaction with azides.86 Computational studies suggested that fluorescent activation of this probe was due to an enhancement in oscillator strength of the S0 – S1 transition upon triazole formation. Cyclooctynes may suffer from background labeling of proteins due to reactions with thiols.87 For a typical cyclooctyne fluorophore conjugate, this will result in fluorescence background. On the other hand, Fl-DIBO did not exhibit fluorescence turn-on due to this side reaction. Importantly, both coumBARAC and Fl-DIBO showed reactivity with azides comparable to their parent cyclooctynes.
Nitrile Imine-Alkene Cycloadditions
Nitrile imines react readily with alkenes under physiological conditions to form pyrazolines (Fig. 12A). These nitrile imines can be generated by photolysis of tetrazoles (“photo-click” chemistry), with the expulsion of nitrogen gas.88,89 The initial photolysis occurs with good efficiency, but a drawback is the requirement of UV activation (originally 302 nm for 27), which leads to phototoxicity after prolonged exposure. Modifications of the structure of these tetrazoles allow for longer wavelength activation, such as at 365 or 405 nm (28, Fig. 12B).90 Tetrazole 29, which can be activated using two-photon excitation with a 700 nm laser, further minimizes this limitation.91 Alternatively, the intermediate nitrile imines can also be generated spontaneously by decomposition of hydrazonoyl chlorides such as 30 at neutral pH.92–94 This is advantageous in situations where photolysis of the tetrazole may result in damage to biological samples, although the benefits of photoactivation, such spatial and temporal control, are lost.
Fig. 12.

The nitrile imine-alkene cycloaddition reaction. (A) The intermediate nitrile imine can be generated by photolysis of a tetrazole or by spontaneous decomposition from hydrazonoyl chlorides. The nitrile imine can then undergo cycloaddition with alkenes to form a stable pyrazoline product. (B) Structures of selected tetrazoles and hydrazonyl chlorides that produce fluorescent pyrazolines upon nitrile imine formation and alkene cycloaddition: an electron rich tetrazole (27), a 365 nm activatable tetrazole (28), a two-photon activatable tetrazole (29), a hydrazonyl chloride (30), and a tetrazole that produces long-wavelength emitting pyrazolines (31). R, R’ = solubilizing groups. (C) Various alkene reaction partners that undergo rapid cycloadditions with nitrile imines: acrylamides, norbornenes, cyclopropenes, and spirohexenes.
Increasing the electron density of the nitrile imine, decreasing the electron density of the alkene, or increasing the strain of the alkene all result in an enhancement in the rate of cycloaddition. Recent reports have shown that by increasing the strain of the alkene, through the use of cyclopropenes or spirohexenes, the rate of the cycloaddition can increase dramatically, greater than 104 M−1s−1 for spirohexenes in 1:1 acetonitrile/chloride-free phosphate buffer (Fig. 12C).95,96
A notable feature of this reaction is that, unlike the starting tetrazole or hydrazonoyl chlorides, the product pyrazolines are highly fluorescent, which makes these probes inherently fluorogenic. The resulting fluorophores typically have excitation maxima in the ultraviolet and emit cyan to green fluorescence. While the intermediate nitrile imine is unstable and can form byproducts from hydrolysis or dimerization, these compounds are not fluorescent, providing an excellent strategy to visualize alkene-tagged molecules, including intracellular targets, in living systems. Some fluorescence background, however, may result from weakly fluorescent nitrile imine intermediates.97
To shift the emission wavelengths of the resulting pyrazoline fluorophores to the red or near infrared, Lin and coworkers conjugated the tetrazole to heteroaromatic systems such as thiophenes. These oligothiophene-conjugated tetrazoles, such as 31, form pyrazoline products with emission wavelengths greater than 600 nm.98
The Tetrazine Ligation
Tetrazines are electron deficient heterocycles that undergo rapid inverse electron-demand Diels-Alder reactions with strained alkenes and alkynes (Fig. 13).99,100 After cycloaddition with alkenes, nitrogen is extruded via a retro Diels-Alder reaction to yield a stable dihydropyridazine adduct. These reactions can have second-order rate constants greater than 104 M−1s−1 in organic solvents, depending on the tetrazine and alkene/alkyne reaction partners used, permitting efficient and quantitative conversion to product with low concentrations of reagents. In general, monosubstituted tetrazines (R = aryl, R’ = H) show the highest reactivity, but also suffer from stability issues in the presence of water or biologically relevant nucleophiles. Tetrazine stability is dramatically enhanced by changing the substituents, for example, to R’ = Me or R,R’ = 2-pyridyl groups.99,101 In terms of dienophile reaction partner, trans-cyclooctenes afford the highest kinetics, which can be further enhanced by cyclopropane ring fusion (Fig. 13A).102 A potential limitation of these highly reactive trans-cyclooctenes is isomerization to the more stable but less reactive cis isomer in the presence of thiols.102 More recently, cyclopropenes have been described that combine small size and stability with good kinetics, greater than 10 M−1s−1 in 7:1 H2O/DMSO (Fig. 13B).103,104 Finally, tetrazines also undergo cycloadditions with sterically unhindered cyclooctynes to form pyridazine adducts (Fig. 13C).105
Fig. 13.

The tetrazine ligation. Tetrazines react rapidly with (A) strained trans-cyclooctenes (B) cyclopropenes, or (C) cyclooctynes to yield stable adducts.
