

Abstract
Transcription factors are essential for the differentiation of human induced pluripotent stem cells (iPS) into specialized cell types. Embryoid body (EB) formation promotes the differentiation of iPS cells. We sought to establish an efficient method of transfection and rotary culture to generate EBs that stably express two genes. The pMetLuc2-Reporter vector was transfected using FuGENE HD (FuGENE), Lipofectamine LTX (LTX), X-tremeGENE, or TransIT-2020 transfection reagents. The media was analyzed using a Metridia luciferase (MetLuc) assay. Transfections were performed on cells adherent to plates/dishes (adherent method) or suspended in the media (suspension method). The 201B7 cells transfected with episomal vectors were selected using G418 (200 μg/mL) or hygromycin B (300 μg/mL). Rotary culture was performed at 2.5 or 9.9 rpm. Efficiency of EB formation was compared among plates and dishes. Cell density was compared at 1.6×103,×104, and×105 cells/mL. The suspended method of transfection using the FuGENE HD reagent was the most efficient. The expression of pEBMulti/Met-Hyg was detected 11 days posttransfection. Double transformants were selected 6 days posttransfection with pEBNK/EGFP-Neo and pEBNK/Cherry-Hyg. Both EGFP and CherryPicker were expressed in all of the surviving cells. EBs were formed most efficiently from cells cultured at a density of 1.6×105 cells/mL in six-well plates or 6 cm dishes. The selected cells formed EBs. FuGENE-mediated transfection of plasmids using the suspension method was effective in transforming iPS cells. Furthermore, the episomal vectors enabled us to perform a stable double transfection of EB-forming iPS cells.
Introduction
Human induced pluripotent stem (iPS) cells are a useful tool for drug discovery and regenerative medicine because they differentiate into somatic cells.1 Somatic cells from humans have limitations. For example, primary cultured hepatocytes are useful for the prediction of effects and toxicity of drugs when they are applied for therapy.2,3 It is reported that primary cultured hepatocytes are transplanted into patients with liver failure.4 Cultured hepatocytes are prone to apoptosis and damage.5 Moreover, they do not proliferate.6 If hepatocytes are produced from iPS cells, sufficient of them would be provided. Another example of drug discovery is myodystrophy. Somatic cells from the patients with the disease are useful for drug discovery. Sufficient amount of them are not obtained from the patients. If muscle cells could be differentiated from iPS cells, they would be used for investigation.7 Ethical issues and graft-versus-host disease are circumvented with the use of human iPS cells because they can be established in each patient individually. Human iPS cells may, therefore, be an ideal therapeutic cell source for patients. Embryonic stem cells or iPS cells assemble themselves with a hanging drop method, and express receptors of adhesion molecules.8 These cell aggregates are called embryoid bodies (EBs). EBs mimic 3D structure of early development and promote the differentiation of iPS cells into specialized cell types, such as hepatocytes or cardiomyocytes.9–11 The 3D structure of EBs promotes differentiation more efficiently than cells in 2D and is proposed to be the mechanism underlying this phenomenon.12
Sequential stimulation with growth factors and introduction of growth factors promotes human iPS cells to produce hepatocyte-like cells.13–16 EBs that express the genes necessary for differentiation would be expected to differentiate more efficiently. Thus, it is necessary to establish a method to form EBs that expresses genes of interest.
The introduction of transcription factors is a key step in the differentiation of human iPS cells into specialized cell types.17,18 Introduction of multiple transcription factors to iPS cells promote differentiation to target somatic cells.19 Transcription factors are introduced into iPS cells through plasmids or viral vectors.3,18,20,21 Transfection reagents or electroporation are conventionally used to introduce genes of interest into iPS cells.22,23 The efficiency of transfection reagents is low and selection of the transfected cells is necessary.21 Electroporation is problematic because it can induce cell damage.24 Viral vectors are a more effective method to facilitate the introduction of a gene of interest; however, the construction of these vectors is complicated and cumbersome. Construction of adenovirus requires multiple steps and several months.16 Adenoviral vectors provoke immunological response because they are foreign bodies.25 Furthermore, gene expression is transient with adenoviral vectors. Retroviral vectors harbors tumorigenicity because they integrate host genome.26 On the other hand, plasmid DNA does not induce immune response because they do not produce exogenous protein.27 Plasmid DNA is not associated with tumorigenicity because they do not integrate host genome.28
Episomal vectors contain a latent origin of plasmid replication (oriP) and the Epstein-Barr virus nuclear antigen (EBNA-1) gene that encodes a replication factor. Together, oriP and EBNA-1 enables the persistence and mitotic segregation of Epstein-Barr viral episomes.29 Episomal vectors are distributed to daughter cells through episomal replication. Episomal vectors can be used to establish stable cell lines expressing multiple transgenes in 1 week.30,31 Therefore, we investigated various methods to form EBs that express genes transfected with episomal vectors.
Our goal was to establish a method of formation of EBs that express 100% reporter genes transcribed from episomal vector. First, transfection methods were compared in search for the most efficient ones. Second, optimum condition to select cells transfected with two reporter plasmids. Efficient methods of formation of EBs were investigated. Finally, EBs were formed with cells expressing two reporter plasmids.
