

Abstract
We previously reported encouraging in vitro and in vivoanti-cancer activity of N-((3-chloro-2-hydroxy-5-nitrophenyl)carbamothioyl)benzamide (termed PITENIN-1). In the current work, we describe the structure-activity relationship study of PIT-1 series, based on the replacement of central thiourea unit with a 1,2,3-triazole, which leads to increased liver microsomal stability, drug likeness and toxicity towards cancer cells.
Overactivation of PI3K signaling is a common feature of many types of human cancer.1 Increased activation of a variety of PI3K effectors, through their binding to the PI3K product, lipid PIP3, provides multiple growth, survival, migration and metabolic advantages to cancer cells. Not surprisingly, small molecule targeting of the PI3K pathway, including direct inhibition of PI3K isoforms as well as its multiple effectors (Akt, mTOR, PDK1, etc.) has attracted major attention. While many approaches focused on targeting enzymatic activities in the PI3K network, we and others have recently described a new approach aimed at targeting a universal central step in PI3K signal transduction, i.e. binding of PIP3 to PH domains of effector proteins.2 In particular, we have developed two new classes of small molecule antagonists of PIP3, termed PIT-1 [N-((3-chloro-2-hydroxy-5-nitrophenyl)carbamothioyl)benzamide] and PIT-2 [(Z)-5-(2-benzyl-5-hydroxy-4-nitrobenzylidene)-2-thioxothiazolidin-4-one] (Figure 1).3,4 These two structurally dissimilar molecules displayed very similar activities in cancer cells, including induction of apoptosis and metabolic stress and inhibition of cell migration and invasion. Furthermore, both PIT-1 and PIT-2 displayed synergistic toxicity with TRAIL in human glioblastoma U87MG cells. These activities have been linked to the inhibition of Akt signaling and actin remodeling by ARF6, two pathways regulated by PI3K.3,4 These in vitro activities of PITs translated into the significant inhibition of tumor growth and lung metastasis formation in 4T1 and B16-F10 syngeneic xenograft models by the dimethyl analog of PIT-1.3,4
Figure 1.

Structures of the two classes (PIT-1 and PIT-2) of inhibitors of phopshotidylinositol-3 kinase (PI3K) signaling pathway, termed PITENINs (PITs) and the newly designed 2nd generation triazole-PITs
Despite the promising initial results, PITs displayed obvious limitations, including high micromolar activity as well as multiple non-drug-like features. In particular, nitrophenyl and thiourea moieties of PIT-1 represent potential toxicity concerns and metabolic liabilities. Initial analysis of PIT-1 series revealed surprisingly specific SAR for a micromolar compound suggesting several changes to the molecule, leading to some increase in activity and changes in targeting different PH domains.3,4 In particular, the addition of two methyl groups to the phenyl ring in PIT-1 (DM-PIT-1, Figure 1) resulted in some increase in activity and improved incorporation into long-circulating PEG-PE micelles for in vivo delivery.3,5 Furthermore, changes to the nitrophenyl ring were identified. However, these results did not address the main limitations of PIT-1 series, which was the target of our current work.
In the processes of refining the initially identified structural scaffold of the PIT-1, our first concern was to replace the susceptible thiourea unit with a stable bioisostere.6 The 1,2,3-triazole structural motif has attracted our attention in this regard considering the fact that the triazole is a safe bioequivalent surrogate for the amide bond and that this concept has found its application in the area of anticancer agents as well as in developing non-nucleoside reverse transcriptase inhibitors.7,8 As shown in Figure 1, triazole-PITs have been designed as the structural mimics of DM-PIT-1 and as potential second-generation PITenins (PITs), in anticipation of better antitumor activity.
Scheme 1 reveals the salient features of the synthesis of the newly designed trizole PITENINS. The alkynone precursors 3a–3e have been prepared by following a two-step sequence consisting the addition ethynylmagnesium chloride to corresponding aldehydes 2a–2e and subsequent IBX oxidation. The azidophenols 4a–4f have been synthesized from the corresponding aminophenols by following the standard azidation procedures and are used immediately. The copper catalyzed [3+2] cycloaddition reaction of alkynones 3 with azidophenols 4 has been carried out under established click reaction conditions (20 mol% CuSO4, 20 mol% Na-ascorbate in tert-BuOH-water, at rt) to obtain the requisite 1,2,3-triazole PITENINS.9 Table 1 summarizes the details of the compounds synthesized. All the new compounds have been characterized completely with the help of spectral and analytical data.
Scheme 1.
