

Abstract
Formation of a 6-layered cortical plate and axon tract patterning are key features of cerebral cortex development. Abnormalities of these processes may be the underlying cause for a range of functional disabilities seen in human neurodevelopmental disorders. To identify mouse mutants with defects in cortical lamination or corticofugal axon guidance, N-ethyl-N-nitrosourea (ENU) mutagenesis was performed using mice expressing LacZ reporter genes in layers II/III and V of the cortex (Rgs4-lacZ) or in corticofugal axons (TAG1-tau-lacZ). Four lines with abnormal cortical lamination have been identified. One of these was a splice site mutation in reelin (Reln) that results in a premature stop codon and the truncation of the C-terminal region (CTR) domain of reelin. Interestingly, this novel allele of Reln did not display cerebellar malformation or ataxia, and this is the first report of a Reln mutant without a cerebellar defect. Four lines with abnormal cortical axon development were also identified, one of which was found by whole-genome resequencing to carry a mutation in Lrp2. These findings demonstrated that the application of ENU mutagenesis to mice carrying transgenic reporters marking cortical anatomy is a sensitive and specific method to identify mutations that disrupt patterning of the developing brain.
Keywords: cerebral cortex, cortical lamination, corticofugal axon, ENU mutagenesis, reelin
Introduction
Cortical lamination and formation of corticofugal axon projections are key processes in the development of the cerebral cortex. Proper development of the 6 layers of the cortex is crucial for correct assembly of cortical circuitry (Douglas and Martin 2004). In rodents, cortical lamination is achieved by radial migration during late embryonic and early postnatal periods (Rakic 2007). Post-mitotic neurons born from the ventricular and subventricular zones (VZ/SVZ) migrate along radial glia fibers. Later-born neurons migrate through the layers of earlier-born neurons, forming the cortex in an “inside-out” fashion. Finally, neurons reach their correct position below the marginal zone (MZ) and stop migrating. Cortical neurons develop their projections during late embryogenesis (Lopez-Bendito and Molnar 2003; Jacobs et al. 2007; Grant et al. 2012), while cortical lamination is still in process. Corticofugal axons, from projection neurons in layers V and VI, are classified by their subcortical destinations into corticothalamic, corticotectal, corticospinal and corticopontine tracts (Molyneaux et al. 2007). All of these corticofugal tracts initially take a similar route upon leaving the cortex as they extend through the internal capsule penetrating the striatum. These axons are directed by a number of guidance cues and by the interaction with proper reciprocal projections to cross several boundary zones and reach their target area (Price et al. 2006; Molyneaux et al. 2007; Canty and Murphy 2008; Grant et al. 2012; Molnar et al. 2012).
Humans and rodents share the basic structural organization and developmental principles in the cortex. The characterization of mutant mice with neurodevelopmental abnormalities and the discovery of the causal genes have greatly contributed to our understanding of the genetic regulation of cortical development and function. The best-known example is the identification of reelin (Reln) from the ataxic reeler mutant that develops inverted and disorganized cortical layers, as well as a small, poorly foliated cerebellum (Falconer 1951; D'Arcangelo et al. 1995; Hirotsune et al. 1995). Discovery of reelin facilitated identification of the molecular pathways required for neuronal migration and enabled ascertainment of their role in human neurodevelopmental disorders (Hong et al. 2000; Rice and Curran 2001; Knuesel 2010). However, spontaneous mutation only occurs at low frequency, and spontaneous mutants that are as similarly informative as reeler are rarely identified.
Mutagenesis in mice using N-ethyl-N-nitrosourea (ENU) is a means to maximize the efficiency of a phenotype-based forward genetic analysis (Justice et al. 1999; Anderson 2000; Hatten and Heintz 2005; Stottmann and Beier 2010). While identifying the single-base mutations caused by ENU has historically been challenging, the rapid advances in methods of genomic analysis has made positional cloning relatively straightforward (Moran et al. 2006; Fairfield et al. 2011; Leshchiner et al. 2012). Since many human mutations that cause abnormal phenotypes are single-base pair variants, ENU-induced mutations may be a better model of human mutations compared with other gene disruption methods in mice. ENU mutagenesis may help illuminate a function of the specific domain in known genes, as well as the function of novel genes. Furthermore, single-base substitution can generate mutants with hypomorphic or hypermorphic phenotypes. Therefore, phenotypes that were unappreciated in previous studies of a gene of interest (perhaps due to early embryonic lethality) may be ascertained in unbiased screens designed to focus on a specific phenotype.
Productive ENU mutagenesis screens for neurodevelopmental abnormalities have been performed by us and others (Zarbalis et al. 2004; Mar et al. 2005; Lewcock et al. 2007; Wong et al. 2009; Merte et al. 2010; Dwyer et al. 2011; Stottmann et al. 2011). Phenotypes examined in the previous screens included gross embryonic forebrain morphology (Stottmann et al. 2011), tangential migration of cortical interneurons (Zarbalis et al. 2004), thalamocortical axon development (Dwyer et al. 2011), cranial nerve patterning (Mar et al. 2005), spinal motor neuron pathfinding (Lewcock et al. 2007), dorsal root ganglia nerve patterning (Merte et al. 2010), and axonal and neuromuscular synaptic phenotypes (Wong et al. 2009). While cortical lamination has been recognized as one of the most important aspects of cortical development, screening specifically for lamination mutants has not previously been done. Encouraged by the success of experiments that utilized neuroanatomic reporters, we designed a novel screening strategy focusing on cortical lamination utilizing Rgs4-lacZ as a reporter. Using this strategy, we identified a novel hypomorphic allele of Reln without cerebellar malformation and ataxia, which still retains cortical lamination defects. We also tested TAG1-tau-lacZ as a reporter to screen for corticofugal axon guidance defects (Poliak et al. 2003). This screening method highlighted axon guidance defects in an Lrp2 mutant, whose most pronounced phenotype is holoprosencephaly. We also observed an abnormal VZ in a Phr1 mutant. In addition, we report multiple mutant lines with heritable cortical and other developmental defects. Thus, the incorporation of these reporter transgenes into our ENU mutagenesis strategy has improved our ability to recover mutations that disrupt cortical patterning.
Materials and Methods
Mice
Wild-type C57BL6/N and A/J mice were purchased from Taconic, Inc. and the Jackson laboratory, respectively. Rgs4-lacZ reporter mice, generated by homologous recombination inserting LacZ-neo cassette into Rgs4 exon 1, were purchased from Jackson laboratory (B6;129P2-Rgs4tm1Dgen/J). TAG1-tau-lacZ mice were generated by A.F. and provided by Dr T. Sakurai. In TAG1-tau-lacZ mice, exon 2 containing the start codon and 4 downstream exons were replaced with tau-lacZ reporter (Poliak et al. 2003). Efr3aGt(YHD369)Byg (referred to as Efr3a-lacZ hereafter), was generated by blastocyst injection from a genetrap ES cell line made by BayGenomics (Stryke et al. 2003). Animals were maintained in accordance with guidelines of National Institutes of Health and the Harvard Medical School Center for Animal Resources and Comparative Medicine.
ENU Injection and Screening
Eight-week-old A/J male mice (G0) were treated weekly with fractionated dose of ENU (90, 95, or 100 mg/kg in ethanol, Sigma) by intraperitoneal injections for 3 weeks (Stottmann et al. 2011). Twenty A/J males were injected, and after several weeks of recovery period, were mated with C57BL6/N mice or Rgs4-lacZ reporter mice. Ten G0 males recovered fertility, and 100 G1 males were generated. G1 males were screened using a three-generation breeding strategy as previously described (Beier and Herron 2004; Stottmann and Beier 2010). Briefly, G1 males were outcrossed with C57BL/6N or LacZ reporter mice, and G2 females were backcrossed to their fathers to generate G3 animals. G3 animals were examined at E18.5 or P21 depending on the screening design.
Whole-Mount X-Gal Staining and Histology
Whole-mount X-gal staining was modified from our previously described screening protocol (Dwyer et al. 2011). G3 animals were euthanized, decapitated, and their brains were dissected. Then, brains were cut coronally or in an angled plane with a razor blade, then fixed briefly for 15 min with 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS). The fixed brains were incubated in X-gal staining solution (1 mg/mL X-gal, 5 mM ferricyanide, 5 mM ferrocyanide, 2 mM MgCl2, 0.01% deoxycholate, 2% NP-40 in 0.1 M phosphate buffer [pH 7.3]) for overnight at 4°C. After the staining, the brains were examined with a stereomicroscope and fixed in 4% PFA for overnight before imaging.
For histology of brains, the whole heads (E18.5) or dissected brain (P21) were fixed in Bouin's fixative for several days. The fixed heads were embedded in paraffin, and sectioned at a thickness of 10–12 μm. The sections were mounted on the glass slides and stained with hematoxylin and eosin (H&E). The brains or sections were examined using Leica MZ-6 or MZ-12.5 stereomicroscopes and imaged using a Leica DC500 digital camera. For high-magnification images of histological sections, Zeiss Axio Imager Z1 microscope was used for imaging.
For immunohistochemistry (IHC), the brains were dissected from P7 animals, fixed in 4% PFA in PBS for overnight, and incubated in 30% sucrose for overnight. The OCT-embedded brains were cryosectioned at a thickness of 12 μm, mounted on the glass slides, washed with PBS, and immunostaining was performed. The brain sections were incubated with blocking solution (3% goat serum, 3% bovine serum albumin, 0.3% Triton X-100 in PBS) for 1 h, with rabbit anti-Cux1 (Santa Cruz, sc13024, 1:100) and rat anti-Ctip2 (Abcam, ab18465, 1:500) antibodies for overnight at 4°C, with the secondary antibodies (Alexa Fluor 488 goat anti-rabbit IgG and Alexa Fluor 568 goat anti-rat IgG) for 2 h at room temperature, and then with Hoechst for 5 min. The immunostained sections were imaged at the similar brain region with the identical configuration using Zeiss Axio Imager Z1 microscope.