Tetrazines absorb visible light, with an absorption maximum of around 520 nm. Weissleder and coworkers showed that a series of green-to-red fluorophores are efficiently quenched by tetrazines via FRET, with up to a 20-fold enhancement in fluorescence after destruction of the tetrazine upon Diels-Alder cycloaddition (32 to 33, Fig. 14A).106 In separate work, Chin and coworkers demonstrated that tetrazines can also quench longer wavelength fluorophores such as tetramethylrhodamine (34), although the fluorescence enhancement was less pronounced, possibly due to increased tetrazine-fluorophore separation (Fig. 14B).107
Fig. 14.

Smart tetrazine conjugates containing flexible linkers and well-suited for imaging alkenes in biological systems. (A) BODIPY-tetrazine conjugate 32. As the tetrazine is consumed by cycloaddition to form 33, FRET quenching is relieved and fluorescence is restored. (B) A tetramethylrhodamine-tetrazine conjugate. (C) Carboxyfluorescein diacetate-tetrazine conjugate 35. The acetates are cleaved by intracellular esterases to reveal the free fluorescein 36. Spectroscopic properties correspond to cycloaddition products of the corresponding tetrazine probes.
Weissleder and coworkers next evaluated a panel of these tetrazine-fluorophore conjugates for live cell imaging.108 While these conjugates demonstrated good fluorogenic properties in vitro, their performance in the context of visualizing trans-cyclooctene-functionalized small molecules bound to intracellular protein targets varied. They found that carboxyfluorescein diacetate (CFDA) tetrazine conjugate 35 performed best for intracellular imaging in live cells (Fig. 14C). This compound is virtually non-fluorescent, as acetylation causes the fluorophore to exist in a spirolactonized form. The acetates are cleaved by intracellular esterases, revealing free fluorescein 36 (which remains quenched by tetrazine until cycloaddition). Other groups have also had success in applying this CFDA-tetrazine probe for intracellular labeling of trans-cycloctene and cyclooctyne functionalized proteins in living cells.109,110
By decreasing the distance of the tetrazine to the fluorophore, FRET efficiency can increase dramatically. The close proximity of the tetrazine may allow for other quenching pathways, such as through bond energy transfer (TBET), which may not be possible if the tetrazine is connected to the fluorophore via a flexible linker.111–112 The first realization of this strategy was a series of BODIPY-tetrazine conjugates where the tetrazine was directly attached off the BODIPY core with a rigid phenyl linker (36–38, Fig. 15A).113 Notably, compound 37, the best of the three, underwent a 1600-fold enhancement in brightness upon reaction with trans-cyclooctene. To demonstrate that TBET was operative, BODIPY-tetrazine 38 was synthesized. This compound arranges the BODIPY and tetrazine groups such that their transition dipoles are nearly perpendicular. While this alignment should prevent efficient FRET, a 120-fold enhancement in fluorescence was still observed upon reaction with trans-cyclooctenes, suggesting that other quenching pathways were at play. Finally, minimal influence of solvent polarity on fluorescence ruled out PeT. After cycloaddition, the fluorescence quantum yields of these compounds reached up to 0.8, indicating that quenching was fully relieved upon tetrazine ligation. The suitability of this fluorophore for biological applications was demonstrated by imaging trans-cyclooctene-functionalized antibodies on both fixed and live cells.
Fig. 15.

Rigidly linked smart tetrazine probes: tetrazine-functionalized (A) BODIPYs (36–38), (B) coumarins (39–42), and (C) xanthenes (43–44). Spectroscopic properties of compounds 36 to 42 correspond to the products derived from cycloaddition with trans-cyclooctene. For compounds 43 and 44, spectroscopic properties correspond to products derived from cycloaddition with cyclopropene or with trans-cyclooctene (and subsequent oxidation), respectively.
To expand on this approach in the context of multicolor imaging, Weissleder and coworkers synthesized a panel of rigidly-linked coumarin-tetrazine conjugates (39–42, Fig. 15B).114 Since TBET permits donors to excite significantly red-shifted acceptors, tetrazines may efficiently quench shorter-wavelength emitting fluorophores such as the blue-emitting coumarins. The coumarin-tetrazine conjugates were designed such that transition dipoles of the coumarin fluorophore and the tetrazine aligned significantly to maximize contributions from FRET in addition to TBET. This resulted in extremely efficient fluorescent quenching which was relieved upon tetrazine ligation, resulting in an up to an impressive 11,000-fold enhancement in fluorescence to final quantum yields of 0.3–0.5. These compounds, named HELIOS probes, were suitable for the detection of a variety of trans-cyclooctene-functionalized molecules in fixed and live cells virtually immediately after incubation.