Materials and Methods
Cell culture
The human iPS cell line 201B7 (RIKEN Cell Bank, Tsukuba, Japan) was cultured feeder-free in the ReproFF medium (ReproCELL, Yokohama, Japan) in plates or dishes (Asahi Techno Glass, Funabashi, Japan) with a thin coating of matrigel (Becton Dickinson, Franklin Lakes, NJ). Cells were kept in 5% CO2 at 37°C in a humidified chamber and harvested using Accutase (Innovative Cell Technologies, Inc., San Diego, CA). Cells were spread onto 24- and 96-well plates to perform the Metridia luciferase assays, respectively. The dishes and plates were thinly coated by spreading a mixture of matrigel (0.3 mL) and DMEM-F12 medium (8.7 mL). The dishes were incubated at room temperature for 3 h. The morphological features of the cells were examined using a CKX41N-31PHP microscope (Olympus, Tokyo, Japan). To determine the conditions for selection, G418 (Wako Pure Chemicals, Osaka, Japan) and/or hygromycin B (Wako Pure Chemicals) were added to the medium and cells were monitored under a microscope after 7 days. To select cells that had been double transfected, G418 and hygromycin B at 200 and 300 μg/mL, respectively, were added to the medium 4–6 days posttransfection. The number of cells was counted using Trypan Blue staining and a hemocytometer.
Rotary culture
Cells (1.6×103,×104, and×105 cells/mL) were suspended in ReproFF medium supplemented with 10 μM Y-27632 (Wako Pure Chemicals) and rotary cultured using the CSM03020 mild cell shaker (Taitec, Koshigaya, Japan) in 5% CO2 at 37°C in a humidified chamber for 24 h. Cell suspension volumes of 50, 100, 300, and 500 μL were applied to the wells of 96-, 24-, 12-, and 6-well plates, respectively. Cell suspension volumes of 1, 5, and 5 mL were applied to 6 and 10 cm tissue culture dishes, and 10 cm Petri dishes, respectively. Y-27632 was added to avoid apoptosis of 201B7 inhibiting Rho-associated coiled-coil forming kinase.32 To compare the effect of surface treatment on the optimum cell anchorage and growth, 10 cm tissue culture and 10 cm Petri dishes were compared. The rotation speed was set to 9.9 or 2.5 rpm to analyze the effect of rotation speed. The length (longer diameter) and width (shorter diameter) were measured to analyze the EB size and shape.
Plasmid construction
The EGFP coding sequence (Clontech, Mountain View, CA), was amplified from pEGFP-N1 using the LA PCR Kit Ver. 2.1 (Takara, Kyoto, Japan) and subcloned into the 5′-KpnI-XbaI-3′ sites of the pBluescript II SK(-) (Agilent Technologies, Santa Clara, CA) to generate pBlue/EGFP. The sequence was validated (Bio Matrix Research, Inc., Nagareyama, Japan). The coding region of CherryPicker (CherryPicker1) (Clontech) was amplified using the LA PCR Kit Ver. 2.1 and subcloned into the 5′-KpnI-XbaI-3′ sites of the pBluescript II SK(-) to generate pBlue/Cherry. The sequence was validated (Bio Matrix Research, Inc.). The forward and reverse primers used to clone EGFP and CherryPicker were: 5′-ATCGTCTAGAATGGTGAGCAAGGGCGA GGA-3′ and 5′-ATCGGGTACCTTACTTGTACAGCTCGT CCA-3′, and 5′-ATCGTCTAGAATGATGGTGGACGGCG ACAA-3′ and 5′-ATCGGGTACCTCATTTGTACAGCTC GTCCA-3′, respectively. PCR was performed as follows: 20 s denaturation at 98°C and 3 min annealing/extension at 68°C for 30 cycles. EGFP was subcloned from pBlue/EGFP to pEBNK-Neo using 5′-NotI-KpnI-3′ restriction sites, generating pEBNK/EGFP-Neo. CherryPicker was subcloned from pBlue/Cherry to pEBNK-Hyg using 5′-NotI-KpnI-3′ restriction sites, generating pEBNK/Cherry-Hyg. pEBMulti-Neo and pEBMulti-Hyg were obtained from Wako Pure Chemicals. The restriction sites in pEBMulti-Neo and pEBMulti-Hyg are reversed, 5′-KpnI-NotI-3′. If the 5′-NotI-KpnI-3′ fragments of EGFP and CherryPicker were subcloned into the two plasmids, the inserted fragments would be in the reverse direction. To insert the 5′-NotI-KpnI-3′ fragments of EGFP and CherryPicker in the correct direction, the multiple cloning site was changed to 5′-NotI-KpnI-3′. To achieve this, linker sequences 5′-AGCGGCCGCGAGCTCGGTACCTA and 5′-GGCCTAGGTACCGAGCTCGCGGCCGCTGTAC were digested with NotI and KpnI, and subcloned into pEBMulti-Neo and pEBMulti-Hyg to make pEBNK-Neo and pEBNK-Hyg. Metridia luciferase was subcloned from the pMetLuc2-Reporter (Clontech) to pEBMulti-Hyg using 5′-SalI-NotI-3′ restriction sites, generating pEBMulti/Met-Hyg.