Mapping and Genotyping
Genomic DNA was prepared from tails using the Puregene core kit A (Qiagen). SNP genotyping was performed at the Centre for Applied Genomics in the Hospital for Sick Children (Toronto, ON, Canada) using Illumina medium-density SNP panels, or at Geneseek, Inc. (Lincoln, NE, USA) using the Mouse Universal Genotyping Array (MUGA), and the analysis was performed as previously described (Moran et al. 2006). Once the map position was identified, mice were genotyped using microsatellite markers and restriction fragment length polymorphisms (RFLP) for efficient colony maintenance and recombinant tests. Polymorphic genetic markers flanking the mapped region and often an additional marker in the middle of the region were selected using Mouse Genome Informatics database or SNP2RFLP (http://genetics.bwh.harvard.edu/snp2rflp/), and used to identify a carrier of A/J haplotype at the map position (Beckstead et al. 2008).
Sequencing of Candidate Genes
The candidate genes were identified by Mouse Genome Informatics database search. For candidate gene sequence analysis using genomic DNA, all exons including coding sequence and at least 25 bp flanking sequence including the splice junctions were sequenced bidirectionally from tail DNA of mutants and wild-type A/J mice. At least 2 mutants were sequenced for each line. The mutant sequences were compared with both A/J control sequence and the reference C57BL/6 sequence from UCSC Genome Browser. Sequencing primers were designed either using Exon Primer function in UCSC Genome Browser or manually using Primer3Plus (www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi). Sequence alignment and variation analysis were done using Sequencher 5.0 software (Gene Codes Corporation).
For sequence analysis of cDNA, total RNA was isolated from phenotypically identified mutant tissue using TRIZOL reagent (Invitrogen), and cDNA was synthesized using Superscript III first-strand kit (Invitrogen). PCR primers were designed to amplify overlapping 800–1000 bp fragments covering the entire transcript, and the same primers were used for bidirectional sequencing. The sequences were aligned and compared with the reference cDNA sequence from Ensembl database. Genomic DNA from mutants and wild-type A/J mice were sequenced to confirm that the mutation is not present in the parental strain. We also confirmed that the mutations are not previously known SNPs identified from MGI database or Sanger Institute Mouse Genome Project. For reverse transcription-polymerase chain reaction (RT-PCR) analysis of Reln transcript, forward primers against exon 62 (5′-AGCCGAAGGACTTCACACAA-3′) and reverse primers against exon 65 (5′-GAAGTGCCTGAGCCCATGT-3′) were used.
Western Blotting
Brains from P7 mice were homogenized in lysis buffer (20 mM Tris-HCl, 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 1% sodium deoxycholate, 1 mM PMSF, 1X protease, and phosphatase inhibitor cocktail) and centrifuged, and the protein concentrations in the supernatant were measured using BCA protein assay kit (Pierce). About 25 μg of samples were separated on 6 or 12% SDS-PAGE gel and transferred onto PVDF membranes. The membranes were incubated with blocking solution (5% milk, 1X TBS, 0.1% Tween-20) for 2 h, with mouse anti-reelin antibodies (G10, 1:4000, Abcam) or anti-GAPDH (D16H11, 1:4000, Cell Signaling Technologies) for overnight at 4°C, then with HRP-conjugated anti-mouse secondary antibodies for 2 h. SuperSignal West Femto substrate (Pierce) was used for detection.
Results
Selection of LacZ Reporter Lines for Efficient Examination of Cortical Layers and Axon Tracts
To identify mutants with developmental defects in the cerebral cortex, we selected 2 LacZ reporter lines for the screening. To identify reporter lines that could potentially be informative with respect to cortical layer patterning, we first examined gene expression pattern data from the Allen Brain Atlas (http://www.brain-map.org/) and the published literature. Selected genes were cross-referenced with already existing LacZ reporter strains using Mouse Genome Informatics database (http://www.informatics.jax.org/). Several reporter lines were tested at different prenatal and postnatal ages to determine the best reporter mice and the timing for the screen.
The Rgs4-lacZ (Regulator of G-protein signaling 4) reporter line was chosen to detect cortical lamination defects. Discrete cortical layers could be readily visualized using whole-mount X-gal staining after a minimal dissection to expose a coronal cross section (Fig. 1A,B). Reporter patterning was examined at postnatal day 7 (P7), P14–15 (see Supplementary Fig. 1A), and P21 (Fig. 1B). The Rgs4-lacZ reporter transgene was expressed in cortical layer II/III and V, but not in layer IV at P21 (Fig. 1B and 2A). The patterning at P21 was more distinct than younger ages, so the screening was done at this age. This expression pattern is consistent with a previous in situ hybridization study of Rgs4 expression (Ingi and Aoki 2002; Ebert et al. 2006). Distinct expression of Rgs4-lacZ in both superficial and deep cortical layers enables detection of lamination defect in all layers of the cortical plates; for example, the upper layer-specific patterning defect can be detected even when the deep layer patterning is relatively normal. Homozygous Rgs4-null mice were viable and fertile, and showed normal gross brain morphology and cortical layer formation (Grillet et al. 2005). To assess the utility of Rgs4-lacZ mice for analysis of cortical lamination, mice homozygous for the spontaneous reeler allele (Relnrl/rl) and heterozygous for Rgs4-lacZ were generated. As anticipated, the cortical defects in Relnrl/rl were readily visualized using the Rgs4-lacZ reporter (Fig. 4). Efr3a-lacZ is another reporter line that we tested, in which a β-geo reporter gene (a fusion of β-galactosidase and neomycin phosphotransferase II) is inserted in the intron 18–19 of Efr3a (Efr3 homolog A). Layer-specific expression pattern in Efr3a-lacZ was observed at P11 (see Supplementary Fig. 1B) and in adult (P56, Allen Brain Atlas). However, the homozygous mice were early embryonic lethal and this line was not used in the screen to avoid potential effects of haploinsufficiency.
Figure 1.
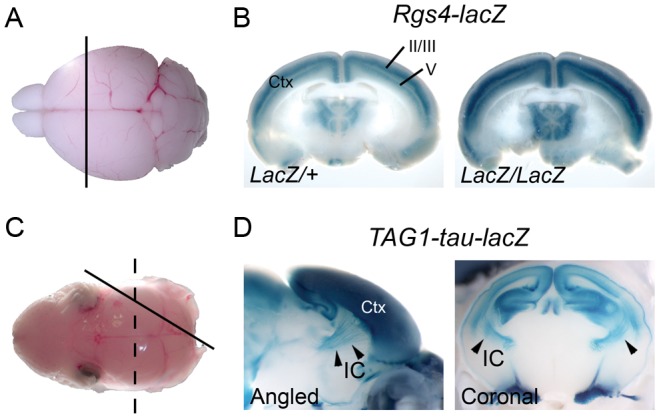
LacZ reporters used for ENU screening. (A) A brain from a P21 mouse was dissected out and coronally sectioned to expose the cross section of the cortex. (B) Reporter expression pattern in the cortex (Ctx) of Rgs4-lacZ mice visualized by whole-mount X-gal staining. Note that superficial layers II/III and deeper layer V shows strong X-gal staining. (C) A brain from P0 pups were dissected out and sectioned in an angled plane (solid line) or coronally (dashed line) to expose the cross section of the cortex and striatum. (D) Reporter expression pattern in the brain of TAG1-tau-lacZ mice. TAG1-tau-lacZ marks the corticofugal axons of the cortex (Ctx) which fasciculate at the internal capsule (IC). Arrowheads indicate the internal capsule.
Figure 2.
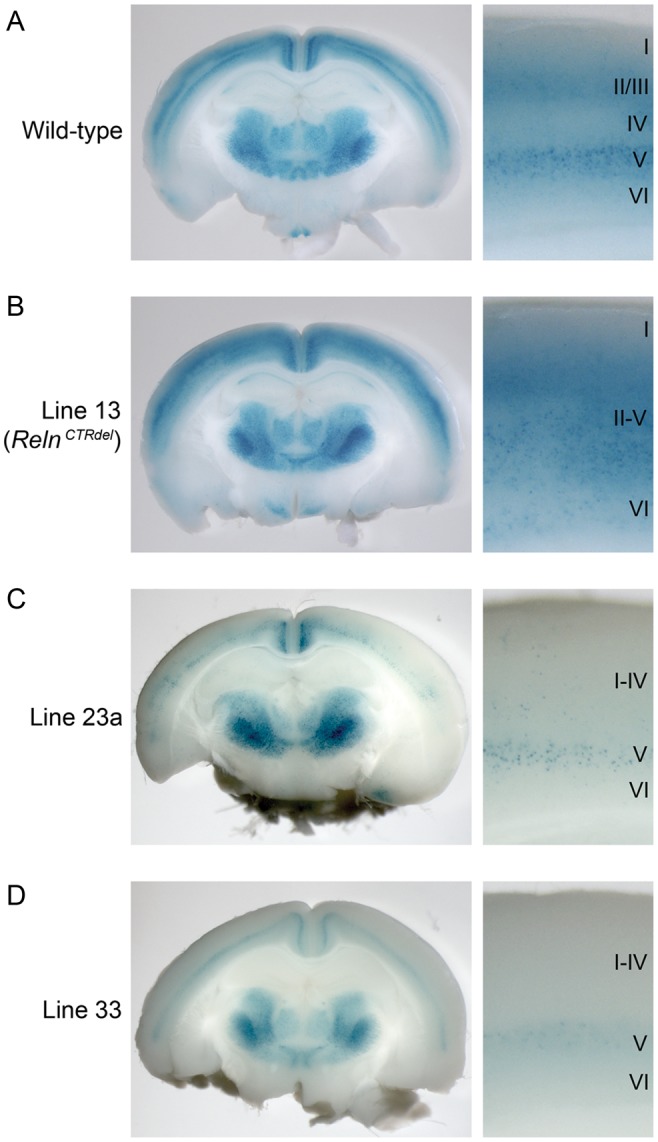
Mutants with cortical lamination defects discovered using an Rgs4-lacZ transgenic reporter. Coronal-sections of whole-mount X-gal stained P21 brains and high-magnification images of the cortical layers are shown. (A) In wild-type cortex, layers II/III and V were stained as 2 distinct bands, and this allows identification of all 6 layers. (B) In line 13 mutant (RelnCTRdel), reporter expression pattern in the cortex was disorganized, and there was no distinction between superficial and deep layers (II–V). (C) Line 23a mutant brain, which has weak reporter expression, with the superficial layers more severely affected. Layers I–IV cannot be distinguished, and layer V is weakly stained. (D) Line 33 mutant brain, which has a similar phenotype as line 23a.