In another implementation of this strategy, Devaraj and coworkers developed a surrogate for vinyl-tetrazines, which could be attached to iodine-functionalized xanthene fluorophores via Heck coupling reactions.115 Using this strategy, they prepared Oregon Green and tetramethyl rhodamine fluorophores conjugated to tetrazines via a rigid styrenyl linker (43–44, Fig. 15C). The Oregon Green probe 43 underwent a 135-fold fluorescence enhancement in pH 7.4 PBS upon reaction with cyclopropenes. Notably, the red-emitting tetramethylrhodamine was also efficiently quenched; a 42-fold enhancement in fluorescence was observed for 44 upon reaction with cyclopropene in ethanol. Importantly, these styrenyl tetrazines remained highly reactive, with rate constants of over 100 M−1s−1 in 1:1 DMF/pH 7.4 PBS for reactions with trans-cyclooctene. Rigid xanthene-tetrazine conjugates can also be prepared using other synthetic strategies, although increased donor-acceptor distance decreases the efficiency of fluorescence quenching.116
Interestingly, the dihydropyridazine adduct from reaction of 43 or 44 with trans-cyclooctene was partially oxidized to the pyridazine. Devaraj and coworkers completed this oxidation by treatment with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) before performing fluorescence measurements. This DDQ oxidized product was more fluorescent than the cycloadducts with cyclopropene – a 400-fold enhancement in fluorescence was observed for 43 while a 76-fold enhancement in fluorescence was observed for 44. In principle, these products can also arise from reaction with cyclooctynes instead of trans-cyclooctenes. The superior fluorescence enhancement upon alkyne-tetrazine vs. alkene-tetrazine cycloaddition was also recapitulated in a separate study of NIR-fluorophore tetrazine conjugates.117 Although the product from tetrazine-cyclooctene cycloaddition resulted in no fluorescence enhancement, consistent with previous observations, the product from the tetrazine-cyclooctyne cycloaddition resulted in a 1.6-fold enhancement in fluorescence. The origin of these differences may be worthy of further investigation.
One potential limitation of these probes is that tetrazines may degrade by other pathways besides the bioorthogonal reaction, which will also relieve FRET quenching. Methyl tetrazines, in contrast to monosubstituted tetrazines, have been shown to be highly stable in serum.100 In addition, Devaraj and coworkers showed that their xanthene-tetrazine conjugates were stable in both aqueous buffer and in the presence of a large excess of cysteine, demonstrating that these degradation pathways should not contribute to background fluorescence for their fluorogenic tetrazine probes.115
The scope of smart tetrazine fluorophores utilizing FRET or TBET is inherently limited by the absorption wavelengths of tetrazines. This may be addressed through modification of the tetrazine to change its absorption properties, which will expand the scope of fluorophores that can be efficiently quenched via these strategies.
A recent report from Robillard and coworkers described a trans-cyclooctene that releases amine-functionalized cargo upon reaction with tetrazine (45, Fig. 16). After cycloaddition with tetrazine, the dihydropyridazine product 46 undergoes a β-elimination of carbamate, which liberates the free amine after decarboxylation.118 The cycloaddition rate of 45 was diminished 20-fold compared to the the parent trans-cyclooctene due to the steric hindrance from the carbamate. Still, both cycloaddition and elimination steps were sufficiently rapid such that a majority of the cargo was released within minutes after the addition of sub-millimolar concentrations of tetrazine. While this strategy was used for drug delivery, attachment of a fluorescent probe and a quencher or simply a caged fluorophore will produce a smart trans-cyclooctene imaging probe, similar to the smart Staudinger ligation probes. One caveat, however, is that this strategy requires the use of a tetrazine as a reporter group, which has not been as widely used as strained alkenes.119
Fig. 16.

A smart trans-cyclooctene probe (45) for drug delivery. Cycloaddition with tetrazines to form dihydropyridazine 46 results in elimination of the carbamate group to release its amine-functionalized cargo.
Selected Applications of Bioorthogonal Smart Probes
As bioorthogonal smart probes are activated only when they encounter their reaction partner, the signal from these probes can be used as a proxy for reaction progress. Thus, these probes, particularly fluorophores, have been useful tools for monitoring and optimizing bioorthogonal reactions. For example, smart probes have been used to track the progress of small-molecule conjugation reactions onto biomolecules and glass surfaces.120,121 Additionally, smart azide and alkyne-functionalized fluorophores have been used in the evaluation of new ligands and reaction partners for the copper-catalyzed azide-alkyne click reaction.52,53,63,76,77 Reaction partners with higher rate constants activate smart probes more rapidly. The signal from these probes can therefore be used to identify the fastest reagent combinations, particularly in a high-throughput fashion.
The rate of activation of bioorthogonal smart probes not only depends on reaction rate constant but also on reagent concentration. The signal from these probes can consequently be used to determine the concentration of its reaction partner in solution. This enables, for example, the rapid determination of concentrations of tagged metabolites retrieved from biological systems.
More interestingly, reaction rate also depends on the effective concentration of the two reagents. If the two bioorthogonal reaction partners are held in close proximity, their effective concentration and thus reaction rate can increase dramatically.122 Ligand-receptor and protein-protein binding interactions, among others, are mediators of signaling processes in biology. Our understanding of these processes can be greatly augmented by technologies that enable the visualization or detection of these interactions in living systems. A bioorthogonal smart probe and its reaction partner conjugated onto the two different binding partners will more rapidly produce signal if binding occurs. Care must be taken, however, in choosing the attachment sites of the bioorthogonal reagents to minimally perturb binding.
Aside from detecting binding interactions, bioorthogonal smart probes can be used to sense specific isoforms and post-translational-modification states of proteins or specific sequences of oligonucleotides. In this approach, effectively a bioorthogonal “AND” switch, the smart probe and its reaction partner are conjugated onto two groups targeted to different sites of a specific biomolecule. If the two targeting groups can bind the same molecule, the bioorthogonal reagents will be held in close proximity and reaction is accelerated.
One implementation of this strategy is the sequence-specific detection of DNA in living systems (Fig. 17).123 The two bioorthogonal reaction partners are conjugated to two different oligonucleotides. Only in the presence of a template strand of DNA that can hybridize with both oligonucleotides will the rapid fluorescence activation of the smart probe occur. Examples of this approach use the Staudinger ligation or azide-alkyne/tetrazine-cyclopropene cycloadditions.124–126 Of these examples, only tetrazine- and cyclopropene-functionalized oligonucleotides have been shown to capably detect template DNA inside of living mammalian cells. Importantly, mismatches in the hybridized DNA significantly reduce reaction rate, resulting in sequence-specific detection.