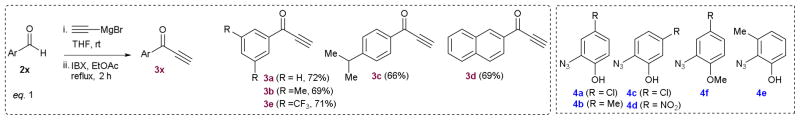
Synthesis of alkynones 3a–3e (eq.1) and their Cu-catalyzed [3+2]-cycloaddition (eq.2 ) with selected 2-azidophenols 4a–4e
Table 1.
The synthesis of and screening results with 1,4-disubstituted 1,2,3-triazolesa
| |||
|---|---|---|---|
| S.No | Triazole (Yield, Code) | EC50 (μmol) | |
| alone | with TRIALb | ||
| 1 |
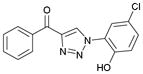 1aa (87%, YK-NCL-176) |
103.6 | 51.6 |
| 2 |
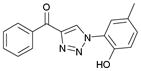 1ab (91%, YK-NCL-184) |
>100 | >100 |
| 3 |
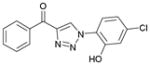 1ac (87%, YK-NCL-191) |
48.9 | 42.3 |
| 4 |
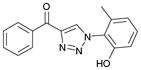 1ae (84%, YK-NCL-185) |
8.1 | 8.0 |
| 5 |
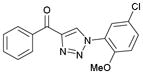 1af (82%, YK-NCL-178) |
>100 | >100 |
| 6 |
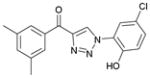 1ba (83%, YK-NCL-190) |
57.8 | 30.6 |
| 7 |
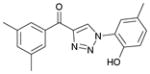 1bb (85%, YK-NCL-193) |
>100 | >100 |
| 8 |
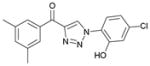 1bc (91%, YK-NCL-183) |
>100 | 64.4 |
| 9 |
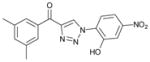 1bd (86%, YK-NCL-194) |
89.1 | 70.5 |
| 10 |
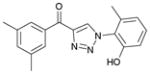 1be (91%, YK-NCL-192) |
104.0 | 98.4 |
| 11 |
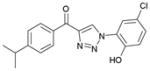 1ca (86%, YK-NCL-195) |
64.1 | 41.6 |
| 12 |
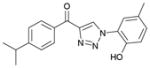 1cb (83%, YK-NCL-197) |
>100 | >100 |
| 13 |
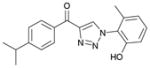 1ce (82%, YK-NCL-196) |
>100 | >100 |
| 14 |
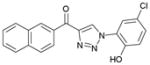 1da (87%, YK-NCL-186) |
>100 | >100 |
| 15 |
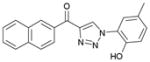 1db (91%, YK-NCL-188) |
>100 | >100 |
| 16 |
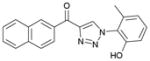 1de (84%, YK-NCL-187) |
85.2 | 74.9 |
| 17 |
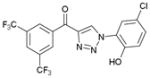 1ea (82%, YK-NCL-240) |
11.99 | 3.15 |
| 18 |
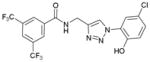 1fa (85%, YK-NCL-234) |
16.2 | 14.0 |
Experiments were performed in A2780 cells as described in the Electronic Supplementary Information
TRIAL- Tumor necrosis factor–Related Apoptosis–Inducing Ligand
The initial screening of 1aa against growth of A2780 human ovarian cancer cells revealed that 1aa has comparable activity with the corresponding PIT-1 analogue.10 In addition, the corresponding methyl ether 1af was completely inactive consistent with the importance of free –OH group in the original thiourea series.3 In this course of SAR studies, we have installed the dimethyl groups and isopropyl groups on the ring A along with variable functional groups on the ring C. However, this approach was not effective. Replacing the aryl ring A with a napthyl unit has been also examined, albeit with no improvement in the activity. In contrast, we found that the addition of the -CF3 group at the C-3 and C-5 positions on ring A in combination with a chloro group at the C-4 position of the ring C (1ea) showed excellent improvement (20 times higher than that of the DM-PIT-1) in anti-tumor activity.
In order to examine whether the addition of an extra hydrogen bond donor in between the triazole and ring A will have any effect, the compound 1fa has been synthesized (Scheme 2). The compound 1fa has showed good activity, which, however is lower than that of 1ea.
Scheme 2.