Figure 4.

A complementation assay and cerebellar morphology of RelnCTRdel. (Left) A compound heterozygous mutant of line 13 and reeler (RelnCTRdel/Relnrl) develops lamination defect that appears similar to a homozygous mutant from line 13 (RelnCTRdel/RelnCTRdel), with undistinguishable layers II–V. As controls, brains from heterozygote (Relnrl/Reln+) and homozygote reeler mutants (Relnrl/Relnrl) are shown. (Right) RelnCTRdel/RelnCTRdel and RelnCTRdel/Relnrl show normal cerebellar size and foliation in contrast to Relnrl/Relnrl with abnormal cerebellum.
TAG1-tau-lacZ was used as an axonal reporter line to detect abnormal development of corticofugal axon tracts (Furley et al. 1990; Poliak et al. 2003). TAG1 (Transiently expressed Axonal surface Glycoprotein 1) is a cell adhesion molecule that is expressed in the cell body and developing axons of many subpopulations of neurons in the central and peripheral nervous system (Furley et al. 1990; Wolfer et al. 1994). Corticofugal axons, including corticothalamic, corticospinal, and corticopontine tracts, express TAG1 during late embryonic and early postnatal periods (Wolfer et al. 1994). TAG1 deficiency cause aberrant sensory afferents from dorsal root ganglion to spinal cord (Law et al. 2008), defect in cerebellar granule neuron development (Xenaki et al. 2011), and some behavioral abnormalities reflecting myelination defect (Savvaki et al. 2008). However, it was reported that TAG1-tau-lacZ homozygous mice only showed a subtle phenotype in the cerebral cortex and did not show gross morphological abnormalities (Denaxa et al. 2005). To test if TAG1-tau-lacZ can be used for the screening, embryonic day 18.5 (E18.5) or P0 mouse brains were sectioned in an angled plane or coronal orientation, and whole-mount X-gal staining was performed (Fig. 1C,D). Corticofugal axon tracts in the internal capsule passing through the striatum were clearly visualized, and the axon patterning appeared similar to that shown by TAG1 IHC (Wolfer et al. 1994), even in homozygous reporter mice (data not shown). However, due to possibility of the genetic interaction between TAG1 deficiency and ENU-induced mutations, the screening strategy was designed so that G3 embryos examined are heterozygous TAG1-tau-lacZ carriers.
Forward Genetic Screens Using ENU Mutagenesis
Rgs4-lacZ and TAG1-tau-lacZ reporter mice were incorporated into a three-generation ENU screening strategy to identify recessive mutations. We used an outcross strategy to utilize genetic polymorphism between strains for haplotype-based mapping (Beier and Herron 2004; Moran et al. 2006). Male A/J mice were mutagenized with ENU; these are Generation 0 (G0) mice. Because LacZ reporter lines were on C57BL/6 background, G1 males were generated from outcross of G0 males to C57BL/6 females. The reporter mice were introduced into each line at either G0 or G1 outcross mating to generate LacZ-positive G1 or G2 mice. To generate recessive mutants, LacZ-positive daughters (G2) were backcrossed to their G1 fathers, and G3 offspring were examined. In presumptive mutant lines, G2 males were tested for heritability of the phenotype using the same backcross strategy or by intercross to siblings.
67 G1 males were tested, of which 59 were fertile and were screened using reporter mice (Table 1). Twenty-seven lines were screened using Rgs4-lacZ at P21, making this the first reported mutagenesis experiment that screened for brain patterning defects in adults. Thirty-two lines were screened at E18.5 using TAG1-tau-lacZ. At least 4 litters from independent LacZ-positive G2 mothers were examined for each line, if possible, to maximize the possibility of the mutant discovery with the minimum number of dissections (Stottmann and Beier 2010). A total of 472 G3 litters (more than 3000 progeny) have been examined. The screening strategy for each line was modified based on the initial phenotype observed. For example, if the average litter size was <5 pups in lines screened postnatally, E18.5 embryos were examined again for embryonic or perinatal lethality (line 11, 44, and 50). Of the 59 lines screened, a total of 18 mutant lines were identified (Tables 1 and 2). Reporter patterning defects were detected in 7 lines: line 13, 23a, and 33 using Rgs4-lacZ reporter, and line 7, 27, 48, and 61 using TAG1-tau-lacZ reporter. Importantly, the patterning abnormalities in most of these 7 lines would have been undetectable without using reporters. This clearly demonstrated the utility of LacZ reporters for identification of mutants with brain patterning defects in the absence of gross morphological defects. Five additional lines with brain phenotypes were identified without using reporters: line 8 listed under the cortical layer formation phenotype, plus 4 lines (line 16, 42, 55, and 67) with abnormal brain morphology. Therefore, total 12 lines (7 with patterning defects and 5 with morphological defects) were identified as mutants with brain malformation. In addition, 4 lines (line 44, 49, 50, and 68) with neural tube closure defects were identified, and the number of neurodevelopmental mutants from this screen adds up to total 16. The remaining 2 lines had other developmental defects, and it is not known if they have any neurodevelopmental defects.
Table 1.
Summary of screening
| Type of screening | Age | Number of lines tested | Total litters examined | Total G3 animals examined | Mutants (reporter patterning) | Mutants (other defects) | Mapped |
|---|---|---|---|---|---|---|---|
| Rgs4-lacZ | P21 | 27 | 198 | 1116 | 3 | 3 | 3 |
| Small litter size at P21a | E18 | 8 | 49 | 355 | – | 3b | 3 |
| TAG1-tau-lacZ | E18 | 32 | 204 | 1506 | 4 | 5c | 6 |
| Total | 59 | 472 | 3102 | 7 | 11 | 12 |
aThese lines were initially screened at P21 using Rgs4-lacZ, but screened again for embryonic phenotypes due to small litter size, which suggested prenatal or postnatal lethality.
bThe other 5 lines showed embryonic lethality, but these were not pursued further.
cSix additional lines repeatedly showed embryonic lethality, but these were not pursued further.
Table 2.
Summary of mutants
| Line | Phenotype | Age | LacZ reporter | Chr | Interval (Mb) | No. of mutants genotyped for mapping | Heritability | Gene/mutation |
|---|---|---|---|---|---|---|---|---|
| Cortical layer formation | ||||||||
| 8 | Cobblestone-like cortical malformation; cleft palate and craniofacial morphology; omphalocele; stiff leg posture or club foot | E18 | 9 | Cena – 31 | 7 | Y | ||
| 13 | Disorganized cortical layers | P21 | Rgs4 | 5 | Cena – 28 | 11 | Y | Reln CTRdel |
| 23a | Weak reporter expression and thinner or no staining in the superficial cortical layer; ventriculomegaly or hydrocephaly | P21 | Rgs4 | Ambiguous | 2 | Y | ||
| 33 | Weak reporter expression and thinner or no staining in the superficial cortical layer; smaller olfactory bulb; ventriculomegaly or hydrocephaly | P21 | Rgs4 | Ambiguous | 19 (7 lamination mutants) | Y | ||
| Corticofugal axon development | ||||||||
| 7 | Axon guidance defect (absent internal capsule); club foot | E18 | TAG1 | 14 | 97–108 | 6 | Y | Phr1 Arg3936Stop |
| 27 | Axon guidance defect; holoprosencephaly; cleft palate | E18 | TAG1 | 2 | 58–81 | 5 | Y | Lrp2Cys4032Ser |
| 48 | Axon growth or guidance defect | E18 | TAG1 | Ambiguous | 8 | Y | ||
| 61 | Axon growth or guidance defect; ventriculomegaly | E18 | TAG1 | Ambiguous | 3 | Yb | ||
| Other neurodevelopmental phenotypes: brain morphology | ||||||||
| 16 | Microcephaly; growth defect; curved or absent fifth digit or triphalangeal thumb; early embryonic lethality (completely resorbed by E18) | E18 | Ambiguous | 8 (4 mutants with microcephaly) | Y | |||
| 42 | Ventriculomegaly or hydrocephaly; growth defect and postnatal lethality | P21 | 8 | 105–130 | 4 | Y | ||
| 55 | Ventriculomegaly or hydrocephaly; anophthalmia or micropthalmia | P21 | Ambiguous | 4 | Y | |||
| 67 | Ventriculomegaly or hydrocephaly | P21 | 11 | 47–69 | 3 | Y | ||
| Other neurodevelopmental phenotypes: neural tube closure | ||||||||
| 44 | Encephalocele; spina bifida; perinatal lethality (small litter size at P21c) | E18 | 8 | 37–122 | 5 | Y | ||
| 49 | Spina bifida, exencephaly or open neural tube; oligodactyly | E18 | 6 | 121–149 | 5 | Y | Lrp6Trp104Arg | |
| 50 | Spina bifida, exencephaly or open neural tube; oligodactyly; perinatal lethality (small litter size at P21c) | E18 | 6 | 85–140 | 6 | Y | Same as line 49d | |
| 68 | Spina bifida, exencephaly or open neural tube | E18 | 3 | 117–143 | 5 | Y | Sec24bTyr84Stop | |
| Other developmental phenotypes | ||||||||
| 11 | Cleft lip and palate; perinatal lethality (small litter size at P21c) | E18 | 14 | 79–108 | 9 | Y | ||
| 18 | Growth defect; skeletal defect | E18 | 11 | 76–100 | 5 | Y | ||
a“Cen” means the centromere. In these lines, the most centromere-proximal SNP was homozygous for A/J, therefore a recombination breakpoint is not further determined.
bHeritability of the axon defect in line 61 has not been confirmed yet.
cThese lines were initially screened at P21 using Rgs4-lacZ, but screened again for embryonic phenotypes due to small litter size, which suggested prenatal or postnatal lethality.
dThe same mutation was found in 2 different lines, which originate from the same G0 mice.