Transfection of 201B7 cells
In the adherent method, cells were transfected when they were adherent to the bottom of the well, 24 h after spreading. In the suspension method, cells were harvested with Accutase, suspended, and incubated in the transfection reagent for 5 min at room temperature in a hood.17 The transfection reagents, FuGENE HD Transfection Reagent (Promega, Madison, WI), Lipofectamine LTX (Life Technologies, Grand Island, NY), X-tremeGENE Transfection Reagent (Roche, Basel, Switzerland), and TransIT-2020 Transfection Reagent (Mirus Bio, Madison, WI), were used following the manufacturer's instructions.
Transfection of EBs
EBs were formed from 201B7 cells in six-well plates. The EBs were collected by centrifugation at 156 g and the supernatant was discarded. The EBs were suspended in ReproFF medium and transfected. EBs were suspended in 2 mL of ReproFF, and transfected with 1.0 μg of pEBNK/EGFP-Neo and 1.0 μg of pEBNK/Cherry-Hyg with FuGENE.
Metridia luciferase and secreted alkaline phosphatase assays
In a 96-well plate, 100 ng of pMetLuc2-Reporter was transfected into 201B7 cells cultured in 50 μL of medium per well. Transfected cells secrete Metridia luciferase into the medium. After transfection, the medium was replaced with 50 μL of ReproFF each day. The activity of Metridia luciferase was measured using the Ready-To-Glow Secreted Luciferase Reporter Assay (Clontech) following the manufacturer's instructions. The media was changed daily to monitor the luciferase activity. Luciferase activity was measured using the Gene Light (GL-200A) (Microtech CO., Ltd., Funabashi, Japan). To monitor transfection efficiency, the pSEAP2 control vector (Clontech) was transfected 10% of pMetLuc2-Reporter or pEBMulti/Met-Hyg. The transcriptional activity was measured using a secreted embryonic alkaline phosphatase (SEAP) chemiluminescence kit (Clontech) and Gene Light following the manufacturer's instruction. Luciferase activity was calculated with the Metridia luciferase activity divided by the SEAP activity.
Selection of cells expressing EGFP and CherryPicker
Using the suspension method, 201B7 cells were transfected with 1.0 μg of pEBNK/EGFP-Neo and 1.0 μg of pEBNK/Cherry-Hyg using FuGENE and cultured in six-well plates supplemented with Y-27632 at 10 μM G418 (200 μg/mL) and hygromycin B (300 μg/mL) were added 4 days posttransfection. G418 and hygromycin B were removed from the media after 2 days of incubation.
Image analysis
ImageJ 1.41q (National Institutes of Health, Bethesda, MD) was used for image analysis. A region of 201B7 cells was outlined in an image and the area was measured. The image was split into red, green, and blue channels. The red channel (CherryPicker) was selected for further analysis. The area of CherryPicker signal was measured automatically with the software. The area of the CherryPicker signal was divided by the outlined area of interest to calculate the percentage of CherryPicker-positive cells.
Statistical analysis
One-way analysis of variance was applied with the JMP 10.0.2 software (SAS Institute Japan, Cary, NC). p-Value<0.05 was determined as statistical significance. Tukey–Kramer analysis was applied in search for pairs of data with statistical significance.
Results
FuGENE, LTX, X-tremeGENE, and TransIT-2020 transfection reagents were compared to identify the most efficient reagent for transfection. We compared the activities of Metridia luciferase divided by those of SEAP (relative luciferase activity) in adherent and suspended cells. To compare the effect of cell density on transfection efficiency, we plated 1.8×105 and 9.0×105 cells/well in 96-well plates. The experiments were repeated three times. The suspension method of cell transfection using FuGENE was the most effective (p<0.05) at both cell densities, 1.8×105 (Fig. 1A) and 9.0×105 cells/well (Fig. 1B). At 1.8×105 cells/well, suspension or adherent transfection method with FuGENE, and adherent method with LTX showed higher relative luciferase activities (p<0.05). At 9.0×105 cells/well, suspension or adherent transfection methods using FuGENE and LTX yielded the highest four efficiencies (p<0.05). We selected the suspension method using FuGENE or LTX at 9.0×105 cells/well for further investigation.
FIG. 1.

Time-course of transient transfection. Four reagents were used to transfect 201B7 with pMetLuc2-Reporter plasmids. Plasmid expressing secreted alkaline phosphatase was transfected to monitor transfection efficiency. Transfections were carried out on cells adherent to the well surface (broken line) or suspended in the media (solid line). Cells at a density of 1.8×105 (A) or 9.0×105 cells/well (B) were transfected in 96-well plates. Cells (1.8×105 per well) were spread in 96-well plates. Metridia luciferase was secreted into the media. Media was collected and analyzed using a luciferase assay. Activity of metridia luciferase was divided by that of secreted alkaline phosphatase (relative luciferase activity). Metridia luciferase was subcloned into pEBMulti-Hyg (pEBMulti/Met-Hyg) and transfected in to 201B7 cells using LTX or FuGENE HD reagents (C). (●)=FuGENE HD, (■)=Lipofectamine LTX (LTX), (◯)=TransIT 2020, (□)=X-tremeGENE, error bar: standard deviation, *p<0.05, n=3.