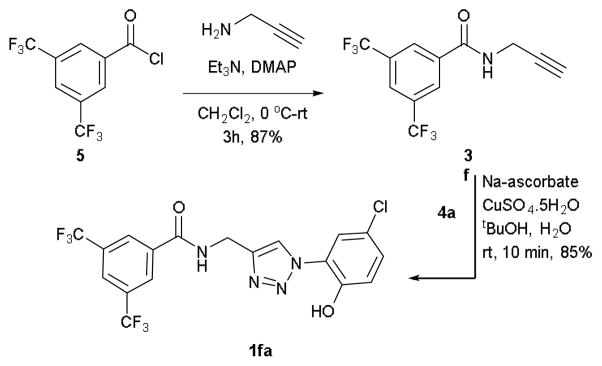
Synthesis of triazole1fa
Having examined a wide range of analogs, compound 1ea has been selected for further progress in the direction of examining its pharmacological properties. The mode of activity of 1ea has been studied by lipid overlay assay, measuring the binding of the AktPH domain to PI(3,4)P2 spotted on nicrocellulose membrane, as previously described.3 Compound 1ea displayed substantially higher activity compared to PIT-1 (Figure 2A). We further examined inhibition of PI3K/Akt signaling in human glioblastoma U87MG and ovarian carcinoma A-2780 cells. Interestingly, we observed very robust inhibition of the TORC1/p70S6K/S6 pathway downstream from Akt, which correlated with cytotoxicity of the compounds and suggested that increased activity of 1ea translated into more specific inhibition of a particular pathway downstream from PI3K/Akt (Figure 2B). Other targets of the compound 1ea in the PI3K/Akt signaling pathway remain to be fully elucidated in the future.
Figure 2.

Increased activity of 1ea. A) Increased inhibition of PI(3,4)P2/Akt-PH domain binding by 1ea.Lipid overlay experiment was performed as performed as described in.3 B) Inhibition of S6 phopshorylation by 1ea. U87MG or A2780 cells were treated with indicated concentrations of inhibitors for 7 hr, followed by Western blotting using phopsho-S6 and total S6 antibodies.C) Increased cytotoxicity of 1ea compared to DM-PIT-1 in multiple cell types. Cells were treated with indicated concentrations of inhibitors alone or in combination with 1 μg/ml TRAIL for 24 hr. Cell viability was determined using CellTiter-Glo assay. D) 1ea efficiently blocks migration of A2780 cells. Wound healing assay was performed as previously described.4 Cell monolayers were treated with indicated concentrations of inhibitors, followed by a scratch wound. Size of the cell free area was photographed at indicated periods of time. Quantification of open wound area is shown on the right.
Subsequently, the cytotoxicity of new PIT analogs in three different cancer cell lines (U87MG, A2780 and T47D) has revealed that 1ea displayed highest activity in all cases (Figure2C). In addition to activation of cell death, PITs displayed several additional important properties. First, we found that PIT-1 analogs reverse resistance of cancer cells to anti-cancer cytokine TRAIL. This led to synergistic cytotoxicity of PIT-1 and TRAIL when applied in combination.3 This useful property was retained with 1ea (Figure 2C and Table 2). Second, PIT-1 analogs not only increased cell death, but also suppressed cell migration and invasion though the attenuation of actin cytoskeleton remodeling.4 Consistently, 1ea displayed significantly increased activity in cell migration assay (Figure 2D).
Finally, one of the major goals of our study was to improve the pharmacological properties of PIT-1/DM-PIT-1. DM-PIT-1 displayed T1/2=1.8 min (CL=1262 μL*min−1*mg−1) in mouse liver microsomal stability assay in vitro. Compound 1ea displayed T1/2=119 min (CL=19.4 μL*min−1*mg−1) in the same assay. Pharmacokinetics analysis following i.v. injection of 1 mg/kg of the drug showed reasonable T1/2=3.22 hr with CL of 930 mL/hr/kg and bioavailability of 85.1% following i.p. administration. Furthermore, 1ea, unlike DM-PIT-1, complied with 4 out of 5 Lipinski rules, with the exception of liphophilicity (calculated LogP = 5.3). Overall, our SAR analysis describes a new analog of PIT-1 with significantly improved anti-cancer activity and pharmacological properties, which may present a promising molecule for further analysis in mouse xenograft models.
Conclusions
In summary, a structure-activity relationship (SAR) study of the N-((3-chloro-2-hydroxy-5-nitrophenyl)carbamothioyl)benzamide (PIT-1) series revealed that increased liver microsomal stability and toxicity towards cancer cells could be achieved by replacing the central thiourea unit with the 1,2,3-triazole heterocycle unit. Secondly, the nitro group of the nitrophenyl moiety can be replaced with chlorine, removing another potential liability. Finally, the addition of two trifluoromethyl moieties to the second phenyl ring of the molecule further increased activity, but also resulted in the increased lipophilicity. Overall, a derivative incorporating all of the modifications (i.e. 1ea) displayed good stability in vitro and in vivo and significantly increased toxicity against a number of cancer cell lines alone and also in combination with TRAIL. Interestingly, compound 1ea displayed particularly robust activity in inhibiting the mammalian Target of Rapamycin Complex 1 (TORC1) signaling downstream from Akt. Mechanism of this inhibition is currently under investigation.