Genetic mapping was performed using whole-genome SNP analysis (Moran et al. 2006). The Illumina medium-density SNP panel containing 1440 SNPs and the Geneseek Mouse Universal Genotyping Array (MUGA) with 9000 SNPs were used for this screening. About 900 and 3000 SNPs, respectively, on the panel were polymorphic between A/J and C57BL/6. To date, 12 lines have been mapped as recessive mutations. Six lines were reproducibly heritable, but showed ambiguous mapping results (Table 2). Of the 12 lines mapped, mutations have been identified in 6 lines (Table 2).
Lamination Mutants Identified Using Rgs4-LacZ Reporter Include a Reln Mutation
Abnormal cortical lamination was detected in 3 lines screened using Rgs4-lacZ reporter mice, validating this approach of using transgenic reporters to highlight cortical patterning (Fig. 2). Compared with the wild type with distinct 6 cortical layers (Fig. 2A), Rgs4-lacZ reporter was unable to distinguish layers II-V in line 13 (Fig. 2B). Heterozygous and homozygous mutants rarely showed weak reporter expression level. Other than the lamination phenotype, the mutant brains appeared grossly normal. To locate the mutation in line 13, genetic mapping using whole-genome SNP analysis was performed (Fig. 3A). SNPs proximal to rs13478154 at 27.4 Mb of chromosome 5 were genotyped as homozygous for A/J alleles in all 6 mutants that were tested. This region contains Reln and Cdk5, both of which can cause lamination defects when disrupted (Caviness 1977; Ohshima et al. 1996). Genomic DNA from the mutants and an A/J control were analyzed for the entire coding sequence of Cdk5 and exon intron junctions, but no mutation was found. Since Reln has 65 exons, Reln cDNA was sequenced instead of genomic DNA. It was found that the Reln transcript from mutant mice had an insertion of 42 bp after sequences corresponding to exon 63 (Fig. 3B). The inserted sequence matches the intron sequence distal to the exon-intron junction of exon 63, and a T → C mutation was found at the junction. The insertion of the short intronic sequence is likely due to a splicing defect as a result of this mutation. The insertion is predicted to add 5 amino acids followed by a premature stop codon (Fig. 3B), which will result in the truncation of C-terminal 33 amino acids, corresponding to the entire C-terminal region (CTR) domain of reelin. Based on the above prediction, line 13 mutants were named “RelnCTRdel”. RT-PCR analysis reveals that mutants do not express wild-type transcript (Fig. 3C), and the abundance of the mutant transcript is similar to wild-type levels, suggesting that there is no significant nonsense-mediated decay of the mutant mRNA. Reelin protein in the cortical lysate was measured using Western blot analysis, and the full-length reelin protein was present in the mutant (Fig. 3D).
Figure 3.

Line 13 has a mutation in Reln. (A) Haplotype-based mapping using a whole-genome SNP panel located the causal mutation on chromosome 5 between the centromere and 27.4 Mb. Genotypes of 6 G3 mutants (M1–M6) in the proximal region of chromosome 5 are shown. SNPs are color-coded: Gray, A/J; black, C57BL/6N (B6); white, heterozygous. (B) Sequencing of Reln cDNA identified a mutation at the exon–intron junction of exon 63. An alignment of Reln cDNA sequence from the mutant (middle) against the sequences of reference cDNA (top) and genomic DNA (bottom) is shown. Between exon 63 and 64, an insertion of intronic sequence was found. Predicted protein sequence from the mutant transcript is shown. (C) Reverse transcription-polymerase chain reaction analysis shows that mutants do not express wild-type transcripts. Mutant transcripts (254 bp) are larger than wild-type transcripts (212 bp). (D) Western blot analysis using anti-reelin antibodies shows that reelin is expressed in the mutant cortex. Arrow indicates full-length reelin. GAPDH was used as loading controls.
To confirm causality of the RelnCTRdel mutation for the lamination defect, a complementation assay was done by crossing it with mice carrying the spontaneous reeler mutation Relnrl (Fig. 4). The compound heterozygote RelnCTRdel/Relnrl has a cortical patterning defect, demonstrating that these mutations are allelic. The cortical lamination phenotype in the homozygous RelnCTRdel mutant appears less severe compared with homozygous Reln rl. In the latter, Rgs4-lacZ reporter expression is equally distributed from the VZ to the MZ, while in the homozygous RelnCTRdel cortex, layers I and VI were still distinguishable. The compound heterozygote RelnCTRdel/Relnrl also displayed a less severe lamination defect similar to the homozygous RelnCTRdel. Surprisingly, both homozygous RelnCTRdel mutants or compound heterozygotes did not show cerebellar malformation (Fig. 4), and did not have ataxia. RelnCTRdel is the first Reln mutant with cortical lamination defect and no cerebellar malformation; all previously reported Reln mutants have cerebellar malformation and ataxia (Falconer 1951; Flaherty et al. 1992; Miao et al. 1994; Hirotsune et al. 1995; Takahara et al. 1996; Royaux et al. 1997; Andersen et al. 2002). This result implies that the CTR domain of reelin is required for proper positioning of cortical neurons but not necessary for the migration of cerebellar neurons. A more detailed analysis of this mutant is underway.
Two additional lines with lamination defects were discovered using Rgs4-lacZ reporter expression screening. Line 23a displayed weak reporter expression and superficial layers were thinner and stained more weakly than deep layers (Fig. 2C). The phenotype was heritable, but with incomplete penetrance. New phenotypes appeared in later generations, including enlarged lateral ventricles (without severe thinning of cortical layers) and hydrocephaly, which were detected more frequently than the lamination defect. As some mice with ventriculomegaly showed normally patterned reporter expression, it is not yet clear if lamination defects and ventriculomegaly are caused by the same mutation.
Line 33 also displayed weak reporter expression and superficial layers were thinner and stained more weakly than deep layers (Fig. 2D). Some mutant brains had smaller olfactory bulb and narrow anterior cortex (data not shown). The lamination defect and abnormal brain morphology phenotype were heritable, but were not seen in Mendelian ratios. Similar to line 23a, ventriculomegaly and hydrocephaly appeared in later generations. The relationship between the lamination defect, small olfactory bulb phenotype, and ventriculomegaly are not clear and genetic mapping analysis is presently inconclusive. To confirm that abnormal reporter patterning seen in line 33 is truly related to defects in cortical layer formation, IHC using layer-specific markers was performed (Fig. 5). In a presumptive mutant obtained from a cross between proven heterozygous parents, the formation of layers II–IV is disturbed as demonstrated by the observation that the Cux1-positive neuronal population is significantly decreased (Fig. 5A). It appears that neurons expressing Cux1 most highly in layer IV are not present in the mutant. Even in the remaining Cux1-positive neurons, the intensity of staining is less than in wild type. Also, the Ctip2-positive neuronal population in layer VI, below the most brightly stained layer V, appears reduced (Fig. 5B). Nuclear stain using Hoechst also confirmed loss of cells in layer IV (Fig. 5C). It is not clear whether the cell number and density were significantly decreased in mutant layer VI, despite of loss of Ctip2 expression. In this presumptive mutant, the cortex was thinner than the wild-type littermates, likely reflecting the absence of a neuronal population. These observations imply that Rgs4-lacZ may be a useful reporter for the detection of mild neurogenesis defects or even cell-type specification defects, not only neuronal migration defects.
Figure 5.
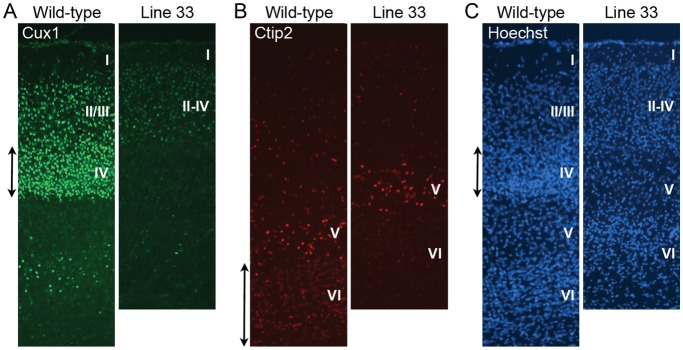
Layer-specific marker analysis of line 33 mutant at P7. (A) Immunohistochemistry using a marker for the superficial layers II–IV, Cux1, is shown. The mutant shows less strong staining and layer IV neurons that are most brightly stained in wild type (double-headed arrow) are missing. (B) Deep layer neurons are visualized using layer V/VI maker Ctip2. There are some layer VI neurons lightly stained with this marker in the wild type (double-headed arrow), and few Ctip2-positive neurons are detected in this layer in the mutant. (C) Nuclear stain using Hoechst is shown. Cortical layers (I–VI) are labeled.
Ventriculomegaly and Hydrocephaly Mutants Additionally Discovered from the Postnatal Brain Screening
Several additional mutant lines with ventriculomegaly (lines 42, 55, and 67) were discovered in the P21 screening using the Rgs4-lacZ reporter (Fig. 6). The severity of the phenotype varied between mutants even from the same line. Some of these mutants showed domed heads, severe cortical thinning, and postnatal lethality during the third week, while other mutants had moderate ventriculomegaly that was undetectable at P21 prior to brain dissection. Even in mutants with moderate ventriculomegaly, the hippocampal structure was significantly disturbed as the mutants had a smaller, posteriorly displaced hippocampus (Fig. 6). When able to be assayed, cortical lamination was unaffected (e.g., Fig. 6B).
Figure 6.