Fig. 17.

DNA template-accelerated bioorthogonal reactions of smart probes for sequence-specific detection of DNA.
Finally, the nitrile imine-alkene cycloaddition has been used by Lin and coworkers as a platform to develop photoactivatable probes that are suitable for visualizing trafficking processes or for superresolution imaging (47, Fig. 18).127 While the kinetics of the cycloaddition with unfunctionalized terminal alkenes are modest, the reaction rate can be enhanced dramatically by using an intramolecular variant of the reaction. Indeed, these probes were fully converted to fluorescent pyrazoline product 48 within minutes of irradiation with a handheld 302 nm UV lamp. By attaching these probes to a Taxol derivative, Lin and coworkers were able to visualize microtubules in live cells, albeit using conventional microscopy techniques. In principle, other bioorthogonal reactions may use this same strategy for the development of photoactivatable fluorescent probes.128
Fig. 18.

A rapidly photoactivatable tetrazole probe.
Conclusion
While significant progress has been made in the development of new bioorthogonal chemistries, these efforts are aided by the development of better, smarter probes for these reactions. The fluorescent probes described here have enabled the visualization and detection of tagged biomolecules with ever higher sensitivity, while the caged probes hold promise for the selective delivery of drugs or imaging agents. Further advances in probe development, such as probes based off other imaging modalities, and novel applications of these probes will expand the versatility of bioorthogonal chemistry to aid our understanding of biological systems.
Supplementary Material
Acknowledgements
We thank B. Belardi, C. Woo, A. Baldridge, and D. Spiciarich for critical reading of the manuscript. This work was funded by NIH Grant GM058867 to C.R.B.
Notes and References
- 1.Patterson DM, Nazarova LA, Prescher JA. ACS Chem. Biol. 2014;9:592–605. doi: 10.1021/cb400828a. [DOI] [PubMed] [Google Scholar]
- 2.Lang K, Chin JW. ACS Chem. Biol. 2014;9:16–20. doi: 10.1021/cb4009292. [DOI] [PubMed] [Google Scholar]
- 3.Lang K, Chin JW. Chem. Rev. 2014;114:4764–4806. doi: 10.1021/cr400355w. [DOI] [PubMed] [Google Scholar]
- 4.Ramil CP, Lin Q. Chem. Commun. 2013;49:11007–11022. doi: 10.1039/c3cc44272a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Debets MF, van Hest JCM, Rutjes FPJT. Org. Biomol. Chem. 2013;11:6439–6455. doi: 10.1039/c3ob41329b. [DOI] [PubMed] [Google Scholar]
- 6.Mahal LK, Yarema KJ, Bertozzi CR. Science. 1997;276:1125–1128. doi: 10.1126/science.276.5315.1125. [DOI] [PubMed] [Google Scholar]
- 7.Chen I, Howarth M, Lin W, Ting AY. Nat. Methods. 2005;2:99–104. doi: 10.1038/nmeth735. [DOI] [PubMed] [Google Scholar]
- 8.Wang L, Zhang Z, Brock A, Schultz PG. Proc. Natl. Acad. Sci. 2002;100:56–51. doi: 10.1073/pnas.0234824100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tuley A, Lee Y-J, Wu B, Wang ZU, Liu WR. Chem. Commun. 2014;50:7424–7426. doi: 10.1039/c4cc02000f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeng Y, Ramya TNC, Dirksen A, Dawson PE, Paulson JC. Nat. Methods. 2009;6:207–209. doi: 10.1038/nmeth.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rannes JB, Ioannou A, Willies SC, Grogan G, Behrens C, Flitsch SL, Turner NJ. J. Am. Chem. Soc. 2011;133:8436–8439. doi: 10.1021/ja2018477. [DOI] [PubMed] [Google Scholar]
- 12.Carrico IS, Carlson BL, Bertozzi CR. Nat. Chem. Biol. 2007;3:321–322. doi: 10.1038/nchembio878. [DOI] [PubMed] [Google Scholar]
- 13.Crisalli P, Kool ET. J. Org. Chem. 2013;78:1184–1189. doi: 10.1021/jo302746p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalia J, Raines RT. Angew. Chem. Int. Ed. 2008;47:7523–7526. doi: 10.1002/anie.200802651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Agarwal P, van der Weijden J, Sletten EM, Rabuka D, Bertozzi CR. Proc. Natl. Acad. Sci. 2013;110:46–51. doi: 10.1073/pnas.1213186110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agarwal P, Kudirka R, Albers AE, Barfield RM, de Hart GW, Drake PM, Jones LC, Rabuka D. Bioconj. Chem. 2013;24:846–851. doi: 10.1021/bc400042a. [DOI] [PubMed] [Google Scholar]
- 17.In quenching via ICT, the excited singlet state of the fluorophore may quickly convert into a twisted charge transfer state where electron density is transferred from donor to acceptor moiety. This state rapidly returns to the ground state via non-radiative decay pathways, quenching fluorescence. Smart probes based off ICT processes typically show changes in absorption or emission wavelengths after bioorthogonal reaction. See also: de Silva AP, Gunaratne HQN, Gunnlaugsson T, Huxley AJM, McCoy CP, Rademacher JT, Rice TE. Chem. Rev. 1997;97:1515–1566. doi: 10.1021/cr960386p.