Supplementary Material
Acknowledgments
We would like to thank Dr. Jinbo Lee for helpful discussions. CVR and YK thank CSIR (India) for funding this project (12 FYP ORIGIN program, CSC0108) and for a fellowship to YK. This work was supported in part by NIH/NCI U54CA151881 grant to V.T. and A.D.
Footnotes
Electronic Supplementary Information (ESI) available: For synthesis and characterization data and spectra of all the new compounds see DOI: 10.1039/c000000x/
References
- 1.Wong KK, Engelman JA, Cantley LC. Curr Opin Genet Dev. 2010;20:87–90. doi: 10.1016/j.gde.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McNamara CR, Degterev A. Future Med Chem. 2011;3:549–565. doi: 10.4155/fmc.11.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miao BC, Skidan I, Yang JS, Lugovskoy A, Reibarkh M, Long K, Brazell T, Durugkar KA, Maki J, Ramana CV, Schaffhausen B, Wagner G, Torchilin V, Yuan JY, Degterev A. Proc Natl Acad Sci U S A. 2010;107:20126–20131. doi: 10.1073/pnas.1004522107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miao B, Skidan I, Yang J, You Z, Fu X, Famulok M, Schaffhausen B, Torchilin V, Yuan J, Degterev A. Oncogene. 2012;31:4317–4332. doi: 10.1038/onc.2011.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skidan I, Miao B, Thekkedath RV, Dholakia P, Degterev A, Torchilin V. Drug Deliv. 2009;16:45–51. doi: 10.1080/10717540802517951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Tron GC, Pirali T, Billington RA, Canonico PL, Sorba G, Genazzani AA. Med Res Rev. 2008;28:278–308. doi: 10.1002/med.20107. [DOI] [PubMed] [Google Scholar]; b) Patani GA, LaVoie EJ. Chem Rev. 1996;96:3147–3176. doi: 10.1021/cr950066q. [DOI] [PubMed] [Google Scholar]; c) Jochim AL, Miller SE, Angelo NG, Arora PS. Bioorg Med Chem Lett. 2009;19:6023–6026. doi: 10.1016/j.bmcl.2009.09.049. [DOI] [PubMed] [Google Scholar]; d) Brik A, Alexandratos J, Lin YC, Elder JH, Olson AJ, Wlodawer A, Goodsell DS, Wong CH. ChemBioChem. 2005;6:1167–1169. doi: 10.1002/cbic.200500101. [DOI] [PubMed] [Google Scholar]; e) Angelo NG, Arora PS. J Am Chem Soc. 2005;127:17134–17135. doi: 10.1021/ja056406z. [DOI] [PubMed] [Google Scholar]; f) Palmer MH, Findlay RH, Gaskell AJ. J Chem Soc Perkin Trans 1. 1974;4:420–428. [Google Scholar]
- 7.a) Thirumurugan P, Matosiuk D, Jozwiak K. Chem Rev. 2013;113:4905–4979. doi: 10.1021/cr200409f. [DOI] [PubMed] [Google Scholar]; b) Hou JL, Liu XF, Shen J, Zhao GL, Wang PG. Expert Opin Drug Discov. 2012;7:489–501. doi: 10.1517/17460441.2012.682725. [DOI] [PubMed] [Google Scholar]; c) Agalave SG, Maujan SR, Pore VS. Chem Asian J. 2011;6:2696–2718. doi: 10.1002/asia.201100432. [DOI] [PubMed] [Google Scholar]
- 8.Selected papers on use of 1,2,3-triazole unit in the development of anti-cancer/anti-viral therapeutics: Jayaprakash KN, Peng CG, Butler D, Varghese JP, Maier MA, Rajeev KG, Manoharan M. Org Lett. 2010;12:5410–5413. doi: 10.1021/ol102205j.Alvarez R, Velazquez S, Sanfelix A, Aquaro S, Declercq E, Perno CF, Karlsson A, Balzarini J, Camarasa MJ. J Med Chem. 1994;37:4185–4194. doi: 10.1021/jm00050a015.
- 9.a) Himo F, Lovell T, Hilgraf R, Rostovtsev VV, Noodleman L, Sharpless KB, Fokin VV. J Am Chem Soc. 2005;127:210–216. doi: 10.1021/ja0471525. [DOI] [PubMed] [Google Scholar]; b) Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 10.The synthesis of compound 1aa and its activity as potassium channel activator has been reported earlier by: Calderone V, Giorgi I, Livi O, Martinotti E, Mantuano E, Martelli A, Nardi A. Eur J Med Chem. 2005;40:521–528. doi: 10.1016/j.ejmech.2005.01.010.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.