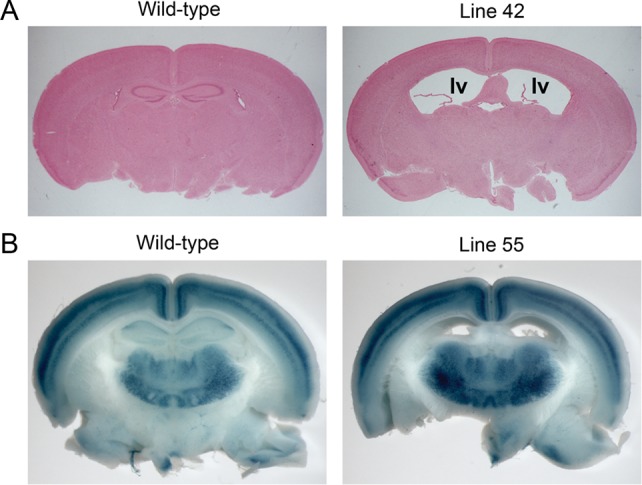
Ventriculomegaly mutants were identified during the screening using a Rgs4-lacZ reporter. (A) Histology of P21 brains from line 42. “lv” indicates lateral ventricles. (B) Whole-mount X-gal stained brains from line 55. Mutant brain showed ventriculomegaly with normal lamination.
Mutants with Abnormal Cortical Axons Identified Using TAG1-tau-LacZ
To identify mutants with defects in corticofugal axon tract formation and patterning, 32 lines were screened at E18.5 using the TAG1-tau-lacZ reporter (Table 1). Abnormal axon development was detected in 4 lines (Fig. 7). Line 7 displayed aberrant LacZ reporter patterning in the striatum; corticofugal axon tracts are not parallel to the angle of section as in control mice, and it seemed as if the cross section of the axon tract appears in the ventral striatum (Fig. 7A). The axon defects of line 7 mutants also could be visualized histologically in mutants that are not carrying the LacZ reporter; the internal capsule was absent in the mutant (Fig. 8A), and the anterior commissure was also absent (Fig. 8B left and middle). In the anterior sections, the cross-sectioned internal capsule is found absent (Fig. 8B right). A complete or partial midline crossing defect in the corpus callosum (Probst bundle) was also occasionally observed but not in all mutants (Fig. 8C). In addition, the lateral ventricles in the mutant brains appeared larger than the control. Genetic mapping using a whole-genome SNP analysis located the mutation on chromosome 14 between 97 and 108 Mb. Phr1 (Mycbp2) was identified as a candidate gene based on the report that knockout mice have the same phenotype (Bloom et al. 2007). Sequence analysis of Phr1 identified a nonsense mutation in exon 69 on chromosome 14 at 103 538 585 bp (C → T, Arg3936Stop). While the axon defect of line 7 is very similar to the characterized knock-out mutants, an abnormal VZ/SVZ was also observed, which has not been previously reported. In the mutant cortex, the axons in the white matter were unusually bundled (Fig. 8D left and middle) and disturbed the VZ/SVZ, dispersing cells in the SVZ and creating a thinner-looking layer of VZ (Fig. 8D right). The mutant cortex was thinner than wild type, and this may partially contribute to the larger ventricular size.
Figure 7.

Mutants with corticofugal axon defects discovered using a TAG1-tau-lacZ reporter. Cross sections of whole-mount X-gal stained E18.5 brains and high-magnification images of the axon tracts are shown. (A) Wild-type and mutant brains from line 7 (Phr1Arg3936Stop) were sectioned in an angled plane. The internal capsule (IC) in the wild-type brain is indicated (arrowheads). In the mutant striatum, aberrant axons are apparent. (B) Wild-type and mutant brains from line 27 (Lrp2Cys4032Ser) were sectioned coronally. In the wild-type brain, corticofugal axon tracts extend toward the thalamus (Th). In the mutant brain, the direction of axon tracts is different from wild type (arrowheads). (C) Wild-type and mutant brains from line 48 were sectioned in an angled plane. High-magnification image of the striatum (Str) shows fewer and misguided corticofugal axons. (D) Wild-type and mutant brains from line 61 were sectioned coronally at E17. In the mutant striatum (Str), fewer axons with aberrant patterning are observed.
Figure 8.

Histology of wild-type and mutant E18.5 brains from line 7 (Phr1Arg3936Stop). (A) The internal capsule (IC, arrowheads in the wild-type images) is absent in the striatum (Str) of the mutant. Enlarged images of striatum are shown on the right. (B, left and middle) Anterior commissure (arrows) is absent in the mutant brain. Instead, cross sections of the aberrant axon tracts (arrowheads) are seen. (B, right) The boxed region in the striatum in B (left) is enlarged and shown. In the wild type, the round-shaped cross sections of the axon tracts are seen, while they are absent in the mutant. (C) Midline-crossing defect of corpus callosum (CC, arrow) is seen in the mutant (Pb; Probst bundle, arrow). (D) Abnormally bundled axon tracts (double-headed arrow) are observed in the mutant cortex. This disturbed the ventricular zone (VZ) and made it thinner in the mutant compared with the wild type. Enlarged images of the VZ are shown on the right.
Line 27 was initially identified by examination of TAG1-tau-lacZ reporter expression; the corticothalamic axon tracts failed to turn toward the thalamus and projected ventrally (Fig. 7B). This line also had variable expression of craniofacial abnormalities, including cleft palate, abnormal snout shape, abnormal eye positioning, and anopthalmia (see Supplementary Fig. 2). Gross morphological defects of the brain, including smaller or single olfactory bulb and abnormal interhemispheric fissure, were often observed. Histological analysis showed the partial or complete absence of the septal nuclei, incompletely divided forebrain hemispheres, fused lateral ventricles, and abnormally developed nasal septum (see Supplementary Fig. 2B and data not shown). Overall, the phenotypes were consistent with holoprosencephaly. Histological analysis of the mutant brain revealed the midline crossing defects in commissural axons; the presence of aberrant anterior commissure in the undivided anterior forebrain (Fig. 9A), midline crossing defects in anterior commissure (Fig. 9B) and corpus callosum (Fig. 9C), and occasionally, the absence of the hippocampal commissure were observed (Fig. 9C). Mutants shown in Figure 9 are not carrying TAG1-tau-lacZ; therefore, the axon defects seen in line 27 mutants are not due to a genetic interaction between the ENU-induced mutation and the reporter locus. After initially mapping the mutation to chromosome 2 using SNP genotyping, whole-genome resequencing was performed for further mapping and cloning; this analysis has been reported elsewhere (Leshchiner et al. 2012). The whole-genome sequencing resulted in the discovery of an A/T transversion in Lrp2 exon 65 on chromosome 2 at 69 280 866 bp, which introduced a missense mutation in a highly conserved amino acid (Cys4032Ser) within LDL-receptor class B domain (Fig. 9D). It has been shown that Lrp2 deficiency causes holoprosencephaly (Willnow et al. 1996), resulting from early patterning defects in the ventral telencephalon and diencephalon (Spoelgen et al. 2009; Christ et al. 2012). However, the only axon defect reported previously was the absence of the corpus callosum (Willnow et al. 1996). Prolapse of choroid plexus or exencephaly, reported in other Lrp2 mutants (Zarbalis et al. 2004), has not been observed in line 27 mutants at E18.5.
Figure 9.

Abnormal midline crossing of commissural axons in line 27 (Lrp2Cys4032Ser) brain. Histology of the wild-type and mutant E18.5 brains from line 27. (A) In a mutant brain with fused cortical hemispheres, anterior commissure is often observed in abnormal planes of section. The aberrant anterior commissure displayed a discontinuous pattern (arrowheads). (B) An example of a mutant brain with a midline-crossing defect (arrowheads) in the anterior commissure (AC). (C) A mutant brain with a midline crossing defect in the corpus callosum (CC). High-magnification images of the corpus callosum are shown on the right. In addition, the hippocampal commissure (HC) is absent, and hyperplasia of the choroid plexus is evident. (D) Image capture of the basewise conservation and multiz alignment tracks from UCSC genome browser. A cysteine (Cys) residue, which changes to serine (Ser) by the missense mutation at 69 280 866 bp (T → A), is conserved between species.
Another mutant with abnormalities of TAG1-tau-lacZ reporter expression is line 48, which displayed weak staining and misguided axon tracts in the striatum (Fig. 7C). In some brains analyzed, markedly fewer but normally guided axon tracts were observed. Other than the axon defect, the mutant brains appeared normal. Embryos with more severe phenotypes were used for SNP genotyping, but the map position of line 48 mutation remains ambiguous.
A fourth mutant discovered by examination of TAG1-tau-lacZ reporter expression, line 61, showed weak LacZ staining with abnormal patterning (Fig. 7D). Line 61 mutants also displayed ventriculomegaly and thin cortex at E18.5, which makes them readily distinguishable without LacZ reporter. In summary, 4 of 32 lines assayed using the TAG1-tau-lacZ reporter displayed axon guidance defects, and 2 of these defects would not have been identified without inclusion of the reporter.
Mutants with Other Neurodevelopmental Defects: Cobblestone-Like Cortical Malformation, Microcephaly, and Neural Tube Closure Defects
While screening for axon guidance mutants at E18.5, 6 lines with various neurodevelopmental defects were also discovered. Line 8 mutants featured variably smaller embryo size, omphalocele, abnormal limb morphology, abnormal craniofacial morphology, and cleft palate (Fig. 10). Histological analysis revealed that mutants from this line had variable expression of cobblestone-like cortical malformation in the cerebral cortex (Fig. 10) together with occasional Probst bundle formation in the corpus callosum (data not shown). Despite the variable phenotype, genetic mapping analysis located a region of A/J homozygosity on chromosome 9 in all mutants tested. There is no known gene in the mapped region that is causal for cobblestone-like cortical malformation, and the mutation in line 8 is very likely to be novel.
Figure 10.