A relative luciferase activity time-course analysis was performed to determine the duration of episomal vector expression (Fig. 1C). pEBMulti/Met-Hyg (100 ng) was transfected into 201B7 cells (1.8×105 cells/well) in 96-well plates using the suspension method along with pSEAP2 control vector (10 ng). Relative luciferase activity reached its peak 4 days posttransfection, suggesting that the expression level of genes transfected in the episomal vector also peaked. Relative luciferase activity was higher with FuGENE transfection than LTX (p<0.05). Thus, suspended method with FuGENE was used for further investigation.
201B7 cells were examined under a microscope after G418 or hygromycin B were added to the media to determine their optimum concentration. They were not transfected with any plasmids. After 7 days of incubation, all of the 201B7 cells were damaged because of G418 (200 μg/mL) or hygromycin B (300 μg/mL) treatment. These concentrations were used in subsequent selections of transfected cells (Fig. 2).
FIG. 2.
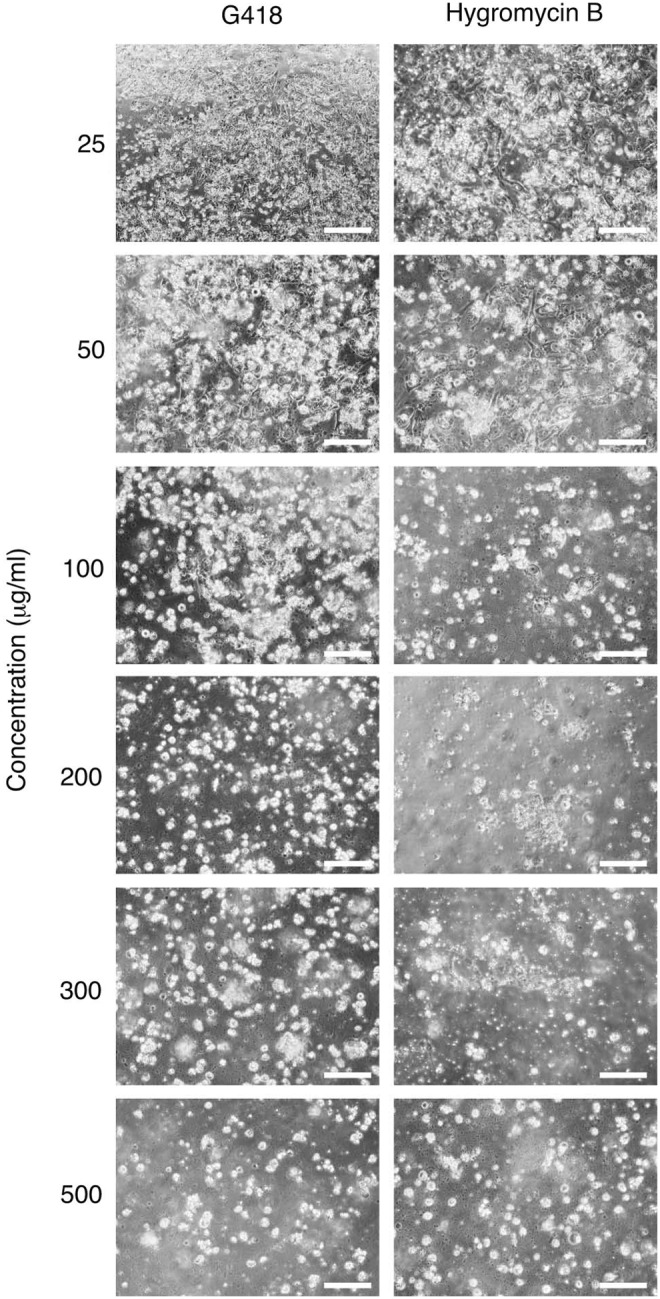
Concentration of G418 and hygromycin B for the selection of transfected cells. In 24-well plates, 201B7 cells were cultured to confluency and G418 or hygromycin B was added. The cells were examined under the microscope 7 days after the addition of the reagents. Magnification: 200×, scale bar: 100 μm.
Using the suspension method, 201B7 cells (4.5×105 cells/well) were transfected with pEBNK/EGFP-Neo and pEBNK/Cherry-Hyg using LTX or FuGENE in six-well plates (Fig. 3). A subset of the cells were positive for EGFP and CherryPicker. Signals from EGFP and CherryPicker were observed in the same cells. G418 and hygromycin B were added 4 days posttransfection, the day of peak luciferase activity (Fig. 1C). No significant changes were observed after 1 day of G418 and hygromycin B incubation. After 2 days of incubation, all of the surviving cells expressed EGFP and CherryPicker indicating that 2 days of G418 and hygromycin B incubation was sufficient for the selection of transfected cells. G418 and hygromycin B were removed from the medium 6 days posttransfection. Signal intensities of EGFP and CherryPicker weakened 11 days posttransfection.