- 18.Gubitz G, Wintersteiger R, Frei RW. J. Liq. Chromatogr. 1984;7:839–854. [Google Scholar]
- 19.Koizumi H, Suzuki Y. J. Chromatogr. 1988;457:299–307. [Google Scholar]
- 20.Uzu S, Kanda S, Nakashima K, Akiyama S. Analyst. 1990;115:1477–1482. [Google Scholar]
- 21.Konarzycka-Bessler M, Bornscheuer UT. Angew. Chem. Int. Ed. 2003;42:1418–1420. doi: 10.1002/anie.200390365. [DOI] [PubMed] [Google Scholar]
- 22.Dilek O, Bane SL. Tet. Lett. 2007;49:1413–1416. doi: 10.1016/j.tetlet.2007.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dilek O, Bane SL. J. Fluoresc. 2011;21:347–354. doi: 10.1007/s10895-010-0723-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Banerjee A, Panosian TD, Mukherjee K, Ravindra R, Gal S, Sackett DL, Bane S. ACS Chem. Biol. 2010;5:777–785. doi: 10.1021/cb100060v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Key JA, Li C, Cairo CW. Bioconj. Chem. 2012;23:363–371. doi: 10.1021/bc200276k. [DOI] [PubMed] [Google Scholar]
- 26.Salahuddin S, Renaudet O, Reymond J-L. Org. Biomol. Chem. 2004;2:1471–1475. doi: 10.1039/b400314d. [DOI] [PubMed] [Google Scholar]
- 27.Kitov PI, Vinals DF, Ng S, Tjhung KF, Derda R. J. Am. Chem. Soc. 2014;136:8149–8152. doi: 10.1021/ja5023909. [DOI] [PubMed] [Google Scholar]
- 28.Ji A, Ren W, Ai H. Chem. Commun. 2014;50:7469–7472. doi: 10.1039/c4cc01551g. [DOI] [PubMed] [Google Scholar]
- 29.Saxon E, Bertozzi CR. Science. 2000;287:2007–2010. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- 30.Lin FL, Hoyt HM, van Halbeek H, Bergman RG, Bertozzi CR. J. Am. Chem. Soc. 2005;127:2686. doi: 10.1021/ja044461m. [DOI] [PubMed] [Google Scholar]
- 31.Schiedel MS, Briehn CA, Bauerle P. Angew. Chem. Int. Ed. 2001;40:4677–4680. doi: 10.1002/1521-3773(20011217)40:24<4677::aid-anie4677>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 32.Lemieux GA, de Graffenried CL, Bertozzi CR. J. Am. Chem. Soc. 2003;125:4708–4709. doi: 10.1021/ja029013y. [DOI] [PubMed] [Google Scholar]
- 33.Hangauer MJ, Bertozzi CR. Angew. Chem. Int. Ed. 2008;47:2394–2397. doi: 10.1002/anie.200704847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang PV, Prescher JA, Hangauer MJ, Bertozzi CR. J. Am. Chem. Soc. 2007;129:8400–8401. doi: 10.1021/ja070238o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cohen AS, Dubikovskaya EA, Rush JS, Bertozzi CR. J. Am. Chem. Soc. 2010;132:8563–8565. doi: 10.1021/ja101766r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Denburg JL, Lee RT, McElroy WD. Arch. Biochem. Biophys. 1969;134:381–394. doi: 10.1016/0003-9861(69)90297-5. [DOI] [PubMed] [Google Scholar]
- 37.Saxon E, Armstrong JI, Bertozzi CR. Org. Lett. 2000;2:2141–2143. doi: 10.1021/ol006054v. [DOI] [PubMed] [Google Scholar]
- 38.Nilsson BL, Kiessling LL, Raines RT. Org. Lett. 2000;2:1939–1941. doi: 10.1021/ol0060174. [DOI] [PubMed] [Google Scholar]
- 39.Azoulay M, Tuffin G, Sallem W, Florent J-C. Bioorg. Med. Chem. Lett. 2006;16:3147–3149. doi: 10.1016/j.bmcl.2006.03.073. [DOI] [PubMed] [Google Scholar]
- 40.Prescher JA, Dube DH, Bertozzi CR. Nature. 2004;430:873–877. doi: 10.1038/nature02791. [DOI] [PubMed] [Google Scholar]
- 41.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew. Chem. Int. Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 42.Tornoe CW, Christensen C, Meldal M. J. Org. Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 43.Agard NJ, Prescher JA, Bertozzi CR. J. Am. Chem. Soc. 2004;126:15046–15047. doi: 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- 44.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Proc. Natl. Acad. Sci. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ning X, Guo J, Wolfert MA, Boons G-J. Angew. Chem. Int. Ed. 2008;47:2253–2255. doi: 10.1002/anie.200705456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jewett JC, Sletten EM, Bertozzi CR. J. Am. Chem. Soc. 2010;132:3688–3690. doi: 10.1021/ja100014q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Debets MF, van Berkel SS, Schoffelen S, Rutjes FP, van Hest JC, van Delft FL. Chem. Commun. 2010;46:97–99. doi: 10.1039/b917797c. [DOI] [PubMed] [Google Scholar]