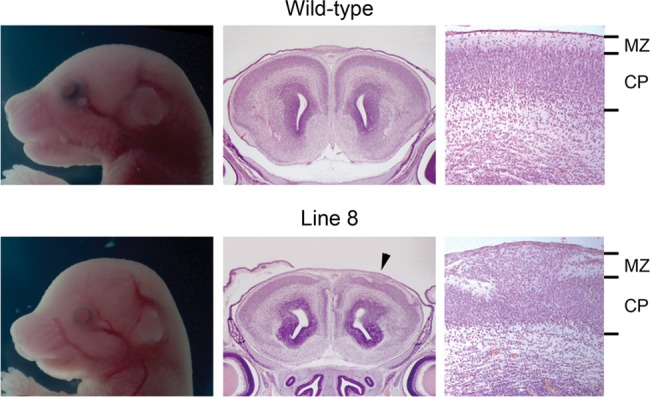
Cranial morphology and histology of the wild-type and mutant E18.5 brains from line 8. Histological analysis of the anterior cerebral cortex revealed the disorganization of cortical plate (CP) and invasion of the marginal zone (MZ), a phenotype called as a cobblestone-like cortical malformation. The arrowhead indicates where the high-magnification image is taken.
Line 16 was initially identified due to their smaller embryo size, digit abnormalities, and frequent resorptions (early embryonic lethality) (see Supplementary Fig. 3). In some mutants that survived until E18.5, disproportionately smaller cortex (microcephaly) was observed (see Supplementary Fig. 3). The abnormalities in this line have been found to be heritable, although a map position has not been determined. This line was maintained until G5 generation by pedigree test and was lost due to poor mating.
Multiple mutant lines with neural tube closure defects were discovered. Mutants from line 49, 50, and 68 showed incomplete neural tube closure, including spina bifida and exencephaly, or craniorachischisis (see Supplementary Fig. 4C,D). The G1 males 49 and 50 were obtained from the same G0 father and their mutations were both mapped to a region of chromosome 6. This region includes the gene Lrp6, and mice carrying a targeted disruption or spontaneous mutation of Lrp6 have a similar phenotype (Pinson et al. 2000; Kokubu et al. 2004; Carter et al. 2005; Zhou et al. 2010). Sequencing analysis revealed that both line 49 and 50 carry a mutation in Lrp6 exon 2 on chromosome 6 at 134 491 809 bp (T → A) that results in a missense change of a highly conserved amino acid (Trp104Arg).
Line 68 also featured mice with neural tube defects (see Supplementary Fig. 4D). Genetic mapping localized the mutation to a 117–143 Mb interval on chromosome 3. This region contains Sec24b, and an ENU-induced mutation in Sec24b causing a similar phenotype has been previously reported (Merte et al. 2010). Sequence analysis of Sec24b revealed a nonsense mutation (Tyr84Stop) in exon 2 on chromosome 3 at 129 744 214 bp (T → A).
Another line with a neural tube anomaly (line 44) showed distinct phenotypes from the other open neural tube mutants identified from this screening. Spina bifida in this mutant was limited to a small opening on the back or neck, and the exposed vertebrae appeared grossly normal (Supplementary Fig. 4B). In addition to spina bifida, omphalocele, limb defects, and oligodactyly were observed, as well as encephalocele, which appeared as a small protrusion of the brain in the center of the head (see Supplementary Fig. 4B).
Discussion
The efficiency of mouse ENU mutagenesis screens focusing on neural development has been well demonstrated in previous studies performed by us and others (Zarbalis et al. 2004; Mar et al. 2005; Lewcock et al. 2007; Wong et al. 2009; Merte et al. 2010; Dwyer et al. 2011; Stottmann et al. 2011). Notably, screens using LacZ reporters have accelerated the identification of loci that have effects on specific neuroanatomic structures (Zarbalis et al. 2004; Dwyer et al. 2011). In this study, we used Rgs4-lacZ, a reporter that allowed us to query the development of cortical lamination in a manner that has not been previously described. Moreover, this is the first ENU screen designed to detect patterning defects in the adult brain. Given that most mutants identified from embryonic screens show perinatal lethality, this postnatal screen is novel in that it enables analyses of functional and behavioral consequences of the mutation in adult mice. These mutations are perhaps superior models of the human patients who survive birth and suffer the consequences of neurological defects. We also used TAG1-tau-lacZ reporter to specifically interrogate the development of corticofugal axon tracts. Overall, 59 lines were screened and 16 with neurodevelopmental phenotypes and 2 other developmental phenotypes were found. Seven mutant lines (12% of the lines screened) with reporter patterning defects were identified. Of note, 4 of these 7 lines would not have been identifiable without the use of the LacZ reporter. The use of reporters for the screening added some inefficiency to the screening, as the reporter locus will not be carried by all progeny. However, we found that this did not significantly increase the effort required for the initial identification of the mutant lines, as examination of 4 G3 litters was productive, which is similar to screening regimens without reporter transgenes (Stottmann et al. 2011). Furthermore, the continuing development of tools for mapping and cloning, such as the whole-genome sequencing analysis we applied for the identification of the mutation in Lrp2 (Leshchiner et al. 2012), offsets any inefficiency in mutant generation.
The mutations we have identified to date include a number of re-mutations of known neurodevelopmental genes; of the 12 mapped mutants, the 6 in which causal genes have been identified are re-mutations. This is in large part a function of ascertainment bias, as the causal genes were recognized as candidates because there are existing mutants with similar phenotypes. While perhaps less striking than identification of a novel gene, re-mutations often provide valuable insight as hypomorphic or neomorphic alleles of known genes. One example is our discovery of RelnCTRdel, a Reln mutant without ataxia; this is the first report of hypomorphic allele of Reln without cerebellar malformation and impaired movement. The RelnCTRdel phenotype suggests that defects in Reln function may differentially affect neuronal migration in a brain region-specific manner. A previous in vitro study demonstrated that the CTR domain is important for reelin function (Nakano et al. 2007), and our observation of RelnCTRdel phenotype provides the first in vivo evidence supporting this idea. A more detailed characterization of RelnCTRdel is in progress, which will provide insight into the role of the CTR domain in reelin function. Further, this mutant will serve as means to study the role of Reln in learning, memory, and aging, which are difficult to analyze using spontaneous reeler mutants due to their movement disorder and reduced viability. In addition, we reported previously unappreciated phenotypes in Phr1 and Lrp2 mutants, and this provided clues that may reveal additional functions of the genes. In line 7 (Phr1Arg3936Stop), abnormal VZ morphology was observed. In line 27 (Lrp2Cys4032Ser), midline axon guidance defects in both corpus callosum and anterior commissure were observed.
Importantly, this screen was intended to specifically focus on two processes that are perturbed in human brain malformation disorders; namely, cortical lamination and corticofugal axon guidance. Our discovery of mutations in Reln, Phr1, and Lrp2 demonstrated that this strategy is very useful for identification of genes related to human brain disorders. Reln is associated with human neurodevelopmental disorders, including lissencephaly, schizophrenia, and autism (Impagnatiello et al. 1998; Hong et al. 2000; Fatemi et al. 2002; Skaar et al. 2005). Axon misguidance phenotypes in Phr1 mutant brain are similar to Dcx/Dclk-deficient mice, and a related corticofugal axon guidance phenotype was observed in the subcortical band of postmortem human brain with double cortex syndrome (Deuel et al. 2006). Lrp2 is a causal gene for Donnai-Barrow syndrome and facio-oculo-acoustico-renal syndrome in human, and craniofacial and brain phenotypes in patients, notably corpus callosum defect, are comparable to our ENU-induced mutant (Kantarci et al. 2007 2008). SNP variants in Lrp2 have recently been associated with autism in humans (Ionita-Laza et al. 2012). In addition to lamination and axon guidance defects, the other phenotypes identified during this screening, such as cobblestone-like cortical malformation, agenesis of corpus callosum and anterior commissure, and microcephaly, are also relevant to human disorders (Mochida and Walsh 2004; Nugent et al. 2012).
Recent developments in sequencing technology have been extraordinarily useful for cloning ENU-induced mutations (Fairfield et al. 2011; Leshchiner et al. 2012). We utilized the whole-genome sequencing and SNPtrack (http://genetics.bwh.harvard.edu/snptrack/) for both mapping and identifying the causal variant of line 27 (Leshchiner et al. 2012). This one-step mapping and cloning strategy greatly reduced a burden of positional cloning. Pooled DNA from 9 affected mice were more than sufficient for mapping and identification of a missense mutation in Lrp2. Increasing availability and affordability of next-gen sequencing technology and development of variant analysis tools will make mouse forward genetics even more practical.
Our previous and current ENU mutagenesis screens have demonstrated that forward genetic screen in mice is a sensitive means to query neurodevelopment. With the growing repository of null alleles available in the mouse (Guan et al. 2010), ENU-induced mutagenesis efforts may increasingly implicate known genes rather than identify genes for which no previous allele has been reported. However, many null mutants will have early developmental defects and gross morphological defects that will preclude evaluation of cortical patterning. ENU mutagenesis will continue to be advantageous to discover gene function in previously unappreciated contexts, especially when used to query specific biological processes or focus on phenotypes resembling human disorders. Unbiased mutagenesis analysis remains a powerful complementary approach for investigation of the patterning of the developing nervous system.
Supplementary Material
Supplementary material can be found at: http://www.cercor.oxfordjournals.org/
Funding
This work was supported by National Institutes of Health (5R01MH081187) to D.R.B.
Supplementary Material
Notes
We thank T. Sakurai (Mount Sinai Hospital) for the TAG1-tau-lacZ mouse line. We thank M. Lun for technical assistance with animal husbandry; H. Qiu, G. Talbott, and Z. Bhaiwala with genotyping; M. Prysak, G. Talbott, and T. Lun with histology. We thank M. Johnson for consultation with immunohistochemistry. Conflict of Interest: None declared.