FIG. 3.

Time-course of transfected cells. In a six-well plate, 201B7 cells were transfected with pEBNK/EGFP-Neo and pEBNK/Cherry-Hyg using suspended method with Lipofectamine LTX or FuGENE HD at 4.5×105 cells/well. G418 and hygromycin B were added 4–6 days posttransfection. W: white light, E: EGFP, C: CherryPicker. Magnification: 200×, scale bar: 100 μm. Color images available online at www.liebertpub.com/tea
Photos of the same experiments in Figure 3 were analyzed to determine the incubation period with G418 and hygromycin B to obtain the cells 100% expressing the reporter genes. After 2 days of incubation with G418 and hygromycin B (6 days posttransfection), 100% of the cells were positive for CherryPicker (Fig. 4). CherryPicker was analyzed because the cells expressed EGFP and CherryPicker at the same time based on Figure 3. This percentage decreased as cells approached 11 days posttransfection. It was suggested that 2 days incubation with G418 and hygromycin B was suitable to select cells expressing reporter genes.
FIG. 4.
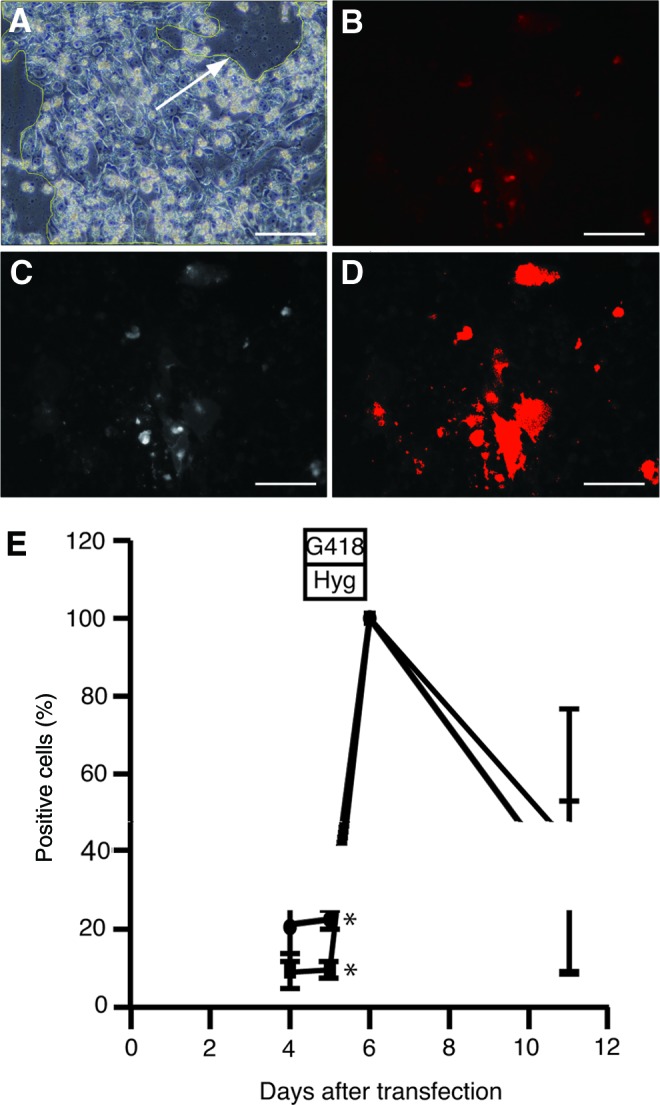
Percentage of EGFP and CherryPicker cells over time. Photos of the same experiments in Figure 3 were analyzed to determine the incubation period with G418 and hygromycin B to obtain the cells 100% expressing the reporter genes. Using ImageJ 1.42q regions of cells were selected and outlined (arrow) (A). Images of CherryPicker-positive cells (B) were split into red (C), green, and blue channels. The threshold of the signal was set and the area of CherryPicker signal (red) was measured. The area of the CherryPicker-positive cells (D) was divided by the area of the outlined area (A) to calculate the percentage of positive cells (E). Arrow: outline of cells, Magnification: 200×, scale bar: 100 μm, (●)=FuGENE HD, (■)=Lipofectamine LTX (LTX), error bar: standard deviation, *p<0.05, n=3. Color images available online at www.liebertpub.com/tea
We next investigated different methods to form EBs from 201B7 cells in search for optimum methods (Fig. 5). Size and shape of EBs were analyzed as well. Cells at a density of 1.6×105 cells/mL were cultured to form EBs in six-well plates rotated at 9.9 (Fig. 5A) and 2.5 rpm (Fig. 5B). The length and width of EBs were measured to analyze their size and shape (Fig. 5C). No EBs were formed from cells plated at a density of 1.6×103 cells/well. The greatest number of EBs formed from cells plated in 10 cm tissue culture and Petri dishes (p<0.05) (Fig. 5D). We speculated that this was the result of culturing more cells in 5 mL of cell suspension. The efficiency of EB formation was calculated as the number of EBs formed divided by the number of cells cultured (Fig. 5E). EB formation was most efficient in six-well plates and 6 cm dishes than any other plate/dish (p<0.05). Rotation at 9.9 rpm was more efficient than 2.5 rpm in promoting EB formation. In every dish/plate type, EBs generated from cells plated at a density of 1.6×105 were larger at 2.5 rpm than 9.9 rpm (Fig. 5F). These data suggested that the lower rotation speed was more effective for the formation of EBs. To analyze the shape of the EBs, we calculated their width–length ratio (Fig. 5G). Perfectly round EBs have a ratio of 1.0. The ratio did not significantly differ depending on plates/dishes, cell density, or rotation speed.