- 48.Dommerholt J, Schmidt S, Temming R, Hendriks LJA, Rutjes FPJT, van Hest JCM, Lefeber DJ, Friedl P, van Delft FL. Angew. Chem. Int. Ed. 2010;49:9422–9425. doi: 10.1002/anie.201003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.de Almeida G, Sletten EM, Nakamura H, Palaniappan KK, Bertozzi CR. Angew. Chem. Int. Ed. 2012;51:2443–2447. doi: 10.1002/anie.201106325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hong V, Steinmetz NF, Manchester M, Finn MG. Bioconj. Chem. 2010;21:1912–1916. doi: 10.1021/bc100272z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Presolski SI, Hong V, Cho S-H, Finn MG. J. Am. Chem. Soc. 2010;132:14570–14576. doi: 10.1021/ja105743g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soriano Del Amo D, Wang W, Jiang H, Besanceney C, Yan AC, Levy M, Liu Y, Marlow FL, Wu P. J. Am. Chem. Soc. 2010;132:16893–16899. doi: 10.1021/ja106553e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Besanceney-Webler C, Jiang H, Zheng T, Feng L, Soriano del Amo D, Wang W, Klivansky LM, Marlow FL, Liu Y, Wu P. Angew. Chem. Int. Ed. 2011;50:8051–8056. doi: 10.1002/anie.201101817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Le Droumaguet C, Wang C, Wang Q. Chem. Soc. Rev. 2010;39:1233–1239. doi: 10.1039/b901975h. [DOI] [PubMed] [Google Scholar]
- 55.Jarowski PD, Wu Y-L, Schweizer WB, Diederich F. Org. Lett. 2008;10:3347–3350. doi: 10.1021/ol801253z. [DOI] [PubMed] [Google Scholar]
- 56.Scrafton DK, Taylor JE, Mahon MF, Fossey JS, James TD. J. Org. Chem. 2008;73:2871–2874. doi: 10.1021/jo702584u. [DOI] [PubMed] [Google Scholar]
- 57.Schweinfurth D, Hardcastle KI, Bunz UHF. Chem. Commun. 2008;2008:2203–2205. doi: 10.1039/b800284c. [DOI] [PubMed] [Google Scholar]
- 58.Bag SS, Kundu R. J. Org. Chem. 2011;76:3348–3356. doi: 10.1021/jo200231k. [DOI] [PubMed] [Google Scholar]
- 59.Er JC, Tang MK, Chia CG, Liew H, Vendrell M, Chang Y-T. Chem. Sci. 2013;4:2168–2176. [Google Scholar]
- 60.Sivakumar K, Xie F, Cash BM, Long S, Barnhill HN, Wang Q. Org. Lett. 2004;6:4603–4606. doi: 10.1021/ol047955x. [DOI] [PubMed] [Google Scholar]
- 61.Beatty KE, Xie F, Wang Q, Tirrell DA. J. Am. Chem. Soc. 2005;127:14150–14151. doi: 10.1021/ja054643w. [DOI] [PubMed] [Google Scholar]
- 62.Beatty KE, Liu JC, Xie F, Dieterich DC, Schuman EM, Wang Q, Tirrell DA. Angew. Chem. Int. Ed. 2006;45:7364–7367. doi: 10.1002/anie.200602114. [DOI] [PubMed] [Google Scholar]
- 63.Sawa M, Hsu T-L, Itoh T, Sugiyama M, Hanson SR, Vogt PK, Wong C-H. Proc. Natl. Acad. Sci. 2006;103:12371–12376. doi: 10.1073/pnas.0605418103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang C, Xie F, Suthiwangcharoen N, Sun J, Wang Q. Sci. China Chem. 2012;55:125–130. [Google Scholar]
- 65.Herner A, Nikic I, Kallay M, Lemke EA, Kele P. Org. Biomol. Chem. 2013;11:3297–3306. doi: 10.1039/c3ob40296g. [DOI] [PubMed] [Google Scholar]
- 66.Herner A, Girona GE, Nikic I, Kallay M, Lemke EA, Kele P. Bioconj. Chem. 2014;25:1370–1374. doi: 10.1021/bc500235p. [DOI] [PubMed] [Google Scholar]
- 67.Horner A, Volz D, Hagendorn T, Furniss D, Greb L, Ronicke F, Nieger M, Schepers U, Brase S. RSC Advances. 2014;4:11528–11534. [Google Scholar]
- 68.In quenching via PeT for the probes described here, the excited singlet state of the fluorophore moiety accepts an electron from a nearby electron donor. The diradical intermediate relaxes back down to the ground state via back electron transfer. Fluorescence is activated when the donor is no longer capable of transferring electrons to the acceptor fluorophore. As the fluorophore moiety remains untouched upon activation of PeT probes, absorption and emission wavelengths should remain unchanged. See also Ref. 17
- 69.Xie F, Siovakumar K, Zeng Q, Bruckman MA, Hodges B, Wang Q. Tetrahedron. 2008;64:2906–2914. [Google Scholar]
- 70.Shieh P, Hangauer MJ, Bertozzi CR. J. Am. Chem. Soc. 2012;134:17428–17431. doi: 10.1021/ja308203h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Urano Y, Kamiya M, Kanda K, Ueno T, Hirose K, Nagano T. J. Am. Chem. Soc. 2005;127:4888–4894. doi: 10.1021/ja043919h. [DOI] [PubMed] [Google Scholar]
- 72.Shieh P, Siegrist MS, Cullen AJ, Bertozzi CR. Proc. Natl. Acad. Sci. 2014;111:5456–5461. doi: 10.1073/pnas.1322727111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Koide Y, Urano Y, Hanaoka K, Terai T, Nagano T. ACS Chem. Biol. 2011;6:600–608. doi: 10.1021/cb1002416. [DOI] [PubMed] [Google Scholar]
- 74.Shie J-J, Liu Y-C, Lee Y-M, Lim C, Fang J-M, Wang C-H. J. Am. Chem. Soc. 2014;136:9953–9961. doi: 10.1021/ja5010174. [DOI] [PubMed] [Google Scholar]