References
- Anderson KV. Finding the genes that direct mammalian development : ENU mutagenesis in the mouse. Trends Genet. 2000;16:99–102. doi: 10.1016/s0168-9525(99)01921-6. [DOI] [PubMed] [Google Scholar]
- Andersen TE, Finsen B, Goffinet AM, Issinger OG, Boldyreff B. A reeler mutant mouse with a new, spontaneous mutation in the reelin gene. Brain Res Mol Brain Res. 2002;105:153–156. doi: 10.1016/s0169-328x(02)00389-3. [DOI] [PubMed] [Google Scholar]
- Beckstead WA, Bjork BC, Stottmann RW, Sunyaev S, Beier DR. SNP2RFLP: a computational tool to facilitate genetic mapping using benchtop analysis of SNPs. Mamm Genome. 2008;19:687–690. doi: 10.1007/s00335-008-9149-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier DR, Herron BJ. Genetic mapping and ENU mutagenesis. Genetica. 2004;122:65–69. doi: 10.1007/s10709-004-1437-5. [DOI] [PubMed] [Google Scholar]
- Bloom AJ, Miller BR, Sanes JR, DiAntonio A. The requirement for Phr1 in CNS axon tract formation reveals the corticostriatal boundary as a choice point for cortical axons. Genes Dev. 2007;21:2593–2606. doi: 10.1101/gad.1592107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canty AJ, Murphy M. Molecular mechanisms of axon guidance in the developing corticospinal tract. Prog Neurobiol. 2008;85:214–235. doi: 10.1016/j.pneurobio.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Carter M, Chen X, Slowinska B, Minnerath S, Glickstein S, Shi L, Campagne F, Weinstein H, Ross ME. Crooked tail (Cd) model of human folate-responsive neural tube defects is mutated in Wnt coreceptor lipoprotein receptor-related protein 6. Proc Natl Acad Sci U S A. 2005;102:12843–12848. doi: 10.1073/pnas.0501963102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caviness VS., Jr The reeler mutant mouse: a genetic experiment in developing mammalian cortex. Soc Neurosci Symp. 1977;2:27–46. [Google Scholar]
- Christ A, Christa A, Kur E, Lioubinski O, Bachmann S, Willnow TE, Hammes A. LRP2 is an auxiliary SHH receptor required to condition the forebrain ventral midline for inductive signals. Dev Cell. 2012;22:268–278. doi: 10.1016/j.devcel.2011.11.023. [DOI] [PubMed] [Google Scholar]
- D'Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- Denaxa M, Kyriakopoulou K, Theodorakis K, Trichas G, Vidaki M, Takeda Y, Watanabe K, Karagogeos D. The adhesion molecule TAG-1 is required for proper migration of the superficial migratory stream in the medulla but not of cortical interneurons. Dev Biol. 2005;288:87–99. doi: 10.1016/j.ydbio.2005.09.021. [DOI] [PubMed] [Google Scholar]
- Deuel TA, Liu JS, Corbo JC, Yoo SY, Rorke-Adams LB, Walsh CA. Genetic interactions between doublecortin and doublecortin-like kinase in neuronal migration and axon outgrowth. Neuron. 2006;49:41–53. doi: 10.1016/j.neuron.2005.10.038. [DOI] [PubMed] [Google Scholar]
- Douglas RJ, Martin KA. Neuronal circuits of the neocortex. Annu Rev Neurosci. 2004;27:419–451. doi: 10.1146/annurev.neuro.27.070203.144152. [DOI] [PubMed] [Google Scholar]
- Dwyer ND, Manning DK, Moran JL, Mudbhary R, Fleming MS, Favero CB, Vock VM, O’'Leary DD, Walsh CA, Beier DR. A forward genetic screen with a thalamocortical axon reporter mouse yields novel neurodevelopment mutants and a distinct emx2 mutant phenotype. Neural Dev. 2011;6:3. doi: 10.1186/1749-8104-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert PJ, Campbell DB, Levitt P. Bacterial artificial chromosome transgenic analysis of dynamic expression patterns of regulator of G-protein signaling 4 during development. I. Cerebral cortex. Neuroscience. 2006;142:1145–1161. doi: 10.1016/j.neuroscience.2006.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairfield H, Gilbert GJ, Barter M, Corrigan RR, Curtain M, Ding Y, D'Ascenzo M, Gerhardt DJ, He C, Huang W, et al. Mutation discovery in mice by whole exome sequencing. Genome Biol. 2011;12:R86. doi: 10.1186/gb-2011-12-9-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falconer DS. Two new mutants, "trembler" and "reeler" with neurological actions in the house mouse (Mus musculus L.) J Genet. 1951;50:192–205. doi: 10.1007/BF02996215. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Stary JM, Egan EA. Reduced blood levels of reelin as a vulnerability factor in pathophysiology of autistic disorder. Cell Mol Neurobiol. 2002;22:139–152. doi: 10.1023/A:1019857620251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty L, Messer A, Russell LB, Rinchik EM. Chlorambucil-induced mutations in mice recovered in homozygotes. Proc Natl Acad Sci U S A. 1992;89:2859–2863. doi: 10.1073/pnas.89.7.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furley AJ, Morton SB, Manalo D, Karagogeos D, Dodd J, Jessell TM. The axonal glycoprotein TAG-1 is an immunoglobulin superfamily member with neurite outgrowth-promoting activity. Cell. 1990;61:157–170. doi: 10.1016/0092-8674(90)90223-2. [DOI] [PubMed] [Google Scholar]
- Grant E, Hoerder-Suabedissen A, Molnar Z. Development of the corticothalamic projections. Front Neurosci. 2012;6:53. doi: 10.3389/fnins.2012.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillet N, Pattyn A, Contet C, Kieffer BL, Goridis C, Brunet JF. Generation and characterization of Rgs4 mutant mice. Mol Cell Biol. 2005;25:4221–4228. doi: 10.1128/MCB.25.10.4221-4228.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan C, Ye C, Yang X, Gao J. A review of current large-scale mouse knockout efforts. Genesis. 2010;48:73–85. doi: 10.1002/dvg.20594. [DOI] [PubMed] [Google Scholar]
- Hatten ME, Heintz N. Large-scale genomic approaches to brain development and circuitry. Annu Rev Neurosci. 2005;28:89–108. doi: 10.1146/annurev.neuro.26.041002.131436. [DOI] [PubMed] [Google Scholar]
- Hirotsune S, Takahara T, Sasaki N, Hirose K, Yoshiki A, Ohashi T, Kusakabe M, Murakami Y, Muramatsu M, Watanabe S, et al. The reeler gene encodes a protein with an EGF-like motif expressed by pioneer neurons. Nat Genet. 1995;10:77–83. doi: 10.1038/ng0595-77. [DOI] [PubMed] [Google Scholar]
- Hong SE, Shugart YY, Huang DT, Shahwan SA, Grant PE, Hourihane JO, Martin ND, Walsh CA. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat Genet. 2000;26:93–96. doi: 10.1038/79246. [DOI] [PubMed] [Google Scholar]
- Impagnatiello F, Guidotti AR, Pesold C, Dwivedi Y, Caruncho H, Pisu MG, Uzunov DP, Smalheiser NR, Davis JM, Pandey GN, et al. A decrease of reelin expression as a putative vulnerability factor in schizophrenia. Proc Natl Acad Sci U S A. 1998;95:15718–15723. doi: 10.1073/pnas.95.26.15718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingi T, Aoki Y. Expression of RGS2, RGS4 and RGS7 in the developing postnatal brain. Eur J Neurosci. 2002;15:929–936. doi: 10.1046/j.1460-9568.2002.01925.x. [DOI] [PubMed] [Google Scholar]
- Ionita-Laza I, Makarov V, Buxbaum JD. Scan-statistic approach identifies clusters of rare disease variants in LRP2, a gene linked and associated with autism spectrum disorders, in three datasets. Am J Hum Genet. 2012;90:1002–1013. doi: 10.1016/j.ajhg.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs EC, Campagnoni C, Kampf K, Reyes SD, Kalra V, Handley V, Xie YY, Hong-Hu Y, Spreur V, Fisher RS, et al. Visualization of corticofugal projections during early cortical development in a tau-GFP-transgenic mouse. Eur J Neurosci. 2007;25:17–30. doi: 10.1111/j.1460-9568.2006.05258.x. [DOI] [PubMed] [Google Scholar]
- Justice MJ, Noveroske JK, Weber JS, Zheng B, Bradley A. Mouse ENU mutagenesis. Hum Mol Genet. 1999;8:1955–1963. doi: 10.1093/hmg/8.10.1955. [DOI] [PubMed] [Google Scholar]
- Kantarci S, Al-Gazali L, Hill RS, Donnai D, Black GC, Bieth E, Chassaing N, Lacombe D, Devriendt K, Teebi A, et al. Mutations in LRP2, which encodes the multiligand receptor megalin, cause Donnai-Barrow and facio-oculo-acoustico-renal syndromes. Nat Genet. 2007;39:957–959. doi: 10.1038/ng2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarci S, Ragge NK, Thomas NS, Robinson DO, Noonan KM, Russell MK, Donnai D, Raymond FL, Walsh CA, Donahoe PK, et al. Donnai-Barrow syndrome (DBS/FOAR) in a child with a homozygous LRP2 mutation due to complete chromosome 2 paternal isodisomy. Am J Med Genet A. 2008;146A:1842–1847. doi: 10.1002/ajmg.a.32381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuesel I. Reelin-mediated signaling in neuropsychiatric and neurodegenerative diseases. Prog Neurobiol. 2010;91:257–274. doi: 10.1016/j.pneurobio.2010.04.002. [DOI] [PubMed] [Google Scholar]
- Kokubu C, Heinzmann U, Kokubu T, Sakai N, Kubota T, Kawai M, Wahl MB, Galceran J, Grosschedl R, Ozono K, et al. Skeletal defects in ringelschwanz mutant mice reveal that Lrp6 is required for proper somitogenesis and osteogenesis. Development. 2004;131:5469–5480. doi: 10.1242/dev.01405. [DOI] [PubMed] [Google Scholar]