FIG. 5.

Rotary culture of 201B7 cells. 201B7 cells were spread on 96-, 24-, 12-, or 6-well plates, and 6 or 10 cm tissue culture dishes, or 10 cm Petri dishes. Cells were cultured using a mild cell shaker and allowed to form embryoid bodies (EBs). EBs formed from cells plated at a density of 1.6×105 in six-well plates rotated at 9.9 (A) and 2.5 rpm (B). The length (larger diameter) (a) and width (smaller diameter) (b) of each EB was measured (C). Cells were plated at densities of 1.6×103 (s), 1.6×104 (m), or 1.6×105 (l) cells/mL. The plates/dishes were rotated at 9.9 (black) or 2.5 rpm (gray). The number of EBs per well/dish (D) was divided by the cell number at the beginning of rotary culture (E) to calculate the efficiency of EB formation. The size of EBs was calculated by multiplying the length (a) by the width (b) (F). The length–width ratio of EBs were calculated by dividing the length (a) by the width (b) (G). Magnification: 100×(A, B) and 200×(C); scale bars: 200 μm (A, B) and 100 μm(C), error bar: standard deviation, *p<0.05, n=3.
Next, we addressed whether EBs can express double transfected genes. Using the suspension method, 201B7 cells at the density of 4.3×105 cells/well were transfected with 1.0 μg of pEBNK/EGFP-Neo and 1.0 μg of pEBNK/Cherry-Hyg using FuGENE in six-well plates. G418 and hygromycin B were added 4 days posttransfection. Surviving 201B7 cells were harvested using Accutase and subjected to rotary culture at 9.9 rpm for 1 day in 12-well plates (Fig. 6B, D, F). Twelve-well plates were used because not enough cells were obtained with selection with G418 and hygromycin B. Control 201B7 cells that had not been transfected were subjected to rotary culture at 9.9 rpm for 1 day in 12-well plates (Fig. 6A, C, E). EBs were formed under white light (Fig. 6A, B). EBs that had not been transfected did not express EGFP (Fig. 6C) or CherryPicker (Fig. 6E). EBs transfected with pEBNK/EGFP-Neo and pEBNK/Cherry-Hyg, on the other hand, expressed both EGFP (Fig. 6D) and CherryPicker (Fig. 6F).
FIG. 6.

Rotary culture of human induced pluripotent stem cells transfected with EGFP and CherryPicker. Nontransfected 201B7 cells were subjected to rotary culture at 9.9 rpm for 1 day in 12-well plates (A, C, E). 201B7 cells cotransfected with pEBNK/EGFP-Neo and pEBNK/Cherry-Hyg were selected using G418 and hygromycin (B). Surviving cells were subjected to rotary culture at 9.9 rpm for 1 day in 12-well plates (B, D, F). Magnification: 200×, scale bar: 100 μm. (A, B) white light; (C, D) EGFP; (E, F) CherryPicker. Color images available online at www.liebertpub.com/tea
We next determined the region where transfection had occurred in EBs and whether it was possible to select the transfected portions. EBs were transfected with pEBNK/EGFP-Neo and pEBNK/Cherry-Hyg, and subjected to selection with G418 and hygromycin B. EBs were observed under white light posttransfection (Fig. 7A). EGFP (Fig. 7B) and CherryPicker (Fig. 7C) signals were observed on the surface of EBs. EBs were completely destroyed after 2 days of incubation with G418 and hygromycin B (Fig. 7D). These data showed that only surface cells were transfected and it was difficult to transfect the inner cells.
FIG. 7.

EBs transfected with EGFP and CherryPicker. EBs were formed from human induced pluripotent stem cells in a six-well plate after 24 h of rotary culture at 9.9 rpm. The EBs were transfected with pEBNK/EGFP-Neo and pEBNK/Cherry-Hyg and examined under the microscope (A) to detect EGFP (B) and CherryPicker (C). G418 and hygromycin B were added (D). Magnification: 100×, scale bar: 200 μm, arrows: expression of EGFP or CherryPicker. Color images available online at www.liebertpub.com/tea
Discussion
The transfection of plasmids with various reagents is simple to manipulate. One significant limitation of plasmid transfection is low transfection efficiency. Previous studies have employed FuGENE HD, lipofectamine PLUS, and lipofectamine 2000 reagents for transfection.17,21,22 For the first time, we compared FuGENE, LTX, X-tremeGENE, and TransIT-2020 reagents to identify a method that would achieve higher transfection efficiency in iPS cells. Transfections are typically performed on cells that are growing and attaching onto plates or dishes. However, transduction of adenovirus vector in dissociated and suspended cells has been shown to be more efficient than in adherent cells.3 Using the adherent method, adenovirus vector transduces into the periphery of iPS cell colonies.3 In our study, we compared the adherent and suspension methods. Consistent with the findings of a previous study, our results show that the suspension method using FuGENE transfection reagent was the most effective.22 Notably, FuGENE achieves the same transfection efficiency as electroporation.20 Therefore, we conclude that FuGENE-mediated transfection using the suspension method is the most suitable for introducing plasmid into iPS cells.