- 75.Staros JV, Bayley H, Standring DN, Knowles JR. Biophys. Res. Commun. 1978;80:568–572. doi: 10.1016/0006-291x(78)91606-6. [DOI] [PubMed] [Google Scholar]
- 76.Uttamapinant C, Tangpeerachaikul A, Grecian S, Clarke S, Singh U, Slade P, Gee KR, Ting AY. Angew. Chem. Int. Ed. 2012;51:5852–5856. doi: 10.1002/anie.201108181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bevilacqua V, King M, Chaumontet M, Nothisen M, Gabillet S, Buisson D, Puente C, Wagner A, Taran F. Angew. Chem. Int. Ed. 2014;53:5872–5876. doi: 10.1002/anie.201310671. [DOI] [PubMed] [Google Scholar]
- 78.Zhou Z, Fahrni CJ. J. Am. Chem. Soc. 2004;126:8862–8863. doi: 10.1021/ja049684r. [DOI] [PubMed] [Google Scholar]
- 79.Key JA, Koh S, Timerghazin QK, Brown A, Cairo CW. Dyes and Pigments. 2009;82:192–203. [Google Scholar]
- 80.Li J, Hu M, Yao SQ. Org. Lett. 2009;11:3008–3011. doi: 10.1021/ol9010344. [DOI] [PubMed] [Google Scholar]
- 81.Qi J, Tung C-H. Bioorg. Med. Chem. Lett. 2011;21:320–323. doi: 10.1016/j.bmcl.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Qi J, Han M-S, Chang Y-C, Tung C-H. Bioconj. Chem. 2011;22:1758–1762. doi: 10.1021/bc200282t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Key JA, Cairo CW. Dyes and Pigments. 2011;88:95–102. [Google Scholar]
- 84.Rong L, Liu L-H, Chen S, Cheng H, Chen C-S, Li Z-Y, Qin S-Y, Zhang X-Z. Chem. Commun. 2013;50:667–669. doi: 10.1039/c3cc47323f. [DOI] [PubMed] [Google Scholar]
- 85.Jewett JC, Bertozzi CR. Org. Lett. 2011;13:5937–5939. doi: 10.1021/ol2025026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Friscourt F, Fahrni CJ, Boons G-J. J. Am. Chem. Soc. 2012;134:18809–18815. doi: 10.1021/ja309000s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.van Geel R, Prujin GJM, van Delft FL, Boelens WC. Bioconj. Chem. 2012;23:392–398. doi: 10.1021/bc200365k. [DOI] [PubMed] [Google Scholar]
- 88.Song W, Wang Y, Qu J, Madden MM, Lin Q. Angew. Chem. Int. Ed. 2008;47:2832–3824. doi: 10.1002/anie.200705805. [DOI] [PubMed] [Google Scholar]
- 89.Song W, Wang Y, Qu J, Lin Q. J. Am. Chem. Soc. 2008;130:9654–9655. doi: 10.1021/ja803598e. [DOI] [PubMed] [Google Scholar]
- 90.Wang Y, Hu WJ, Song W, Lim RKV, Lin Q. Org. Lett. 2008;10:3725–3738. doi: 10.1021/ol801350r. [DOI] [PubMed] [Google Scholar]
- 91.Yu Z, Ohulchanskyy TY, An P, Prasad PN, Lin Q. J. Am. Chem. Soc. 2013;135:16766–16769. doi: 10.1021/ja407867a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kaya E, Vrabel M, Deiml C, Prill S, Fluxa VS, Carrell T. Angew. Chem. Int. Ed. 2012;51:4466–4469. doi: 10.1002/anie.201109252. [DOI] [PubMed] [Google Scholar]
- 93.Lee Y-J, Wu B, Raymond JE, Zeng Y, Fang X, Wooley KL, Liu WR. ACS Chem. Biol. 2013;8:1664–1670. doi: 10.1021/cb400267m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang XS, Lee Y-J, Liu WR. Chem. Commun. 2014;50:3176–3179. doi: 10.1039/c3cc48682f. [DOI] [PubMed] [Google Scholar]
- 95.Yu Z, Pan Y, Wang Z, Wang J, Lin Q. Angew. Chem. Int. Ed. 2012;51:10600–10604. doi: 10.1002/anie.201205352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yu Z, Lin Q. J. Am. Chem. Soc. 2014;136:4153–4156. doi: 10.1021/ja5012542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Song W, Wang Y, Yu Z, Vera CIR, Qu J, Lin Q. ACS Chem. Biol. 2010;5:875–887. doi: 10.1021/cb100193h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.An P, Yu Z, Lin Q. Org. Lett. 2013;15:5496–5499. doi: 10.1021/ol402645q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Blackman ML, Royzen M, Fox JM. J. Am. Chem. Soc. 2008;130:13518–13519. doi: 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Devaraj NK, Weissleder R, Hilderbrand SA. Bioconj. Chem. 2008;19:2297–2299. doi: 10.1021/bc8004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Karver MR, Weissleder R, Hilderbrand SA. Bioconj. Chem. 2011;22:2283–2270. doi: 10.1021/bc200295y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Taylor MT, Blackman ML, Dmitrenko O, Fox JM. J. Am. Chem. Soc. 2011;133:9646–9649. doi: 10.1021/ja201844c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yang J, Seckute J, Cole CM, Devaraj NK. Angew. Chem. Int. Ed. 2012;51:7476–7479. doi: 10.1002/anie.201202122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Patterson DM, Nazarova LA, Xie B, Kamber DN, Prescher JA. J. Am. Chem. Soc. 2012;134:18638–16843. doi: 10.1021/ja3060436. [DOI] [PubMed] [Google Scholar]