- Law CO, Kirby RJ, Aghamohammadzadeh S, Furley AJ. The neural adhesion molecule TAG-1 modulates responses of sensory axons to diffusible guidance signals. Development. 2008;135:2361–2371. doi: 10.1242/dev.009019. [DOI] [PubMed] [Google Scholar]
- Leshchiner I, Alexa K, Kelsey P, Adzhubei I, Austin-Tse CA, Cooney JD, Anderson H, King MJ, Stottmann RW, Garnaas MK, et al. Mutation mapping and identification by whole-genome sequencing. Genome Res. 2012;22:1541–1548. doi: 10.1101/gr.135541.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewcock JW, Genoud N, Lettieri K, Pfaff SL. The ubiquitin ligase Phr1 regulates axon outgrowth through modulation of microtubule dynamics. Neuron. 2007;56:604–620. doi: 10.1016/j.neuron.2007.09.009. [DOI] [PubMed] [Google Scholar]
- Lopez-Bendito G, Molnar Z. Thalamocortical development: how are we going to get there? Nat Rev Neurosci. 2003;4:276–289. doi: 10.1038/nrn1075. [DOI] [PubMed] [Google Scholar]
- Mar L, Rivkin E, Kim DY, Yu JY, Cordes SP. A genetic screen for mutations that affect cranial nerve development in the mouse. J Neurosci. 2005;25:11787–11795. doi: 10.1523/JNEUROSCI.3813-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merte J, Jensen D, Wright K, Sarsfield S, Wang Y, Schekman R, Ginty DD. Sec24b selectively sorts Vangl2 to regulate planar cell polarity during neural tube closure. Nat Cell Biol. 2010;12:41–46. doi: 10.1038/ncb2002. Supp 41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merte J, Wang Q, Vander Kooi CW, Sarsfield S, Leahy DJ, Kolodkin AL, Ginty DD. A forward genetic screen in mice identifies Sema3A(K108N), which binds to neuropilin-1 but cannot signal. J Neurosci. 2010;30:5767–5775. doi: 10.1523/JNEUROSCI.5061-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao GG, Smeyne RJ, D'Arcangelo G, Copeland NG, Jenkins NA, Morgan JI, Curran T. Isolation of an allele of reeler by insertional mutagenesis. Proc Natl Acad Sci U S A. 1994;91:11050–11054. doi: 10.1073/pnas.91.23.11050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida GH, Walsh CA. Genetic basis of developmental malformations of the cerebral cortex. Arch Neurol. 2004;61:637–640. doi: 10.1001/archneur.61.5.637. [DOI] [PubMed] [Google Scholar]
- Molnar Z, Garel S, Lopez-Bendito G, Maness P, Price DJ. Mechanisms controlling the guidance of thalamocortical axons through the embryonic forebrain. Eur J Neurosci. 2012;35:1573–1585. doi: 10.1111/j.1460-9568.2012.08119.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD. Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci. 2007;8:427–437. doi: 10.1038/nrn2151. [DOI] [PubMed] [Google Scholar]
- Moran JL, Bolton AD, Tran PV, Brown A, Dwyer ND, Manning DK, Bjork BC, Li C, Montgomery K, Siepka SM, et al. Utilization of a whole genome SNP panel for efficient genetic mapping in the mouse. Genome Res. 2006;16:436–440. doi: 10.1101/gr.4563306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano Y, Kohno T, Hibi T, Kohno S, Baba A, Mikoshiba K, Nakajima K, Hattori M. The extremely conserved C-terminal region of Reelin is not necessary for secretion but is required for efficient activation of downstream signaling. J Biol Chem. 2007;282:20544–20552. doi: 10.1074/jbc.M702300200. [DOI] [PubMed] [Google Scholar]
- Nugent AA, Kolpak AL, Engle EC. Human disorders of axon guidance. Curr Opin Neurobiol. 2012;22:837–843. doi: 10.1016/j.conb.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna, Pant HC, Brady RO, Martin LJ, Kulkarni AB. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci USA. 1996;93:11173–11178. doi: 10.1073/pnas.93.20.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinson KI, Brennan J, Monkley S, Avery BJ, Skarnes WC. An LDL-receptor-related protein mediates Wnt signalling in mice. Nature. 2000;407:535–538. doi: 10.1038/35035124. [DOI] [PubMed] [Google Scholar]
- Poliak S, Salomon D, Elhanany H, Sabanay H, Kiernan B, Pevny L, Stewart CL, Xu X, Chiu SY, Shrager P, et al. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J Cell Biol. 2003;162:1149–1160. doi: 10.1083/jcb.200305018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DJ, Kennedy H, Dehay C, Zhou L, Mercier M, Jossin Y, Goffinet AM, Tissir F, Blakey D, Molnar Z. The development of cortical connections. Eur J Neurosci. 2006;23:910–920. doi: 10.1111/j.1460-9568.2006.04620.x. [DOI] [PubMed] [Google Scholar]
- Rakic P. The radial edifice of cortical architecture: from neuronal silhouettes to genetic engineering. Brain Res Rev. 2007;55:204–219. doi: 10.1016/j.brainresrev.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice DS, Curran T. Role of the reelin signaling pathway in central nervous system development. Annu Rev Neurosci. 2001;24:1005–1039. doi: 10.1146/annurev.neuro.24.1.1005. [DOI] [PubMed] [Google Scholar]
- Royaux I, Bernier B, Montgomery JC, Flaherty L, Goffinet AM. Reln(rl-Alb2), an allele of reeler isolated from a chlorambucil screen, is due to an IAP insertion with exon skipping. Genomics. 1997;42:479–482. doi: 10.1006/geno.1997.4772. [DOI] [PubMed] [Google Scholar]
- Savvaki M, Panagiotaropoulos T, Stamatakis A, Sargiannidou I, Karatzioula P, Watanabe K, Stylianopoulou F, Karagogeos D, Kleopa KA. Impairment of learning and memory in TAG-1 deficient mice associated with shorter CNS internodes and disrupted juxtaparanodes. Mol Cell Neurosci. 2008;39:478–490. doi: 10.1016/j.mcn.2008.07.025. [DOI] [PubMed] [Google Scholar]
- Skaar DA, Shao Y, Haines JL, Stenger JE, Jaworski J, Martin ER, DeLong GR, Moore JH, McCauley JL, Sutcliffe JS, et al. Analysis of the RELN gene as a genetic risk factor for autism. Mol Psychiatry. 2005;10:563–571. doi: 10.1038/sj.mp.4001614. [DOI] [PubMed] [Google Scholar]
- Spoelgen R, Adams KW, Koker M, Thomas AV, Andersen OM, Hallett PJ, Bercury KK, Joyner DF, Deng M, Stoothoff WH, et al. Interaction of the apolipoprotein E receptors low density lipoprotein receptor-related protein and sorLA/LR11. Neuroscience. 2009;158:1460–1468. doi: 10.1016/j.neuroscience.2008.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stottmann RW, Beier DR. Using ENU mutagenesis for phenotype-driven analysis of the mouse. Methods Enzymol. 2010;477:329–348. doi: 10.1016/S0076-6879(10)77017-8. [DOI] [PubMed] [Google Scholar]
- Stottmann RW, Moran JL, Turbe-Doan A, Driver E, Kelley M, Beier DR. Focusing forward genetics: a tripartite ENU screen for neurodevelopmental mutations in the mouse. Genetics. 2011;188:615–624. doi: 10.1534/genetics.111.126862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stryke D, Kawamoto M, Huang CC, Johns SJ, King LA, Harper CA, Meng EC, Lee RE, Yee A, L'Italien L, et al. BayGenomics: a resource of insertional mutations in mouse embryonic stem cells. Nucleic Acids Res. 2003;31:278–281. doi: 10.1093/nar/gkg064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahara T, Ohsumi T, Kuromitsu J, Shibata K, Sasaki N, Okazaki Y, Shibata H, Sato S, Yoshiki A, Kusakabe M, et al. Dysfunction of the Orleans reeler gene arising from exon skipping due to transposition of a full-length copy of an active L1 sequence into the skipped exon. Hum Mol Genet. 1996;5:989–993. doi: 10.1093/hmg/5.7.989. [DOI] [PubMed] [Google Scholar]
- Willnow TE, Hilpert J, Armstrong SA, Rohlmann A, Hammer RE, Burns DK, Herz J. Defective forebrain development in mice lacking gp330/megalin. Proc Natl Acad Sci U S A. 1996;93:8460–8464. doi: 10.1073/pnas.93.16.8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfer DP, Henehan-Beatty A, Stoeckli ET, Sonderegger P, Lipp HP. Distribution of TAG-1/axonin-1 in fibre tracts and migratory streams of the developing mouse nervous system. J Comp Neurol. 1994;345:1–32. doi: 10.1002/cne.903450102. [DOI] [PubMed] [Google Scholar]
- Wong F, Fan L, Wells S, Hartley R, Mackenzie FE, Oyebode O, Brown R, Thomson D, Coleman MP, Blanco G, et al. Axonal and neuromuscular synaptic phenotypes in Wld(S), SOD1(G93A) and ostes mutant mice identified by fiber-optic confocal microendoscopy. Mol Cell Neurosci. 2009;42:296–307. doi: 10.1016/j.mcn.2009.08.002. [DOI] [PubMed] [Google Scholar]
- Xenaki D, Martin IB, Yoshida L, Ohyama K, Gennarini G, Grumet M, Sakurai T, Furley AJ. F3/contactin and TAG1 play antagonistic roles in the regulation of sonic hedgehog-induced cerebellar granule neuron progenitor proliferation. Development. 2011;138:519–529. doi: 10.1242/dev.051912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarbalis K, May SR, Shen Y, Ekker M, Rubenstein JL, Peterson AS. A focused and efficient genetic screening strategy in the mouse: identification of mutations that disrupt cortical development. PLoS Biol. 2004;2:E219. doi: 10.1371/journal.pbio.0020219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou CJ, Wang YZ, Yamagami T, Zhao T, Song L, Wang K. Generation of Lrp6 conditional gene-targeting mouse line for modeling and dissecting multiple birth defects/congenital anomalies. Dev Dyn. 2010;239:318–326. doi: 10.1002/dvdy.22054. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.