Episomal vectors are different from conventional plasmids because they are delivered to daughter cells. Our results show that episomal vector expression can be detected 11 days posttransfection. Another advantage of the episomal vector is that we were able to achieve the stable transfection of two plasmids. Using conventional plasmids, stable transformants express a single gene.21 Multiple genes would need to be introduced to differentiate iPS cells into specialized cell types. Our data clearly show that stable double transfection is possible with episomal vectors. Using standard methods, 2 weeks of antibiotic selection is required.21,24 In contrast, it took only 6 days to obtain stable transformants in our study. Our results indicate that episomal vectors facilitate stable double transfection in a shorter time frame than conventional plasmids.
In the presence of growth factors, EB formation promotes differentiation of iPS cells.22,33 If the EBs expressed the transcription factors that are indispensable for differentiation, we speculate that target cell types would be attained more efficiently. Our data demonstrated that the episomal vectors allowed the expression of two transfected genes in EB-forming iPS cells. Because EBs were transfected only on the surface, we recommend that iPS cells are transfected before EB formation.
In conclusion, transfection of plasmids by using FuGENE and suspension method was the most suitable method for iPS cells. Furthermore, an episomal vector facilitated the stable double transfection of forming EBs.
Acknowledgment
This work was supported by a Research Grant-in-Aid for Scientific Research (C) (23591002) from the Japan Society for the Promotion of Science (JSPS).
Disclosure Statement
The authors have nothing to declare.
References
- 1.Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131,861, 2007 [DOI] [PubMed] [Google Scholar]
- 2.Romano G., Morales F., Marino I.R., and Giordano A.A commentary on iPS cells: potential applications in autologous transplantation, study of illnesses and drug screening. J Cell Physiol 229,148, 2014 [DOI] [PubMed] [Google Scholar]
- 3.Tashiro K., Kawabata K., Inamura M., Takayama K., Furukawa N., Sakurai F., et al. Adenovirus vector-mediated efficient transduction into human embryonic and induced pluripotent stem cells. Cell Reprogram 12,501, 2010 [DOI] [PubMed] [Google Scholar]
- 4.Strom S.C., Chowdhury J.R., and Fox I.J.Hepatocyte transplantation for the treatment of human disease. Semin Liver Dis 19,39, 1999 [DOI] [PubMed] [Google Scholar]
- 5.Fisher R.L., Hasal S.J., Sanuik J.T., Scott K.S., Gandolfi A.J., and Brendel K.Cold- and cryopreservation of human liver and kidney slices. Cryobiology 30,250, 1993 [DOI] [PubMed] [Google Scholar]
- 6.Mitaka T., Sattler C.A., Sattler G.L., Sargent L.M., and Pitot H.C.Multiple cell cycles occur in rat hepatocytes cultured in the presence of nicotinamide and epidermal growth factor. Hepatology 13,21, 1991 [PubMed] [Google Scholar]
- 7.Abujarour R., Bennett M., Valamehr B., Lee T.T., Robinson M., Robbins D., et al. Myogenic differentiation of muscular dystrophy-specific induced pluripotent stem cells for use in drug discovery. Stem Cells Transl Med 3,149, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bratt-Leal A.M., Carpenedo R.L., and McDevitt T.C.Engineering the embryoid body microenvironment to direct embryonic stem cell differentiation. Biotechnol Prog 25,43, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu H., Wang Z., Zheng Q., Li J.H., Chong X.Q., and Xiao S.D.Efficient differentiation of newly derived human embryonic stem cells from discarded blastocysts into hepatocyte-like cells. J Dig Dis 11,376, 2010 [DOI] [PubMed] [Google Scholar]
- 10.Lim S.Y., Sivakumaran P., Crombie D.E., Dusting G.J., Pebay A., and Dilley R.J.Trichostatin A enhances differentiation of human induced pluripotent stem cells to cardiogenic cells for cardiac tissue engineering. Stem Cells Transl Med 2,715, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tomizawa M., Toyama Y., Ito C., Toshimori K., Iwase K., Takiguchi M., et al. Hepatoblast-like cells enriched from mouse embryonic stem cells in medium without glucose, pyruvate, arginine, and tyrosine. Cell Tissue Res 333,17, 2008 [DOI] [PubMed] [Google Scholar]
- 12.Baharvand H., Hashemi S.M., Kazemi Ashtiani S., and Farrokhi A.Differentiation of human embryonic stem cells into hepatocytes in 2D and 3D culture systems in vitro. Int J Dev Biol 50,645, 2006 [DOI] [PubMed] [Google Scholar]