- 105.Chen W, Wang D, Dai C, Hamelberg D, Wang B. Chem. Commun. 2012;48:1736–1738. doi: 10.1039/c2cc16716f. [DOI] [PubMed] [Google Scholar]
- 106.Devaraj NK, Hilderbrand S, Upadhyay R, Mazitschek R, Weissleder R. Angew. Chem. Int. Ed. 2010;49:2869–2872. doi: 10.1002/anie.200906120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lang K, Davis L, Torres-Kolbus J, Chou C, Deiters A, Chin JW. Nat. Chem. 2012;4:298–304. doi: 10.1038/nchem.1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yang KS, Budin G, Reiner T, Vinegoni C, Weissleder R. Angew. Chem. Int. Ed. 2012;51:6598–6603. doi: 10.1002/anie.201200994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liu DS, Tangpeerachaikul A, Selvaraj R, Taylor MT, Fox JM, Ting AY. J. Am. Chem. Soc. 2012;134:792–795. doi: 10.1021/ja209325n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lang K, Davis L, Wallace S, Mahesh M, Cox DJ, Blackman ML, Fox JM, Chin JW. J. Am. Chem. Soc. 2012;134:10317–10320. doi: 10.1021/ja302832g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dumas-Verdes C, Miomandre F, Lepicier E, Galangau O, Vu TT, Clavier G, Meallet-Renault R, Audebert P. Eur. J. Org. Chem. 2010;2010:2525–2535. [Google Scholar]
- 112.In through-bond energy transfer, the donor transfers energy via twisted π-bonds to an acceptor. Unlike FRET, minimal spectral overlap between the donor and acceptor is required, although the acceptor should still absorb light at a longer wavelength than the donor emits. See also: Jiao G-S, Thoresen LH, Burgess K. J. Am. Chem. Soc. 2003;125:14668–14669. doi: 10.1021/ja037193l.
- 113.Carlson JCT, Meimetis LG, Hilderbrand SA, Weissleder R. Angew. Chem. Int. Ed. 2013;52:6917–6920. doi: 10.1002/anie.201301100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Meimetis LG, Carlson JCT, Giedt RJ, Kohler RH, Weissleder R. Angew. Chem. Int. Ed. 2014;53:7531–7534. doi: 10.1002/anie.201403890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Xu H, Yang J, Seckute J, Devaraj NK. Angew. Chem. Int. Ed. 2014;53:5805–5809. doi: 10.1002/anie.201400135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wieczorek A, Buckup T, Wombacher R. Org. Biomol. Chem. 2014;12:4177–4185. doi: 10.1039/c4ob00245h. [DOI] [PubMed] [Google Scholar]
- 117.Neves AA, Stockmann H, Wainman YA, Kuo JC-H, Fawcett S, Leeper FJ, Brindle KM. Bioconj. Chem. 2013;24:934–941. doi: 10.1021/bc300621n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Versteegen RM, Rossin R, Hoeve Wt, Janssen HM, Robillard MS. Angew. Chem. Int. Ed. 2013;52:14112–14116. doi: 10.1002/anie.201305969. [DOI] [PubMed] [Google Scholar]
- 119.Seitchik JL, Peeler JC, Taylor MT, Blackman ML, Rhoads TW, Cooley RB, Refakis C, Fox JM, Mehl RA. J. Am. Chem. Soc. 2012;134:2898–2901. doi: 10.1021/ja2109745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dirks AJ, Cornelissen JJLM, Nolte RJM. Bioconj. Chem. 2009;20:1129–1138. doi: 10.1021/bc8004667. [DOI] [PubMed] [Google Scholar]
- 121.Wijdeven MA, Nicosia C, Borrmann A, Huskens J, van Delft FL. RSC Advances. 2014;4:10549–10552. [Google Scholar]
- 122.Lewis WG, Green LG, Grynszpan F, Radic Z, Carlier PR, Taylor P, Finn MG, Sharpless KB. Angew. Chem. Int. Ed. 2002;41:1053–1057. doi: 10.1002/1521-3773(20020315)41:6<1053::aid-anie1053>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 123.Other examples of fluorogenic DNA-templated reactions for oligonucleotide detection are discussed in this review: Michaelis J, Roloff A, Seitz O. Org. Biomol. Chem. 2014;12:2821–2833. doi: 10.1039/c4ob00096j.
- 124.Cai J, Li X, Yue X, Taylor JS. J. Am. Chem. Soc. 2004;126:16324–16325. doi: 10.1021/ja0452626. [DOI] [PubMed] [Google Scholar]
- 125.Sun H, Peng X. Bioconj. Chem. 2013;24:1226–1234. doi: 10.1021/bc4001678. [DOI] [PubMed] [Google Scholar]
- 126.Seckute J, Yang J, Devaraj NK. Nuc. Acids Res. 2013;41:e148. doi: 10.1093/nar/gkt540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yu Z, Ho LY, Lin Q. J. Am. Chem. Soc. 2011;133:11912–11915. doi: 10.1021/ja204758c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Poloukhtine AA, Mbua NE, Wolfert MA, Boons G-J, Popik VV. J. Am. Chem. Soc. 2009;131:15769–15776. doi: 10.1021/ja9054096. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.