- 13.Song Z., Cai J., Liu Y., Zhao D., Yong J., Duo S., et al. Efficient generation of hepatocyte-like cells from human induced pluripotent stem cells. Cell Res 19,1233, 2009 [DOI] [PubMed] [Google Scholar]
- 14.DeLaForest A., Nagaoka M., Si-Tayeb K., Noto F.K., Konopka G., Battle M.A., et al. HNF4A is essential for specification of hepatic progenitors from human pluripotent stem cells. Development 138,4143, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Si-Tayeb K., Noto F.K., Nagaoka M., Li J., Battle M.A., Duris C., et al. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology 51,297, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takayama K., Inamura M., Kawabata K., Katayama K., Higuchi M., Tashiro K., et al. Efficient generation of functional hepatocytes from human embryonic stem cells and induced pluripotent stem cells by HNF4alpha transduction. Mol Ther 20,127, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanaka A., Woltjen K., Miyake K., Hotta A., Ikeya M., Yamamoto T., et al. Efficient and reproducible myogenic differentiation from human iPS cells: prospects for modeling Miyoshi Myopathy in vitro. PLoS One 8,e61540, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y., Pak C., Han Y., Ahlenius H., Zhang Z., Chanda S., et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78,785, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ieda M., Fu J.D., Delgado-Olguin P., Vedantham V., Hayashi Y., Bruneau B.G., et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142,375, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eiges R., Schuldiner M., Drukker M., Yanuka O., Itskovitz-Eldor J., and Benvenisty N.Establishment of human embryonic stem cell-transfected clones carrying a marker for undifferentiated cells. Curr Biol 11,514, 2001 [DOI] [PubMed] [Google Scholar]
- 21.Vallier L., Rugg-Gunn P.J., Bouhon I.A., Andersson F.K., Sadler A.J., and Pedersen R.A.Enhancing and diminishing gene function in human embryonic stem cells. Stem Cells 22,2, 2004 [DOI] [PubMed] [Google Scholar]
- 22.Takarada T., Kou M., Nakamichi N., Ogura M., Ito Y., Fukumori R., et al. Myosin VI reduces proliferation, but not differentiation, in pluripotent P19 cells. PLoS One 8,e63947, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liskovykh M., Chuykin I., Ranjan A., Safina D., Popova E., Tolkunova E., et al. Derivation, characterization, and stable transfection of induced pluripotent stem cells from Fischer344 rats. PLoS One 6,e27345, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cao F., Xie X., Gollan T., Zhao L., Narsinh K., Lee R.J., et al. Comparison of gene-transfer efficiency in human embryonic stem cells. Mol Imaging Biol 12,15, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raper S.E., Chirmule N., Lee F.S., Wivel N.A., Bagg A., Gao G.P., et al. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol Genet Metab 80,148, 2003 [DOI] [PubMed] [Google Scholar]
- 26.Miura K., Okada Y., Aoi T., Okada A., Takahashi K., Okita K., et al. Variation in the safety of induced pluripotent stem cell lines. Nat Biotechnol 27,743, 2009 [DOI] [PubMed] [Google Scholar]
- 27.Wells D.J.Electroporation and ultrasound enhanced non-viral gene delivery in vitro and in vivo. Cell Biol Toxicol 26,21, 2010 [DOI] [PubMed] [Google Scholar]
- 28.Ledwith B.J., Manam S., Troilo P.J., Barnum A.B., Pauley C.J., Griffiths T.G. 2nd, et al. Plasmid DNA vaccines: investigation of integration into host cellular DNA following intramuscular injection in mice. Intervirology 43,258, 2000 [DOI] [PubMed] [Google Scholar]
- 29.Yates J.L., Warren N., and Sugden B.Stable replication of plasmids derived from Epstein-Barr virus in various mammalian cells. Nature.313, 812, 1985 [DOI] [PubMed] [Google Scholar]
- 30.Tanaka J., Miwa Y., Miyoshi K., Ueno A., and Inoue H.Construction of Epstein-Barr virus-based expression vector containing mini-oriP. Biochem Biophys Res Commun 264,938, 1999 [DOI] [PubMed] [Google Scholar]
- 31.Shibata M.A., Miwa Y., Morimoto J., and Otsuki Y.Easy stable transfection of a human cancer cell line by electrogene transfer with an Epstein-Barr virus-based plasmid vector. Med Mol Morphol 40,103, 2007 [DOI] [PubMed] [Google Scholar]
- 32.Watanabe K., Ueno M., Kamiya D., Nishiyama A., Matsumura M., Wataya T., et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol 25,681, 2007 [DOI] [PubMed] [Google Scholar]
- 33.Greenhough S., Bradburn H., Gardner J., and Hay D.C.Development of an embryoid body-based screening strategy for assessing the hepatocyte differentiation potential of human embryonic stem cells following single-cell dissociation. Cell Reprogram 15,9, 2013 [DOI] [PubMed] [Google Scholar]